

Abstract
Background
Endocytic dysfunction and neurotrophin signaling deficits may underlie the selective vulnerability of hippocampal neurons during the progression of Alzheimer’s disease (AD), although there is little direct in vivo and biochemical evidence to support this hypothesis.
Methods
Microarray analysis of hippocampal CA1 pyramidal neurons acquired via laser capture microdissection (LCM) was performed using postmortem brain tissue. Validation was achieved using real-time quantitative PCR (qPCR) and immunoblot analysis. Mechanistic studies were performed using human fibroblasts subjected to overexpression with viral vectors or knockdown via siRNA.
Results
Expression levels of genes regulating early endosomes (rab5) and late endosomes (rab7) are selectively up regulated in homogeneous populations of CA1 neurons from individuals with mild cognitive impairment (MCI) and AD. The levels of these genes are selectively increased as antemortem measures of cognition decline during AD progression. Hippocampal qPCR and immunoblot analyses confirmed increased levels of these transcripts and their respective protein products. Elevation of select rab GTPases regulating endocytosis paralleled the down regulation of genes encoding the neurotrophin receptors TrkB and TrkC. Overexpression of rab5 in cells suppressed TrkB expression, whereas knockdown of TrkB expression did not alter rab5 levels, suggesting that TrkB down regulation is a consequence of endosomal dysfunction associated with elevated rab5 levels in early AD.
Conclusions
These data support the hypothesis that neuronal endosomal dysfunction is associated with preclinical AD. Increased endocytic pathway activity, driven by elevated rab GTPase expression, may result in long term deficits in hippocampal neurotrophic signaling and represent a key pathogenic mechanism underlying AD progression.
Keywords: laser capture microdissection, mild cognitive impairment, RNA amplification, qPCR, rab5, rab7, siRNA, TrkB
Introduction
Hippocampal disconnection along with amyloid and neurofibrillary tangle (NFT) deposition and synaptic loss (1, 2) are pathological hallmarks of Alzheimer’s disease (AD). Although hippocampal CA1 pyramidal neurons are particularly vulnerable to neurodegeneration during the early stages of AD (3, 4), the mechanisms underlying their degeneration are poorly understood.
Endosomal-lysosomal system dysfunction is one of the earliest disturbances that occur in AD and may be a potential mechanism underlying neurodegeneration. Specifically, enlargement of rab5-positive endosomes precedes NFT formation, amyloid deposition, and is selective for AD (5, 6). Endosomes are integral mediators of cellular communication and metabolism through signaling and trafficking functions. Functional interrelationships exist between endosomal trafficking and neurotrophin signaling via high affinity Trk receptors. Proper synthesis, binding, internalization, and retrograde transport of Trks are critical for cell survival and neuroplasticity (7, 8). Signaling endosomes contain specific rab GTPases as well as neurotrophin receptor signaling complexes (9–11). Specific rab GTPases, including rab5 and rab7, have been implicated in the regulation of nerve growth factor (NGF) signaling in vitro (9, 11–13). However, systematic evaluation of the rab GTPase family of genes in relation to neurotrophin signaling within selectively vulnerable human neuronal populations is lacking, particularly within the context of cognitive decline during AD progression.
Due to the degeneration of selectively vulnerable neurons, expression profiles obtained from single cells or homogeneous populations of cells, is likely to be more informative than regional mosaics in AD (14–16). We hypothesize that abnormal acceleration of endocytosis and endosome recycling, including up regulation of select rab GTPase genes and other endocytosis-related transcripts occurs in vulnerable neurons at the earliest stage of AD, which, in part, promotes neurodegeneration by impairing neurotrophin receptor signaling. Herein, endosomal-lysosomal markers and Trk receptors were assessed in hippocampal CA1 neurons harvested from subjects who died with a clinical diagnosis of no cognitive impairment (NCI), mild cognitive impairment (MCI), or AD using laser capture microdissection (LCM) and custom-designed microarray analysis. To determine potential mechanistic interactions between specific rab GTPases and Trk receptors, overexpression and small interference RNA (siRNA) studies were subsequently performed in vitro.
