

Summary
Background
Myosin IIA is an essential platelet contractile protein that is regulated by phosphorylation of its regulatory light chain (MLC) on residues (Thr)18 and (Ser)19 via the myosin light chain kinase (MLCK).
Objective
This work was carried out to elucidate the mechanisms regulating MLC (Ser)19 and (Thr)18 phosphorylation and the functional consequence of each phosphorylation event in platelets.
Results
Induction of 2MeSADP-induced shape change occurs within 5 seconds along with robust phosphorylation of MLC (Ser)19 with minimal phosphorylation of MLC (Thr)18. Selective activation of G12/13 produces both slow shape change and comparably slow MLC (Thr)18 and (Ser)19 phosphorylation. Stimulation with agonists that trigger ATP secretion caused rapid MLC (Ser)19 phosphorylation while MLC (Thr)18 phosphorylation was coincident with secretion. Platelets treated with p160ROCK inhibitor Y-27632 exhibited a partial inhibition in secretion and had a substantial inhibition in MLC (Thr)18 phosphorylation without effecting MLC (Ser)19 phosphorylation. These data suggest that phosphorylation of MLC (Ser)19 is downstream of Gq/Ca2+-dependent mechanisms and sufficient for shape change, while MLC (Thr)18 phosphorylation is substantially downstream of G12/13-regulated Rho kinase pathways and necessary, probably in concert with MLC (Ser)19 phosphorylation, for full contractile activity leading to dense granule secretion. Overall, we suggest that the amplitude of the platelet contractile response is differentially regulated by a least two different signaling pathways, which lead to different phosphorylation patterns of myosin light chain, and this mechanism results in a graded response rather than a simple on/off switch.
Keywords: Platelets, Myosin, secretion, Protease-activated receptors, shape change, cytoskeletal rearrangements
Introduction
Platelets undergo a series of coordinated responses following activation, which play an important role in thrombosis and are essential for the maintenance of hemostasis [1, 2]. Physiological agonists such as collagen, thrombin, and ADP are capable of activating platelets, resulting in events including shape change, aggregation, generation of thromboxane, and secretion of granule contents [3-8]. Platelet shape change is the earliest functional response following activation with physiological agonists and is accompanied by rearrangement of the cytoskeleton [9]. Properties that may lead to cytoskeletal rearrangements such as filament assembly, surface membrane folding, and centralization of secretory granules are thought to be mediated by the phosphorylation of Myosin IIA [9-11]. The phosphorylation of myosin light chain (MLC) results in an increased development of actin-activated ATPase activity and reflects the contractile activity of actomyosin [12, 13]. In intact cells, MLC is found to be phosphorylated on residues threonine 18 (Thr)18 or serine 19 (Ser)19 [14, 15] and is regulated through an increase in calcium/calmodulin-mediated myosin light chain kinase (MLCK) activity and/or through the activation of Rho kinase, which can either directly phosphorylate MLC or phosphorylate myosin phosphatase, thereby inhibiting its activity [14, 16]. Diphosphorylation was first decribed by Ikebe and Hartshorne[17]. They showed that (Thr)18 phosphorylation occurs in smooth muscle more slowly than at MLC (Ser)19. They concluded that phosphorylation at MLC (Thr)18 markedly increases the actin-activated ATPase activity of myosin over singly phosphorylated myosin. Reports from Kiss et. al. [18] suggest other platelet cytoskeletal kinases such as zipper-interacting protein kinase (ZIPK) and integrin-linked kinase (ILK) may contribute to the regulation and balance of kinase and phosphatase activity leading to MLC phosphorylation.
It has been previously reported that the phosphorylation of myosin light chains are important for triggering platelet shape change [9]. Studies from our laboratory have demonstrated that platelet shape change is mediated by MLC phosphorylation in a Ca2+-dependent and Ca2+-independent manner through Gq and RhoA pathways respectively [19]. The contribution of these signaling pathways in the regulation of MLC phosphorylation has been investigated with the use of pharmacological inhibitors 5,5′-dimethyl-BAPTA (a calcium chelator), YM-254890 (a Gq inhibitor [20-22], which prevents signaling leading to Ca2+ mobilization), or Y-27632 (a selective inhibitor of P160ROCK [23]).
Rho A signaling has been shown to cause MLC phosphorylation and this event is important for platelet internal contraction [24]. Suzuki et. al. [16] demonstrated that a reduction in ATP secretion as well as MLC phosphorylation occurred in platelets pre-treated with Y-27632, and activated with the thromboxane analog, STA2 (1 μM) or thrombin (0.05 U/mL), implicating a contribution of Rho kinase in ATP release. We have also shown PAR-mediated dense granule release is inhibited upon blockade of Rho A signaling [25] suggesting Rho A or downstream effectors may contribute to dense granule release.
In the present study, we examined the mechanism and the role of phosphorylation of MLC residues (Thr)18 and (Ser)19 in platelet function. The results demonstrate that the phosphorylation of MLC (Ser)19 occurs by Ca2+-dependent and Ca2+-independent signaling and correlates with platelet shape change. Phosphorylation of MLC on (Thr)18 occurs in a Ca2+-independent manner through both a G12/13 and Rho kinase mechanism. We provide evidence that the phosphorylation of MLC (Thr)18 may be important for the full release of ATP following PAR 1 activation.
Materials and methods
Reagents
Apyrase (Type VII), 2MeSADP, α-thrombin, and acetylsalicylic acid (ASA), and indomethacin were obtained from Sigma (St. Louis, MO). Hexapeptide SFLLRN was obtained from New England Peptide (Gardner, MA). AYPGKF was from GenScript Corp. (Piscataway, NJ). Serotonin was from ACROS Organics (Morris Plains, NJ). Convulxin was purified according to the method of Polgar et. al. [26]. DuPONT Instruments luminescence biometer reagent kit was used for detection of secreted ATP. YM-254890 was a gift from Yamanouchi Pharmaceutical Co., Ltd. (Ibaraki, Japan). The calcium chelator dimethyl-BAPTA [bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid] was purchased from Biomol (Plymouth Meeting, PA). AR-C69931MX was a gift from Astra-Zeneca. ROCK inhibitor Y-27632 was from Calbiochem (San Diego, CA). Fura 2 acetoxylmethyl ester (Fura-2 AM) was from Molecular Probes (Eugene, OR, USA). Phospho-myosin light chain 2 (Ser)19 Mouse mAb and p44/42 MAPK (Erk1/2) (3A7) mouse mAb were obtained from Cell Signaling (Beverly, MA) and phospho-specific MLC (Thr)18, horseradish peroxidase (HRP)–conjugated goat anti–mouse or anti–rabbit immunoglobulin G (IgG) were from Santa Cruz (Santa Cruz, CA). Millipore Immobilon Western Chemiluminescent HRP substrate and PVDF membrane was used for all immunoblotting (Billerica, MA).
Isolation of human platelets
Blood was collected from informed healthy volunteers into one-sixth volume of acid/citrate/dextrose (2.5 g sodium citrate, 2 g glucose, 1.5 g citric acid in 100 ml deionized water). Platelet-rich plasma was obtained by centrifugation at 250g for 15 minutes at ambient temperature and incubated with 1 mM aspirin for 30 minutes at 37°C. Platelets were isolated from plasma by centrifugation at 800g for 10 minutes at ambient temperature and resuspended in Tyrode buffer (138 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 0.42 mM NaH2PO4, 5 mM glucose, 10 mM HEPES [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4], and .2 units/mL apyrase). The platelet count was adjusted to 2 × 108/mL. Approval was obtained from the institutional review board of Temple University for these studies. Informed consent was provided prior to blood donation.