Methods and Materials
Brain tissue and clinical pathological assessment
This study was performed under the auspices of the Rush University Medical Center and Nathan Kline Institute/New York University Institutional Review Boards. Table I summarizes the cases included in the microarray studies. Independent cases employed in the qPCR and immunoblot studies are presented in Tables S1 and S2 respectively (see Supplement), with the exception of 2 NCI and 2 AD cases used for qPCR analysis that were also part of the microarray study. Clinical and demographic characteristics {age, educational level, global cognitive score (GCS), Mini-mental state exam (MMSE), and postmortem interval (PMI)} were compared using one-way analysis of variance (ANOVA) or Kruskal-Wallis test, with Bonferroni correction for post-hoc comparisons when appropriate (17–19). Gender and ApoE allele were assessed across clinical conditions using Fisher’s exact test. Neuropathologic classifications (NIA-Reagan, CERAD, and Braak scoring) were compared among clinical diagnostic groups by Kruskal-Wallis test (17, 18). The level of statistical significance was set at 0.05 (two-sided).
Table I.
Microarray analysis: clinical, demographic, and neuropathological characteristics by diagnosis category.
| Clinical Diagnosis |
Comparison by diagnosis group | Pair-wise comparisons* | ||||
|---|---|---|---|---|---|---|
| NCI (n = 11) | MCI (n = 15) | AD (n = 7) | ||||
| Age at death (years) | Mean ± SD (Range) | 82.1 ± 7.9 (67–92) | 86.2 ± 5.2 (80–97) | 86.7 ± 7.7 (80–101) | p = 0.2a | - |
| Number (%) of males | 5 (45%) | 6 (40%) | 2 (29%) | p = 0.9b | - | |
| Educational level | Mean ± SD (Range) | 16.6 ± 4.1 (8–21) | 18.9 ± 2.3 (16–22) | 17.1 ± 1.6 (16–20) | p = 0.1a | - |
| MMSE | Mean ± SD (Range) | 27.7 ± 1.6 (25–30) | 26.7 ± 2.7 (20–30) | 18.4 ± 3.7 (15–25) | p < 0.0001a | (NCI & MCI) > AD |
| GCS | Mean ± SD (Range) | 0.6 ± 0.3 (0.0–0.9) | 0.2 ± 0.2 (−0.2, 0.5) | −0.8 ± 0.3 (−1.2, −0.4) | p < 0.0001a | NCI > MCI > AD |
| ApoE ε4 allele (%) | 2 (18%) | 6 (40%) | 6 (86%) | p = 0.02b | (NCI & MCI) < AD | |
| PMI (hours) | Mean ± SD (Range) | 8.3 ± 8.7 (2.3–33.5) | 6.7 ± 4.2 (3.0–16) | 7.3 ± 4.1 (2.2–12) | p = 0.8a | - |
| Distribution of Braak scores | 0 | 0 | 0 | 0 | ||
| I/II | 5 | 1 | 1 | p = 0.004c | NCI < (MCI & AD) | |
| III/IV | 6 | 10 | 1 | |||
| V/VI | 0 | 4 | 5 | |||
| Distribution of NIA Reagan diagnosis (likelihood of AD) | No AD | 0 | 0 | 0 | ||
| Low | 8 | 3 | 1 | p = 0.003c | NCI < (MCI & AD) | |
| Intermediate | 3 | 10 | 2 | |||
| High | 0 | 2 | 4 | |||
| CERAD diagnosis | No AD | 6 | 1 | 0 | ||
| Possible | 1 | 2 | 0 | p = 0.006 c | NCI < (MCI & AD) | |
| Probable | 3 | 5 | 3 | |||
| Definite | 1 | 7 | 4 | |||
One-way ANOVA
Fisher’s exact test
Kruskal-Wallis test
With Bonferroni correction
Single cell microaspiration and Terminal Continuation (TC) RNA amplification
Immunocytochemistry to identify CA1 neurons for LCM (16, 20) and TC RNA amplification procedures (Fig 1A) have been described in detail previously (21–24) (see Supplement for additional Methods). Individual CA1 pyramidal neurons were microaspirated using LCM instrumentation (Arcturus PixCell IIe, MDS, Sunnyvale, CA) (Fig. 1B, C). Fifty cells were captured for population cell analysis (20, 23). A total of 1–8 microarrays (containing 50 LCM-captured CA1 neurons each) were performed per human brain, with a median of 3–4 microarrays per case.
Figure 1.
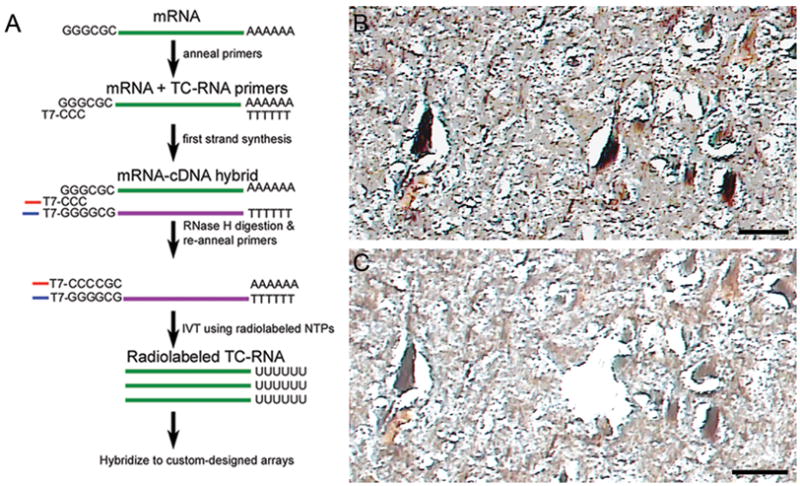
TC RNA amplification procedure and LCM of CA1 hippocampal neurons.
A. Schematic overview of the TC RNA amplification procedure. mRNA extracted from LCM captured cells (green line) and the TC primer serve as templates for the first strand synthesis with poly d(T) acting as a primer. First strand cDNA consists of three portions, the 5′ end comprised of the poly d(T), the mRNA complementary portion in the middle (purple line), and the 3′ end is comprised of the TC primer complementary to the cDNA (denoted as TC′). The TC′ portion hybridizes with the TC primer present in the reaction and forms a double stranded region which provides a functional RNA synthesis promoter for in vitro transcription and robust RNA amplification.
B. Neurofilament-immunoreactive CA1 neurons in the hippocampal pyramidal cell layer from a subject with AD prior to LCM. Tissue sections were dehydrated but not coverslipped to enable proper execution of the LCM process. Scale bar: 50 mm.
C. Slightly larger image in B following LCM of an individual CA1 neuron. Scale bar: 50 mm.
Custom-designed cDNA array platforms and array hybridization
Array platforms consisted of 1 mg of linearized cDNA purified from plasmid preparations adhered to high-density nitrocellulose (Hybond XL, GE Healthcare, Piscataway, NJ). Each cDNA and/or expressed sequence-tagged cDNA (EST) was verified by sequence analysis and restriction digestion. Human and some mouse clones were employed on the custom-designed array. Notably, all of the rab GTPases, neurotrophins, and neurotrophin receptors were derived from human sequences. Approximately 576 cDNAs/ESTs were utilized on the current array platform. Array hybridization is presented in Supplemental Methods (see Supplement).