Measurement of platelet secretion
Platelet secretion was determined by measuring the release of ATP as previously described [25] using the Dupont Instruments luminescence biometer reagent. In experiments where inhibitors were used, the platelet sample was incubated with the inhibitors for 5 min at 37°C prior to the addition of agonists. The secretion was subsequently measured as described above.
Aggregometry
Aggregation of 0.5 ml of washed platelets was analyzed using a lumi-aggregometer (Chrono-log Corp., Havertown, PA). Aggregation was measured using light transmission under stirring conditions (900 rpm) at 37°C. Each sample was allowed to aggregate for the indicated time. The chart recorder (Kipp and Zonen, Bohemia, NY) was set for 0.2 mm/s.
Measurement of intracellular Ca2+ mobilization
PRP was incubated with the fluorescent calcium indicator Fura-2 AM (5 μm) and aspirin (1 mm), fluorescence was measured and the Ca2+ concentration was calculated as previously described using Triton X-100 (0.1%) instead of digitonin for standardization [27].
Western blot analysis
Platelets were stimulated with agonists in the presence of inhibitors or vehicle for the appropriate time under stirring conditions at 37° C and the reaction was stopped by the addition of 0.6 N HClO4. The resulting acid precipitate was collected and chilled on ice. The pellets were centrifuged at 13,000 × g for 10 min followed first by rinsing and then resuspension in 0.5 mL of deionized water. The protein was again pelleted by centrifugation at 13,000 × g for 10 min. The protein pellets were solubilized in sample buffer containing 0.1M Tris, 2% SDS, 1% (v/v) glycerol, 0.1% bromophenol blue, and 100 mM DTT then boiled for 10 minutes. Proteins were separated by 15% SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membrane. Blots were blocked with 5% nonfat milk in TBS-T (TBS + 0.05% Tween) for 1 hour at room temperature and probed overnight at 4°C with appropriate antibodies. Blots were washed 4 times with TBS-T for 10 mins. HRP-conjugated secondary antibody was incubated for 60 minutes at room temperature. Blots were washed an additional 4 times with TBS-T and antigen-antibody complexes were detected using chemiluminescent HRP substrate. Bands were visualized on a Fuji imaging system and densities calculated with Image Gauge software (Fujifilm Medical Systems, Stamford, CT).
Measurement of Percent Myosin light chain phosphorylation
For absolute mass method measurement of myosin light chain phosphorylation, urea gel electrophoresis was used as previously described [19]. In brief, HClO4 acid precipitates were centrifuged at 13,000 × g for 10 min, washed and reconstituted in 50 μl of sample buffer containing 8 M urea, 20 mM Tris-HCl (pH 8.6), 122mM glycerin, 5mM dithiothreitol, with 0.1% bromphenol blue dye. Platelet samples where sonicated in a Branson (Shelton, CT) sonication bath. Electrophoresis was performed using 10% polyacrylamide gels containing 40% (v/v) glycerol with 3.6% polyacrylamide stacking gel containing 8 M urea. The running buffer used in the top chamber was 20 mM Tris, 122 mM glycine at pH 8.6 containing 4 mM urea. The samples were electrophoresed at 8-9 mA per gel and electrophoreses was terminated 1 h after the bromphenol blue marker had come off the bottom of the gel. Gels were either stained with GelCode blue staining reagent or transferred and probed with appropriate antibody.
Statistics
All densitometric analysis was performed using FUJI FILM Science Lab 2003, Image Gauge, Version 4.22 software. Percent MLC phosphorylation was derived first by calculation of the ratio of MLC phosphorylation to total ERK. Second, the zero time point ratio was subtracted from each stimulated sample in all experiments and the highest determination was set to 100%. KaleidaGraph 2003, Version 3.62 by Synergy software and Prism 2007, Version 5 by Graphpad software were used for statistical analysis and graphical representation.
Results
Agonist-induced phosphorylation of specific residues on MLC in human platelets
Actin-myosin interactions play a crucial role in the activation of platelets [9]. These interactions are initiated upon phosphorylation of MLC on (Ser)19 and (Thr)18 residues. Although it has been known that these two MLC residues are phosphorylated in platelets upon agonist stimulation[14], it is not known whether they are phosphorylated by the same signaling pathways. We investigated the regulation of myosin light chain phosphorylation, using the PAR 1 agonist SFLLRN in aspirin-treated platelets. The platelet lysates were then probed for phosphorylation of either MLC (Thr)18 or (Ser)19 using phospho-specific antibodies. Following the stimulation of PAR 1 receptors, platelets undergo shape change, aggregation, and secretion (Fig. 1A), while myosin light chain is phosphorylated on both (Thr)18 and (Ser)19 residues. As shown in Fig. 1B and C, phosphorylation of both residues occur in a time-dependent manner and peak within 15 seconds of the addition of agonist. We also observed a slight difference in the kinetics of phosphorylation of MLC (Thr)18 compared to (Ser)19. Most notably at 5 seconds (Thr)18 was proportionally less phosphorylated than (Ser)19. These data suggest possible differences in the regulation of these two residues of MLC.
Figure 1.
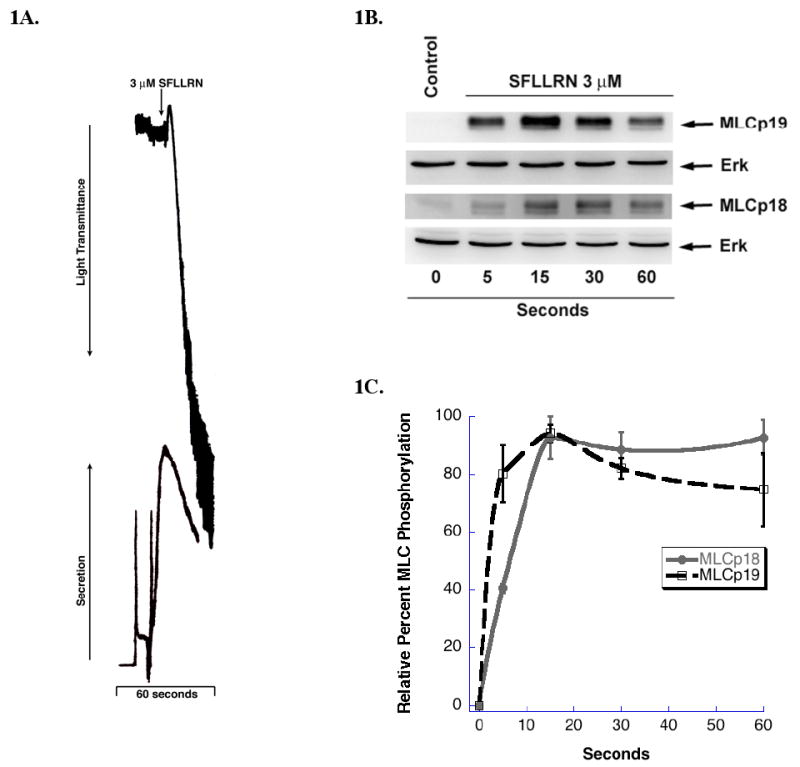
Phosphorylation of MLC by PAR 1 agonist (A) Washed and aspirin-treated human platelets where stimulated with 3 μM SFLLRN at 37° C while stirring for 60 seconds. Platelet aggregation and secretion where measured in a Lumi-aggregometer. The tracings are representative of data from at least three independent experiments. (B) SFLLRN stimulated platelets were subjected to 15% SDS-PAGE and analyzed for MLC phosphorylation by using polyclonal anti-phospho-specific (Ser) 19 or anti-phospho-specific (Thr)18 antibodies. Equal lane loading was assured by probing the samples with total monoclonal Erk antibody. (C) Densitometrical analysis of the western blot was performed. Each data point is the mean ± S.E. percentage value (n = 3) of phosphorylated MLC. The samples that make up each of these data points are derived from platelets from 3 different donors.
Gq-dependent shape change and MLC phosphorylation
Prior studies from our group correlated myosin phosphorylation with the onset of ADP-induced shape change [9]. However, it was not determined whether both residues of MLC were phosphorylated during this response since diphosphorylation of myosin had yet to be described. To evaluate Gq-mediated MLC phosphorylation, we selectively activated the Gq-coupled P2Y1 receptors with 2MeSADP in the presence of a P2Y12 receptor antagonist AR-C69931 MX. As shown in Fig. 2A, platelets activated with 2MeSADP in the presence of AR-C69931 MX undergo rapid shape change, which is completed by 10 seconds. As shown in Fig. 2B, both MLC residues are phosphorylated downstream of selective Gq activation. In each blot, a platelet sample stimulated with SFLLRN was used both as a positive control and as a measure of a full phosphorylation of each residue. Fig. 2C represents the relative percent of MLC phosphorylation of both residues following selective Gq stimulation in relationship to the maximal phosphorylation levels when normalized to the SFLLRN-stimulated sample. The onset of MLC phosphorylation strongly correlates with the timing of platelet shape change. Phosphorylation of MLC (Ser)19 peaks at about 60% of SFLLRN-induced level at about 5 sec and then rapidly declines consistent with our previous studies on total myosin phosphorylation [9, 19]. In contrast, (Thr)18 phosphorylation is only 20% of the SFLLRN level and is sustained for the full time period studied.
Figure 2.
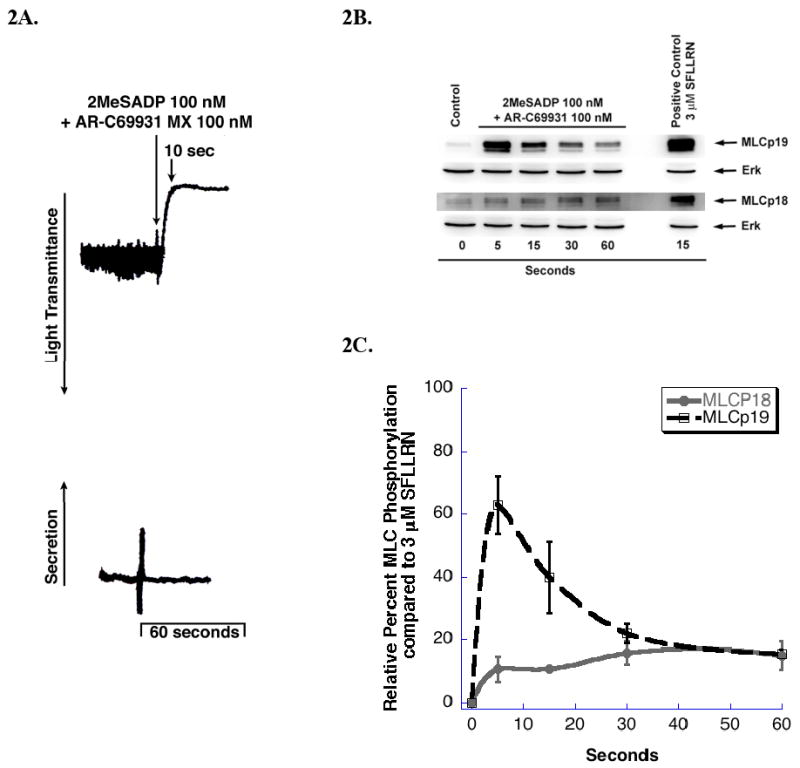
Gq-mediated phosphorylation of MLC (A) Washed and aspirin-treated human platelets where stimulated with 100 nM 2MeSADP in the presence of 100 nM AR-C69931MX. The tracings are representative of data from at least three independent experiments. (B) 2MeSADP stimulated platelets were subjected to western blot analysis as described in Fig.1. (C) Densitometry analysis of the western blots was performed. Each data point is the mean ± S.E. percentage value (n = 3) of phosphorylated MLC and the positive control 3 μM SFLLRN was set to 100% to allow for the assessment of relative amounts of ADP-stimulated Gq-mediated phosphorylation of MLC with respect to the maximal response. The samples that make up each of these data points are derived from platelets from 3 different donors.
ADP-mediated calcium-dependent and calcium –independent MLC phosphorylation
Our previous studies have shown that ADP-induced platelet shape change is mediated by both Ca2+-dependent and Ca2+–independent signaling pathways [19]. To examine whether the ADP-mediated phosphorylation of MLC (Ser)19 occurs by a Ca2+-dependent pathway, we used 5,5′–dimethyl-BAPTA, a high affinity Ca2+ chelator [28]. As shown in Fig. 3A, 2MeSADP-induced MLC (Ser)19 phosphorylation is almost totally inhibited in BAPTA-treated platelets, while the phosphorylation status of MLC (Thr)18 was minimally affected. (Fig 3B.) These results suggest that the Ca2+-dependent pathway plays an important role in the phosphorylation of MLC (Ser)19 but does not regulate the phosphorylation of MLC (Thr)18. These data suggest that this partial MLC (Thr)18 phosphorylation may be regulated by Gq-dependent RhoA activation [25, 29, 30]. Rho-kinase is capable of phosphorylating MLC directly [31]. We have shown that activation of RhoA/P160ROCK pathways downstream of Gq are important for ADP-induced Ca2+-independent shape change [19]. In order to determine whether RhoA-dependent signaling regulates (Thr)18 phosphorylation, we used the Rho kinase inhibitor Y-27632 [23]. As shown in Fig. 3A, in platelets treated with Y-27632, there small inhibition of MLC (Ser)19 phosphorylation at 5 seconds, while by 20 seconds a significant inhibition is observed. MLC (Thr)18 phosphorylation was completely inhibited by Y-27632 (Fig. 3C). Since Y-27632-treated platelets activated with 2MeSADP undergo rapid shape change (Fig. 3D), these results suggest MLC (Thr)18 phosphorylation plays little role in this response. Thus the slight increase in MLC (Thr)18 phosphorylation following stimulation with ADP is probably mediated through a P160ROCK-dependent mechanism.
Figure 3.
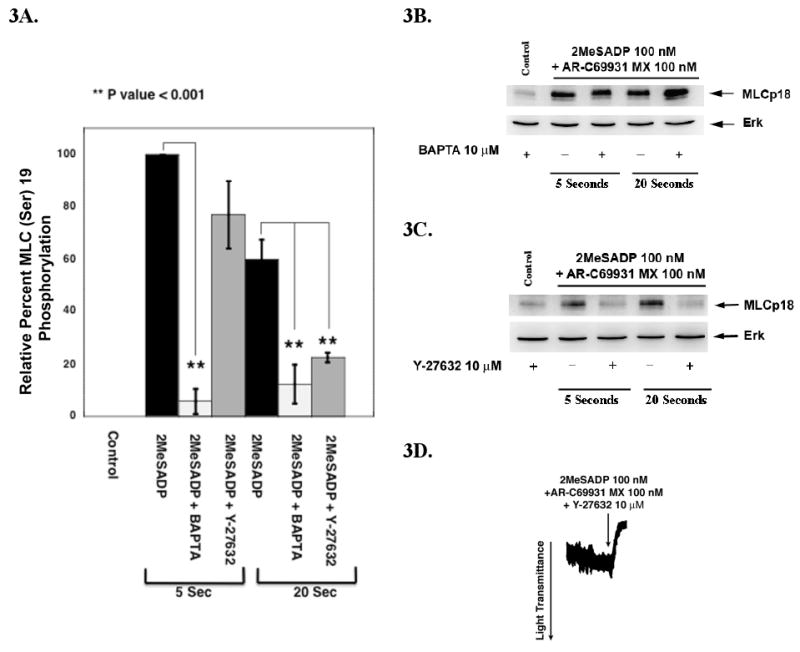
Calcium-dependent and calcium-independent phosphorylation of MLC (A) Densitometrical analysis of MLC (Ser)19 phosphorylation by 2MeSADP. Platelet samples activated with 2MeSADP in the presence of AR-C69931MX, with or without the pre-treatment with dimethyl BAPTA or Y-27632 was subjected to western blot analysis using MLC (Ser)19-phospho specific antibody and analyzed by densitometry. Data are expressed as the mean ± S.E. from three experiments where data was normalized to maximum 2MeSADP MLC (Ser)19 phosphorylation at 5 seconds (taken as 100%). (B) Platelets treated with AR-C69931MX were activated with 2MeSADP in the presence and absence of BAPTA or (C) Y-27632 for 5 and 20 second and subjected to western blot analysis using MLC (Thr)18-phophos specific antibody. (D) Representative shape change tracing of 2MeSADP stimulated platelets in the presence of AR-C69931MX and Y-27632.