Data collection and statistical analysis for custom-designed microarrays
Microarray analysis was performed on hippocampal tissue obtained from 11 NCI, 15 MCI, and 7 mild/moderate AD Religious Orders Study cases (Table I). Microarray data was also available from an additional 5 end-stage AD cases (Table S3 in the Supplement). Relative changes in total hybridization signal intensity and percentage of cDNA clones above negative control were analyzed by mixed models analysis for repeated measures, with random intercept, fixed effect for diagnosis, or other clinical/neuropathological characteristics of interest, Kenward-Roger denominator degrees of freedom, unequal variance assumption as warranted, and unstructured covariance structure (17, 19, 25). Since multiple cells were measured in each subject, between-subjects versus within-subject (between-cell) variation in gene expression levels were assessed by variance component analysis and intraclass correlation coefficient (17). The level of statistical significance was set at the more conservative 0.01 (two-sided) to account for the large number of analyses performed.
qPCR
qPCR was performed on frozen tissue micropunches containing the hippocampal CA1 region obtained from 11 NCI, 6 MCI, and 6 mild/moderate AD cases from the Religious Orders Study (Supplement: Table S1 and Methods). Samples were assayed on a real-time PCR cycler (7900HT, Applied Biosystems) in 96-well optical plates as described previously (17, 23, 24). The ddCT method was employed to determine relative gene level differences with Gapdh qPCR products used as a control (23, 26, 27). Variance component analyses revealed relatively low levels of within-case variability, and the average value of the triplicate qPCR products from each case was used in subsequent analyses. Alterations in PCR product synthesis were analyzed by Kruskal-Wallis test with Bonferroni correction for post-hoc comparison. The level of statistical significance was set at 0.05 (two-sided).
Immunoblot analysis
Frozen microdissected hippocampal tissue samples obtained from 15 NCI, 8 MCI, and 17 AD cases from the Religious Orders Study and other sources (see Table S2 in the Supplement) were homogenized and centrifuged (16, 28) (see Supplement: Methods). Nitrocellulose membranes were blocked in blocking buffer (LiCor, Lincoln, NE) for 1 hour at 4 °C prior to being incubated with antibodies directed against rab5 (rab5A; rabbit polyclonal sc-309; Santa Cruz Biotechnology, Santa Cruz, CA; 1:1,000 dilution), rab7 (rabbit polyclonal sc-10767; Santa Cruz Biotechnology 1:1,000 dilution), or b-tubulin (TUBB; monoclonal antibody; Sigma, 1:1,000 dilution) in blocking buffer overnight at 4°C. Signal intensity of immunoreactive bands was normalized to TUBB immunoreactivity for each assay. Immunoreactive band intensity was compared across clinical diagnostic groups by Kruskal-Wallis test, with Bonferroni correction for post-hoc comparisons. The association between immunoreactive band intensity and Braak staging was assessed by Spearman rank correlation (28). The level of statistical significance was set at 0.05 (two-sided).
rab5 overexpression by infection of human skin fibroblasts
Human forearm skin fibroblasts from normal control subjects (Coriell Cell Repositories, Camden, NJ) were grown as described previously (27, 29). Briefly, herpes simplex virus type 1 (HSV1) amplicons expressing myc-tagged human wild-type rab5 (rab5wt), dominant-positive GTPase-deficient (rab5Q79L), dominant-negative (rab5S34N), or control (LacZ) were generated as previously described (30) (Supplement: Methods). Immunoblot analysis was performed and quantified as above using antibodies directed against TrkB {both full length TrkB (TrkB-FL) and truncated TrkB (TrkB-T1) were detected and quantified; monoclonal antibody 610102; BD Biosciences, San Jose, CA; 1:1,000 dilution}, epidermal growth factor receptor (EGFR; sheep polyclonal; Millipore; 1:1,000 dilution), and TUBB (monoclonal antibody; Sigma, 1:1,000 dilution). A total of 3–4 samples per rab5 viral vector construct were run in duplicate for immunoblot assessments. Immunoreactive band intensities were analyzed by one-way ANOVA with Neumann-Keuls test for post-hoc analysis. qPCR assessment was performed on total RNA extracted from fibroblasts 48 hours after infection using Trizol reagent (Invitrogen). qPCR was performed using primers for TrkA, TrkB, TrkC, and Gapdh (see Supplement: Methods). The ddCT method was employed on a total of 4 samples per viral infection in triplicate, and analyzed by one-way ANOVA with Neumann-Keuls test for post-hoc analysis as described previously (23, 27). The level of statistical significance was set at 0.05 (two-sided).