Effect of G12/13 signaling on MLC phosphorylation
Since activation of Gq failed to cause moderate to high levels of MLC (Thr)18 phosphorylation, we hypothesized that that MLC (Thr)18 phosphorylation is at a great extent downstream of G12/13 signaling. We activated platelets with the PAR 1 agonist SFLLRN in the presence of YM-254890, a selective Gq inhibitor [21]. Such platelets undergo a delayed change in shape and fail to aggregate, secrete, or as shown in Fig. 4A to mobilize intracellular calcium. As shown in Fig. 4B, 4C, and 4D, YM-254890 treated platelets exhibited a significant reduction in MLC (Ser)19 phosphorylation and only a slight decrease in MLC (Thr)18 phosphorylation that is not statistically significant. Similar results were obtained when thrombin-stimulated platelets treated with BAPTA; MLC (Ser)19 phosphorylation was inhibited by about 50% while MLC (Thr)18 phosphorylation was not affected (data not shown). The contribution of G12/13 to the phosphorylation of of MLC Thr(18) was confirmed using a low concentration of U46619 (30 nM) which does not activate Gq pathways (4E)[24, 32, 33]. All these data support the hypothesis that RhoA signaling is essential for MLC (Thr)18 phosphorylation and that G12/13-dependent RhoA signaling provides the greater portion of this phosphorylation with strong agonists, while Gq-mediated RhoA activation only contributes weakly. These results also suggest that both Gq-pathways are important for contributing to significant levels of MLC (Ser)19 phosphorylation during the rapid calcium transient while RhoA pathways contribute to sustained phosphorylation (Ser)19 which can be attributed to inhibition of myosin phosphatase.
Figure 4.
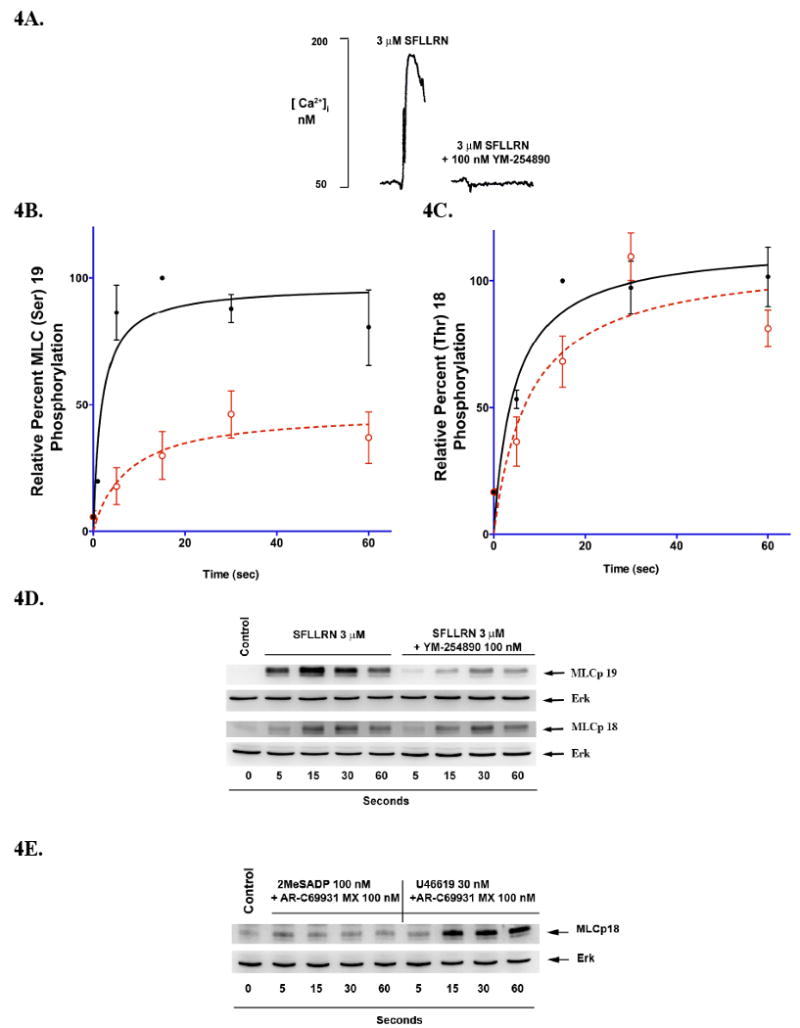
G12/13-mediated phosphorylation of MLC. Aspirin-treated platelets loaded with 5 μM Fura-2AM were activated with SFLLRN (3 μM) in the presence and absence of 100 nM YM-254890 (A). Washed platelets pre-treated with YM-254890 (dotted line) or vehicle DMSO (solid line) for 5 minutes were stimulated with SFLLRN (3 μM) and analyzed for phosphorylation of B,D) MLC (Ser)19 or C,D) MLC (Thr)18 by western blot analysis followed by densitometry. Each data point is the mean ± S.E. percentage value (n = 3) of phosphorylated MLC. The samples that make up each of these data points are derived from platelets from 3 different donors. (D) Representative western blot from platelets stimulated with (3 μM) SFLLRN in the presence and absence of YM-254890 or vehicle for various time points and probed with phospho-specific antibodies. (E) Western blot comparison of MLC (Thr)18 phosphorylation following selective Gq (2MeSADP) or G12/13 (30 nM U46619) activation.
Correlation of MLC (Thr) 18 phosphorylation with dense granule secretion
MLC diphosphorylation have been previously reported to play a role in secretion [34-36]. We correlated MLC phosphorylation levels in aspirin or indomethacin- treated platelets with agonists that either cause or fail to cause secretion. Activation of the PAR or GPVI receptors with peptides SFLLRN, AYPGKF, or thrombin, and convulxin respectively, induced platelet secretion (Fig. 5A), and moderate increase in both MLC (Ser)19 and MLC (Thr)18 phosphorylation levels were observed (Fig. 5B). In comparison, when aspirin or indomethacin-treated platelets were activated with 2MeSADP or serotonin, agonists which failed to cause secretion, MLC (Ser)19 phosphorylation levels are inhibited compared to agonists which caused secretion, and only minimal levels of MLC (Thr)18 phosphorylation were detected (Fig. 5B). As shown in Fig. 5C, when non-aspirin-treated platelets were activated with 2MesADP, these platelets underwent secretion due to the positive feedback of thromboxane on its receptor. Significant levels of MLC (Ser)19 phosphorylation occurred and significant levels of MLC (Thr)18 phosphorylation preceded the initiation of ATP release. These data suggest a threshold level of both MLC (Ser)19 and MLC (Thr)18 may be required for ATP release.
Figure 5.
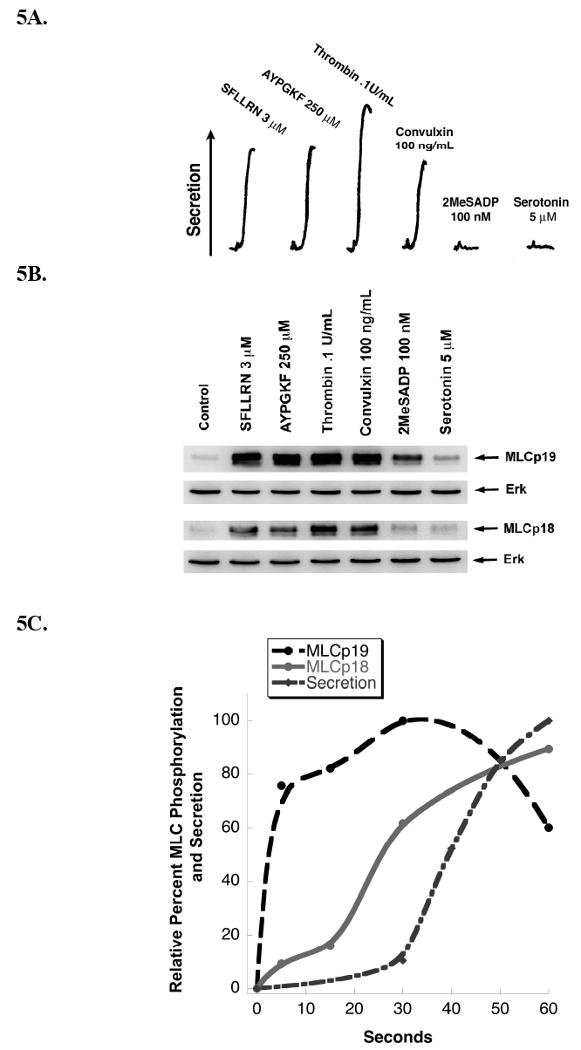
Correlation of agonist-induced MLC phosphorylation with secretion. Washed aspirin-treated platelets were stimulated with various agonists (as indicated) and analyzed for dense granule secretion (Panel A) and MLC phosphorylation (Panel B). The data are representative of at least three independent experiments. (C) Non-aspirinated platelets were activated with 100 nM 2MeSADP and analyzed for dense granule secretion and MLC phosphorylation at various time points. The data were quantitated by densitometry and presented as a correlation with dense granule secretion. Data are representative of three independent experiments.
Role of MLC (Thr)18 phosphorylation in dense granule secretion
Since MLC (Thr)18 phosphorylation occurs through a G12/13/RhoA-dependent mechanism we evaluated ATP release in the presence of a RhoA pathway inhibitor, Y-27632. ATP secretion was partially inhibited in aspirin-treated platelets activated with PAR 1 peptide SFLLRN in the presence of Y-27632 (Fig 6A). As shown in Fig. 6B, there is no significant difference in MLC (Ser)19 phosphorylation levels. However, Y-27632 treatment produces a significant inhibition in MLC (Thr)18 phosphorylation levels (Fig 6C). The inhibition of secretion is similar to the inhibition in MLC (Thr)18 phosphorylation. Y-27632 failed to completely inhibit the MLC (Thr)18 phosphorylation that occurs following PAR 1 activation, suggesting Y-27632 insensitive kinases upstream of P160ROCK may be able to phosphorylate MLC (Thr)18.
Figure 6.
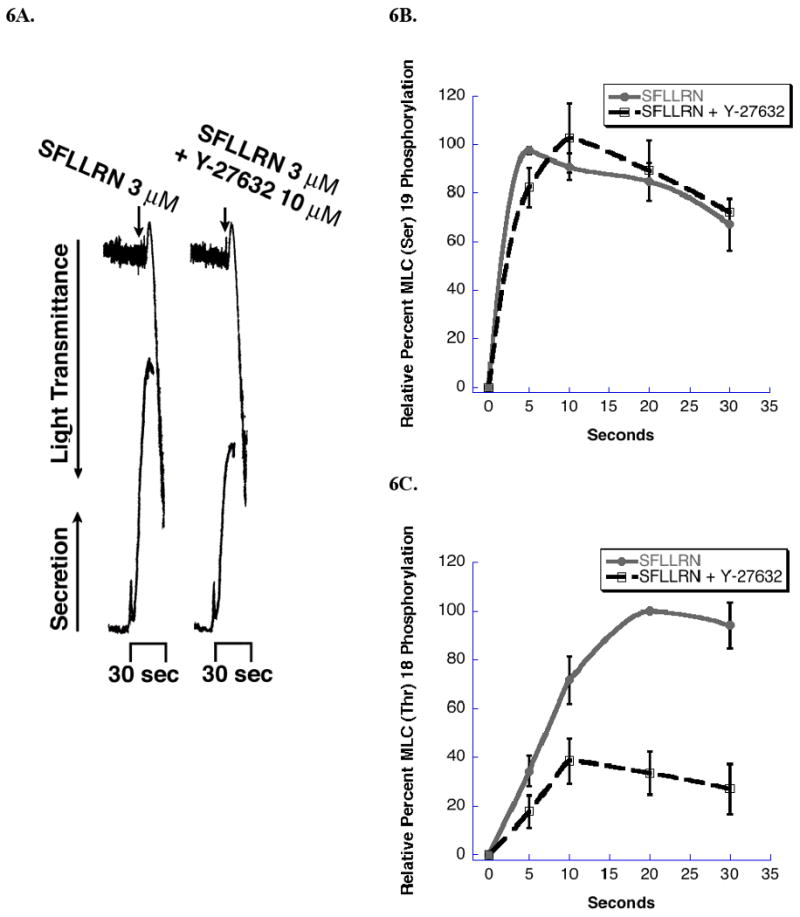
The effect of Y-27632 on dense granule secretion and MLC phosphorylation. Washed and aspirin-treated platelets were stimulated with SFLLRN in the presence of Y-27632 or vehicle and analyzed for A) aggregation and dense granule secretion, B) MLC (Ser)19 phosphorylation, and C) MLC (Thr)18 phosphorylation. Panel A is a representative of three experiments. In Panels B &C, each data point is the mean ± S.E. percentage value (n = 3) of phosphorylated MLC. The samples that make up each of these data points are derived from platelets from 3 different donors.
Effect of YM-254890 or Y-27632 on PAR1-induced Myosin Light Chain Phosphorylation
In order to quantitate the actual percentage of myosin phosphorylation that occurs on each of the two residues of MLC, we used urea gels to detect the shifts in the mobility of the phosphorylated proteins. This method was first used to Perrie et al. [37], who showed that MLC mobility increased on a native acrylamide gel when it become phosphorylated. Platelets were activated with SFLLRN and the proteins resolved on urea gels and then either stained for protein (Figure 7A), or probed with either total MLC antibodies or phospho-specific antibodies for MLC (Ser)19 or (Thr)18 (figures 7B & 7C). There was a large SFLLRN-induced increase in the percentage of MLC (Ser)19 phosphorylation and this change correlates well with the change seen both in the protein staining pattern Figure 7A (70%) and the antibody-dependent detection of total MLC (Fig 7B.1 & C.1). Using an overlay (Fig. 7C.3) of one blot probed for total MLC and the same blot reprobed for (Thr)18 phosphorylation shows a band that runs slightly faster than total phosphorylated light chain. This result indicates that phosphorylation of (Thr)18 adds a slight increase to the mobility of MLC beyond that induced by MLC (Ser)19 phosphorylation alone. Unfortunately, we were not able to detect an increase in mobility of total MLC either by protein staining or by probing for total MLC. These data suggest that only a small but important fraction of MLC becomes phosphorylated on (Thr)18. The other two lanes in each blot show the effect of the Gq inhibitor, YM-254890 or the P160ROCK inhibitor, Y-27632 on these staining patterns. In the presence of YM-254890, the amount of MLC (Ser)19 phosphorylation was dramatically inhibited (Fig. 7B) consistent with Figure 4. Two bands of MLC phosphorylated on (Thr)18 are seen in figure 7C (lane 3) a prominent band probably representing MLC phosphorylation on both (Ser)19 and (Thr)18 and a less prominent band phosphorylated on only (Thr)18. In the presence of Y-27632, (Thr)18 phosphorylation was dramatically reduced, whereas (Ser)19 phosphorylation was close to the stimulated control. Overall these experiments are consistent with the results of our other experiments.