Knockdown of TrkB using siRNA
Predesigned TrkB siRNA constructs were purchased from Applied Biosystems (Silencer TrkB 753 and TrkB 754) (31) and used as described in the Supplemental Methods (see Supplement). Immunoblot analysis was performed and quantified as above using antibodies directed against rab5, TrkB, TUBB, and GAPDH (rabbit polyclonal sc-25778; Santa Cruz Biotechnology; 1:1,000 dilution). A total of 4 samples per condition (TrkB siRNA and vector transfected) were analyzed by one-way ANOVA with Neumann-Keuls test for post-hoc analysis. The level of statistical significance was set at 0.05 (two-sided). Total RNA from siRNA and vector treated cells was extracted 48 hours after transfection and prepared for qPCR as above using previously identified TrkB, rab5, and Gapdh primers.
Results
Microarray analysis identifies selectively altered rab GTPases and Trk receptor expression in early AD
Clinical and neuropathological characteristics of the 33 cases (11 NCI, 15 MCI, 7 mild/moderate AD) included in the microarray analysis are summarized in Table I. No significant differences were observed for age, gender, educational level, and PMI across the 3 groups. The ApoE4 allele was more frequent in AD compared to NCI and MCI cases. Distribution of Braak scores was significantly different across clinical conditions, with NCI cases having lower Braak staging than MCI and AD. NCI cases were classified as Braak stages I–II (45.5%) and III–IV (54.5%). None of the NCI cases were classified as Braak stages V–VI. The MCI cohort displayed Braak stages I–II (6.6%), III–IV (66.7%), and V–VI (26.7%). AD cases were classified as Braak stages I–II (14.3%), III–IV (14.3%), and V–VI (71.4%), respectively (Table I). NIA-Reagan and CERAD criteria significantly differentiated NCI subjects from MCI and AD cases.
Expression profiling was performed on a total of 136 custom-designed microarrays (50 CA1 neurons were analyzed per microarray) following the TC RNA amplification protocol (Fig. 1A). Quantitative analysis revealed differential regulation of several rab GTPases (Fig. 2), including a highly statistically significant up regulation of early endosome effectors rab4 (p < 0.0007; AD>MCI & NCI) and rab5 (p < 0.0003; AD>MCI>NCI), late endosome constituent rab7 (p < 0.0005; AD & MCI>NCI), and the trafficking molecule rab24 (p < 0.003; AD>MCI & NCI). Based on the comparison of expression level differences between MCI and AD cases, alterations in rab5 and rab7 were considered ‘early’ changes, i.e., up regulation was observed in MCI and AD, whereas up regulation of rab4 and rab24 was considered a ‘later’ alteration, as significant changes were found in AD, but not MCI. Significant down regulation of the synaptic-related marker rab3 (p < 0.003; AD<MCI & NCI) was also observed, and considered a ‘later’ alteration occurring in AD. Factor analysis using promax rotation was performed to assess the inter-correlation among these 8 rab GTPase genes. Results identified three subgroups: Group #1 (rab4, rab5, rab7, and rab24; all significantly up regulated genes), Group #2 (rab10 and rab27; no differential regulation of either gene), and Group #3 (rab1 and rab3; this subgroup was somewhat unexpected since rab3 is selectively down regulated and rab1 expression does not differ across the cohort). Furthermore, up regulation of rab4, rab5, rab7, and rab24 was also observed in the end-stage AD subjects (n = 5; Table S3 in the Supplement), consistent with the gene expression changes seen in the mild/moderate AD cases (Fig. 2).
Figure 2.

Differential regulation of select rab GTPases and neurotrophin receptors during the progression of AD.