Figure 7.
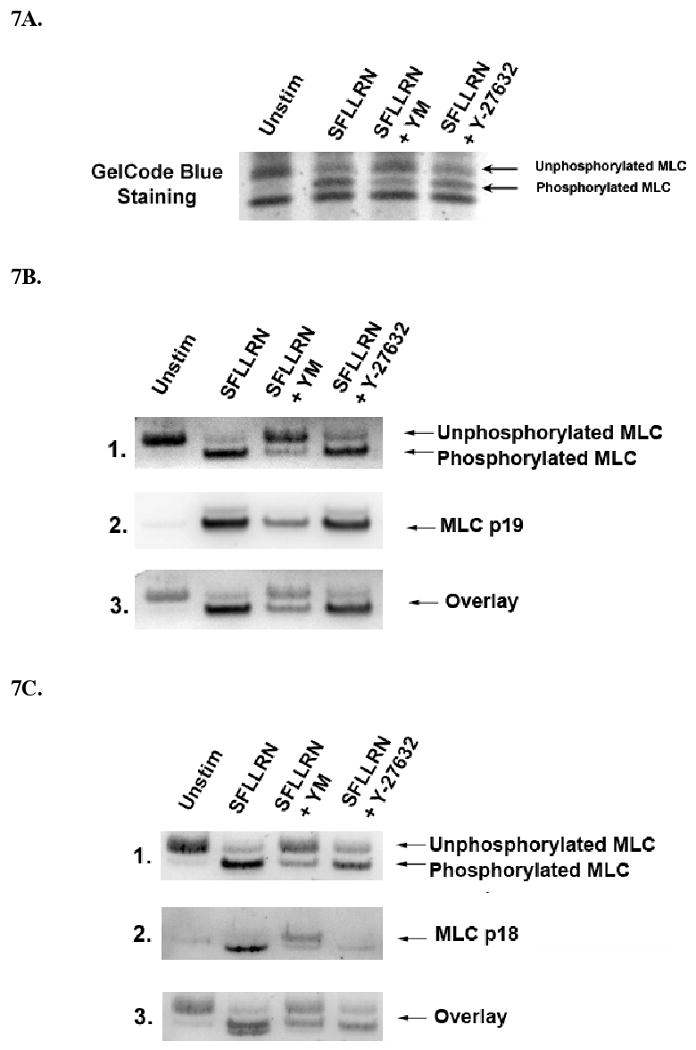
Effect of YM-254890 and Y-27632 on PAR1-induced MLC phosphorylation. (A) Washed and aspirin-treated platelets were activated with 3 μM SFLLRN for 30 seconds in the presence or absence of YM-254890 100 nM or Y-27632 10 μM. The samples were prepared and subjected to urea-PAGE as described in the methods and stained for protein using GelCode blue staining reagent. (B) Samples were subjected to urea-PAGE and transfered to PVDF membranes then probed with MLC phosphospecific antibodies to either (Ser)19 (7B.2.) or (Thr)18 (7C.2.). Blots were then stripped and reprobed for total MLC (7B.1. or 7C.1.) In Fig. (7B.3. and 7C.3.) a 50% overlay of total to phosphorylated MLC is represented.
Discussion
In the present study, we investigated the significance of diphosphorylation of myosin regulatory light chain (MLC) on the functional responses of human platelets. We also addressed the role of Gq and G12/13 mediated signaling pathways regulating agonist-dependent phosphorylation of MLC (Thr)18 and MLC (Ser)19. Previous studies in platelets focused on the gross phosphorylation of MLC without distinguishing the relative contribution of each residue. As a result, little is known about the role each distinct MLC phosphorylation plays in human platelet functional responses. We have demonstrated that when platelets are fully activated by protease-activated receptors (PARs) MLC becomes phosphorylated on both (Thr)18 and (Ser)19.
PARs couple to both heterotrimeric G proteins Gq and G12/13 [38, 39]. We have shown in this work, that activation of the PAR1 receptor under conditions where platelets change shape, aggregate, secrete dense granules [16, 32] undergo a time-dependent increase in both MLC (Ser)19 and (Thr)18 phosphorylation (Fig. 1A and 1B). The kinetics of phosphorylation of MLC (Thr)18 were slightly slower, which is consistent with the differences in kinetics of MLC (Ser)19 and (Thr)18 observed in smooth muscle [17, 40]. The difference in kinetics led us to explore the possibility that MLC (Ser)19 and (Thr)18 may be differentially regulated downstream of Gq and G12/13 pathways and play a role in different functional responses.
Stimulation of aspirinated human platelets with ADP, through P2Y1, causes activation of Gq and PLCβ2, which results in mobilization of intracellular Ca2+ and PKC activation, [41-45]. It has been shown that platelets activated with ADP under these conditions undergo phosphorylation of MLC, which coincides with shape change, suggesting its importance in the initiation of the shape change event [9]. ADP-stimulated platelet myosin phosphorylation is regulated by Ca2+-dependent MLCK activation and Rho/p160ROCK-activated inhibition of the myosin phosphatase, MYPT1, apparently through Gq-mediated mechanisms since ADP does not activate G12/13 [19, 25, 32].
Whether diphosphorylation of MLC is required or plays a role in Ca2+-dependent and –independent ADP-mediated shape change had not been investigated. We show that following activation of Gq pathways by ADP, phosphorylation of MLC (Ser)19 is rapid and robust (Fig. 2B and 2C), but the level of (Thr)18 phosphorylation was relatively weak compared to the level seen following concomitant Gq and G12/13 activation (Fig. 2C). In the presence of the p160ROCK inhibitor, Y-27632, this low level of MLC (Thr)18 phosphorylation is abolished without effecting the initial rate of shape change (Fig. 3D) [19]. These data indicate that MLC (Thr)18 phosphorylation is mediated through Rho kinase and plays little role in initiation of shape change. At 5 seconds the level of MLC (Ser)19 phosphorylation is minimally effected by Y-27632 indicating that shape change depends primarily on a p160ROCK-independent activation of MLC (Ser)19 phosphorylation (Fig. 3A). At later times, MLC (Ser)19 phosphorylation is inhibited by Y-27632, suggesting that maintenance of inactive MYPT1 is required for prolonged MLC (Ser)19 phosphorylation. These conclusions are completely consistent with previous observations [19, 32].
YM-254890 and BAPTA have been used to inhibit Gq-dependent pathways, leaving G12/13 pathways intact. PAR 1-stimulated phosphorylation of MLC (Ser)19 is dependent on both Gq and G12/13-coupled events (Fig. 4B). Since MLC (Thr)18 phosphorylation is not significantly attenuated when Gq is inhibited (Fig. 4B), the majority MLC (Thr)18 phosphorylation must be downstream of G12/13 with a small component dependent of Gq-activated RhoA and not dependent on either Ca2+ or PLCβII activation. Secretion does not occur under selective G12/13 activation because other requirements, such as intracellular Ca2+ mobilization and PKC activation, are not met. At the current time, there is no condition where we can activate platelets to solely cause MLC (Thr)18 phosphorylation independent of MLC (Ser)19 phosphorylation to test whether phosphorylation of MLC (Thr)18 alone could support platelet functional responses. This is probably due to the fact that (Thr)18 phosphorylation may require prior phosphorylation on MLC (Ser) 19 (Figure 7C) [17].
It has been shown that the diphosphorylation of MLC at (Thr)18 and (Ser)19 further increases actin-activated myosin ATPase activity over that of monophosphorylated MLC (Ser)19 [17, 40]. Furthermore in support of a conclusion of our work, diphosphorylation of MLC by MLCK and PKC was required for secretion in RBL-2H3 cells [46]. To evaluate whether MLC diphosphorylation is required for secretion, we activated platelets with strong agonists. Activation of platelet receptors with strong agonists such as thrombin and collagen are known to cause platelet dense granule release (Fig. 5A). However, weaker agonists such as ADP or serotonin are unable to secrete dense granule contents in the absence of thromboxane generation [41-44, 47] possibly due to the inability of these receptors to activate G12/13 pathways and thus provide sufficient MLC phosphorylation (Fig. 5A) [25]. We show that the strong platelet agonists thrombin, SFLLRN, AYPGKF, and convulxin caused moderate to high levels of both MLC (Ser)19 and (Thr)18 phosphorylation, while the weak agonists ADP and serotonin caused only modest levels of MLC (Ser)19 and minimal levels of (Thr)18 phosphorylation when thromboxane generation is blocked (Fig. 5B). In non-aspirinated platelets, stimulation with ADP can cause dense granule secretion due to potentiation by thromboxane A2. Activation of dense granule secretion is delayed under these conditions allowing a clear separation between shape change and onset of secretion. We find that phosphorylation of MLC (Ser)19 correlates with the initial shape change while MLC (Thr)18 phosphorylation is delayed and occurs prior to ATP secretion (Fig. 5C). Finally, when Y-27632 is used in platelets stimulated with a PAR 1 agonist, both ATP secretion and MLC (Thr)18 phosphorylation are attenuated. MLC (Ser)19 phosphorylation is not affected in spite of the fact that a significant portion of MLC (Ser)19 phosphorylation is downstream of G12/13 activation. This suggests that p160ROCK-independent pathways regulate a portion of MLC (Ser)19 and perhaps MLC (Thr)18 phosphorylation. We suggest that either ZIPK or ILK may contribute to the regulation of Gq-independent MLC phosphorylation [18]. One caveat should be noted in that all these data are correlative in nature and do not provide absolute proof of this hypothesis.
From the current study and prior studies, activation of G12/13/Rho A pathways prolongs MLC phosphorylation [48, 49] and this is necessary for irreversible aggregation [49]. Rho A activation and MLC phosphorylation have also been implicated in playing a role in ‘internal contraction’ which could be important for driving dense granule release [24, 50]. We demonstrated that under conditions where intracellular Ca2+ is mobilized and there is PKC activation, the maintenance of moderate levels of both MLC (Ser)19 and MLC (Thr)18 phosphorylation are required for full dense granule secretion. While this is the first proposal of such a mechanism in platelets, a similar conclusion were reached in the case of RBL-2H3 cells [46].
Attempts to measure absolute amounts of MLC (Thr)18 phosporylation by urea gels indicated that only low levels of (Thr)18 phosphorylation occurred under conditions where secretion ensued. This is consistent with the results of Itoh et al. [14] who directly detected low levels of diphosphorylated MLC following thrombin stimulation. We propose this is consistent with a small regulated fraction of MLC that is directly coupled to the release of dense granules.
In conclusion, we have linked MLC (Ser)19 phosphorylation to platelet shape change and have shown that MLC (Ser)19 is phosphorylated downstream of both Ca2+-dependent Gq activation and Ca2+-independent G12/13 pathways. We have also provided evidence that MLC (Thr)18 phosphorylation occurs subsequent to MLC (Ser) 19 primarily through G12/13/Rho A activation and that diphosphorylated MLC may play a role in dense granule secretion
Acknowledgments
This work is supported by HL81322, HL80444 and HL60683 from National Institutes of Health to SPK. TG is supported by NIH training grant in Thrombosis (HL07777).
Footnotes
Disclosure of Conflict of Interests: The authors declare that there is no conflict of interest.
References
- 1.Packham MA. Role of platelets in thrombosis and hemostasis. Can J Physiol Pharmacol. 1994;72:278–84. doi: 10.1139/y94-043. [DOI] [PubMed] [Google Scholar]
- 2.Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: the platelet paradigm. Blood. 1998;91:2645–57. [PubMed] [Google Scholar]
- 3.Hourani SM, Cusack NJ. Pharmacological receptors on blood platelets. Pharmacol Rev. 1991;43:243–98. [PubMed] [Google Scholar]
- 4.Mills DC. ADP receptors on platelets. Thromb Haemost. 1996;76:835–56. [PubMed] [Google Scholar]
- 5.Molino M, Bainton DF, Hoxie JA, Coughlin SR, Brass LF. Thrombin receptors on human platelets. Initial localization and subsequent redistribution during platelet activation. J Biol Chem. 1997;272:6011–7. doi: 10.1074/jbc.272.9.6011. [DOI] [PubMed] [Google Scholar]
- 6.Brass LF. More pieces of the platelet activation puzzle slide into place. J Clin Invest. 1999;104:1663–5. doi: 10.1172/JCI8944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999;96:11023–7. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coughlin SR. Protease-activated receptors in vascular biology. Thromb Haemost. 2001;86:298–307. [PubMed] [Google Scholar]
- 9.Daniel JL, Molish IR, Rigmaiden M, Stewart G. Evidence for a role of myosin phosphorylation in the initiation of the platelet shape change response. J Biol Chem. 1984;259:9826–31. [PubMed] [Google Scholar]
- 10.Siess W. Molecular mechanisms of platelet activation. Physiol Rev. 1989;69:58–178. doi: 10.1152/physrev.1989.69.1.58. [DOI] [PubMed] [Google Scholar]
- 11.Fox JE. Regulation of platelet function by the cytoskeleton. Adv Exp Med Biol. 1993;344:175–85. doi: 10.1007/978-1-4615-2994-1_13. [DOI] [PubMed] [Google Scholar]
- 12.Adelstein RS. Calmodulin and the regulation of the actin-myosin interaction in smooth muscle and nonmuscle cells. Cell. 1982;30:349–50. doi: 10.1016/0092-8674(82)90232-x. [DOI] [PubMed] [Google Scholar]
- 13.Ikebe M, Koretz J, Hartshorne DJ. Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J Biol Chem. 1988;263:6432–7. [PubMed] [Google Scholar]
- 14.Itoh K, Hara T, Shibata N. Diphosphorylation of platelet myosin by myosin light chain kinase. Biochim Biophys Acta. 1992;1133:286–92. doi: 10.1016/0167-4889(92)90049-h. [DOI] [PubMed] [Google Scholar]
- 15.Ikebe M. Phosphorylation of a second site for myosin light chain kinase on platelet myosin. Biochemistry. 1989;28:8750–5. doi: 10.1021/bi00448a011. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki Y, Yamamoto M, Wada H, Ito M, Nakano T, Sasaki Y, Narumiya S, Shiku H, Nishikawa M. Agonist-induced regulation of myosin phosphatase activity in human platelets through activation of Rho-kinase. Blood. 1999;93:3408–17. [PubMed] [Google Scholar]
- 17.Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem. 1985;260:10027–31. [PubMed] [Google Scholar]
- 18.Kiss E, Muranyi A, Csortos C, Gergely P, Ito M, Hartshorne DJ, Erdodi F. Integrin-linked kinase phosphorylates the myosin phosphatase target subunit at the inhibitory site in platelet cytoskeleton. Biochem J. 2002;365:79–87. doi: 10.1042/BJ20011295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul BZ, Daniel JL, Kunapuli SP. Platelet shape change is mediated by both calcium-dependent and -independent signaling pathways. Role of p160 Rho-associated coiled-coil-containing protein kinase in platelet shape change. J Biol Chem. 1999;274:28293–300. doi: 10.1074/jbc.274.40.28293. [DOI] [PubMed] [Google Scholar]
- 20.Taniguchi M, Nagai K, Arao N, Kawasaki T, Saito T, Moritani Y, Takasaki J, Hayashi K, Fujita S, Suzuki K, Tsukamoto S. YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J Antibiot (Tokyo) 2003;56:358–63. doi: 10.7164/antibiotics.56.358. [DOI] [PubMed] [Google Scholar]
- 21.Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, Kobori M. A novel Galphaq/11-selective inhibitor. J Biol Chem. 2004;279:47438–45. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Jin J, Kunapuli SP. Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood. 2006;107:947–54. doi: 10.1182/blood-2005-07-3040. [DOI] [PubMed] [Google Scholar]
- 23.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 24.Johnson GJ, Leis LA, Krumwiede MD, White JG. The critical role of myosin IIA in platelet internal contraction. J Thromb Haemost. 2007;5:1516–29. doi: 10.1111/j.1538-7836.2007.02611.x. [DOI] [PubMed] [Google Scholar]
- 25.Jin J, Mao Y, Thomas D, Kim S, Daniel JL, Kunapuli SP. RhoA downstream of G(q) and G(12/13) pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem Pharmacol. 2009;77:835–44. doi: 10.1016/j.bcp.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polgar J, Clemetson JM, Kehrel BE, Wiedemann M, Magnenat EM, Wells TN, Clemetson KJ. Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (tropical rattlesnake) venom via the p62/GPVI collagen receptor. J Biol Chem. 1997;272:13576–83. doi: 10.1074/jbc.272.21.13576. [DOI] [PubMed] [Google Scholar]
- 27.Daniel JL, Dangelmaier C, Strouse R, Smith JB. Collagen induces normal signal transduction in platelets deficient in CD36 (platelet glycoprotein IV) Thromb Haemost. 1994;71:353–6. [PubMed] [Google Scholar]
- 28.Jen CJ, Chen HI, Lai KC, Usami S. Changes in cytosolic calcium concentrations and cell morphology in single platelets adhered to fibrinogen-coated surface under flow. Blood. 1996;87:3775–82. [PubMed] [Google Scholar]
- 29.Gratacap MP, Payrastre B, Nieswandt B, Offermanns S. Differential regulation of Rho and Rac through heterotrimeric G-proteins and cyclic nucleotides. J Biol Chem. 2001;276:47906–13. doi: 10.1074/jbc.M104442200. [DOI] [PubMed] [Google Scholar]
- 30.Moers A, Wettschureck N, Gruner S, Nieswandt B, Offermanns S. Unresponsiveness of platelets lacking both Galpha(q) and Galpha(13). Implications for collagen-induced platelet activation. J Biol Chem. 2004;279:45354–9. doi: 10.1074/jbc.M408962200. [DOI] [PubMed] [Google Scholar]
- 31.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–9. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 32.Bauer M, Retzer M, Wilde JI, Maschberger P, Essler M, Aepfelbacher M, Watson SP, Siess W. Dichotomous regulation of myosin phosphorylation and shape change by Rho-kinase and calcium in intact human platelets. Blood. 1999;94:1665–72. [PubMed] [Google Scholar]
- 33.Dorsam RT, Kim S, Jin J, Kunapuli SP. Coordinated signaling through both G12/13 and G(i) pathways is sufficient to activate GPIIb/IIIa in human platelets. J Biol Chem. 2002;277:47588–95. doi: 10.1074/jbc.M208778200. [DOI] [PubMed] [Google Scholar]
- 34.Daniel JL, Holmsen H, Adelstein RS. Thrombin-stimulated myosin phosphorylation in intact platelets and its possible involvement secretion. Thromb Haemost. 1977;38:984–9. [PubMed] [Google Scholar]
- 35.Haslam RJ, Lynham JA. Relationship between phosphorylation of blood platelet proteins and secretion of platelet granule constituents. I. Effects of different aggregating agents. Biochem Biophys Res Commun. 1977;77:714–22. doi: 10.1016/s0006-291x(77)80037-5. [DOI] [PubMed] [Google Scholar]
- 36.Bennett WF, Belville JS, Lynch G. A study of protein phosphorylation in shape change and Ca++-dependent serotonin release by blood platelets. Cell. 1979;18:1015–23. doi: 10.1016/0092-8674(79)90214-9. [DOI] [PubMed] [Google Scholar]
- 37.Perrie WT, Perry SV. An electrophoretic study of the low-molecular-weight components of myosin. Biochem J. 1970;119:31–8. doi: 10.1042/bj1190031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paul BZ, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J Biol Chem. 1999;274:29108–14. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 39.Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99:3629–36. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- 40.Ikebe M, Hartshorne DJ, Elzinga M. Identification, phosphorylation, and dephosphorylation of a second site for myosin light chain kinase on the 20,000-dalton light chain of smooth muscle myosin. J Biol Chem. 1986;261:36–9. [PubMed] [Google Scholar]
- 41.Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci U S A. 1998;95:8070–4. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183–6. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 43.Jin J, Daniel JL, Kunapuli SP. Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J Biol Chem. 1998;273:2030–4. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]
- 44.Fabre JE, Nguyen M, Latour A, Keifer JA, Audoly LP, Coffman TM, Koller BH. Decreased platelet aggregation, increased bleeding time and resistance to thromboembolism in P2Y1-deficient mice. Nat Med. 1999;5:1199–202. doi: 10.1038/13522. [DOI] [PubMed] [Google Scholar]
- 45.Leon C, Hechler B, Freund M, Eckly A, Vial C, Ohlmann P, Dierich A, LeMeur M, Cazenave JP, Gachet C. Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y(1) receptor-null mice. J Clin Invest. 1999;104:1731–7. doi: 10.1172/JCI8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi OH, Adelstein RS, Beaven MA. Secretion from rat basophilic RBL-2H3 cells is associated with diphosphorylation of myosin light chains by myosin light chain kinase as well as phosphorylation by protein kinase C. J Biol Chem. 1994;269:536–41. [PubMed] [Google Scholar]
- 47.Roevens P, de Chaffoy de Courcelles D. Desensitization and resensitization of human platelets to 5-hydroxytryptamine at the level of signal transduction. Biochem J. 1995;307(Pt 3):775–82. doi: 10.1042/bj3070775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilde JI, Retzer M, Siess W, Watson SP. ADP-induced platelet shape change: an investigation of the signalling pathways involved and their dependence on the method of platelet preparation. Platelets. 2000;11:286–95. doi: 10.1080/09537100050129305. [DOI] [PubMed] [Google Scholar]
- 49.Missy K, Plantavid M, Pacaud P, Viala C, Chap H, Payrastre B. Rho-kinase is involved in the sustained phosphorylation of myosin and the irreversible platelet aggregation induced by PAR1 activating peptide. Thromb Haemost. 2001;85:514–20. [PubMed] [Google Scholar]
- 50.White JG, Burris SM. Morphometry of platelet internal contraction. Am J Pathol. 1984;115:412–7. [PMC free article] [PubMed] [Google Scholar]