Boxplots (NCI, green; MCI, blue; AD, red) indicating relative expression levels of select rab GTPases and neurotrophin receptors. Significant up regulation of rab4, rab5, and rab7 is depicted along with significant down regulation of rab3, TrkB, and TrkC.
Additional endosomal-lysosomal markers displaying significant differential regulation including an up regulation of cathepsin D (Ctsd; p < 0.0001; AD & MCI>NCI), early endosome antigen 1 (Eea1), an early endosomal marker (p < 0.007; AD & MCI>NCI, extracellular signal-regulated kinase 1 (p44) (Erk1; p < 0.0001; AD>MCI & NCI), lysosomal-associated membrane protein 1 (Lamp1; p < 0.0001; AD>MCI & NCI), and dynein cytoplasmic light chain 1 (Dnclc1; p < 0.003; AD>NCI & MCI). Significant down regulation was observed for the calpain inhibitor calpastatin (Cast; p < 0.0001; AD & MCI<NCI), late endosomal/lysosomal marker Niemann-Pick disease type C1 (Npc1; p < 0.0001; AD & MCI<NCI), and the vacuolar proton pump homolog 1 (Vpp1; p < 0.0001; AD & MCI<NCI). No differential regulation was observed for other genes including several cathespsins (Ctsb, Ctsc, Ctse, Ctsg, Ctsh, Ctsl, Ctsz), cystatin B (Cstb), cystatin C (Cstc), Erk2, Lamp2, mannose-6-phosphate receptors Cimpr (cation independent) and Cdmpr (cation dependent), Npc2, and several rab GTPases that are not regulators of endosomes (rab1, rab2, rab6, rab10, and rab27). Moreover, we did not observe significant regulation of more traditional AD-related genes across the clinical groups examined. For example, within CA1 pyramidal neurons, no expression level changes were found for amyloid-b precursor protein (App), amyloid-b precursor-like protein 1 (Aplp1), amyloid-b precursor-like protein 2 (Aplp2), beta-site APP-cleaving enzyme 1 (Bace1), presenilin 1 (Psen1), presenilin 2 (Psen2), low density lipoprotein-related protein 1 (Lrp1), high density lipoprotein binding protein (Hdlbp), alpha-2-macroglobulin (A2m), and beta-2-microglobulin (B2m), among other AD-related genes.
In contrast to the significant up regulation of select rab GTPase genes, marked down regulation of specific high-affinity neurotrophin receptors was found within CA1 neurons including a significant down regulation of the high affinity brain derived neurotrophic factor (BDNF) receptor TrkB (p < 0.0001; AD & MCI<NCI), considered an ‘early’ gene expression alteration (Fig. 2), and a down regulation of the high affinity neurotrophin-3 receptor TrkC (p < 0.006; AD<MCI & NCI), considered a ‘later’ alteration based on the lack of differential expression levels in MCI. We also observed a trend for a down regulation of the high affinity NGF receptor TrkA (p < 0.02; AD<NCI; MCI did not differ significantly from AD or NCI). No significant alterations were found for gene expression of the low-affinity pan NGF receptor p75NTR across the three groups. Evaluation of additional classes of transcripts within hippocampal CA1 neurons will comprise a separate report due to the extensive amount of data.
Consistent with comparisons across the three clinical diagnostic groups, postmortem expression profiling of the key endosome-associated rab GTPases obtained from CA1 pyramidal neurons correlated with antemortem cognitive measures obtained from the same cases. A strong negative correlation was found between cognitive scores (MMSE and GCS) and rab4, rab5, rab7, and rab24 CA1 neuron expression levels (Fig. 3A–D). On the other hand, there was a strong positive correlation between cognitive scores and rab3, TrkB, and TrkC CA1 expression levels (Fig. 3E–G). Differentially regulated rab GTPases (notably rab7, and to a lesser degree, rab4, rab5, rab24, and rab3) and the neurotrophin receptor TrkB were also associated with neuropathological indices including Braak staging, NIA-Reagan diagnosis, and CERAD diagnosis (Table S4 in the Supplement).
Figure 3.
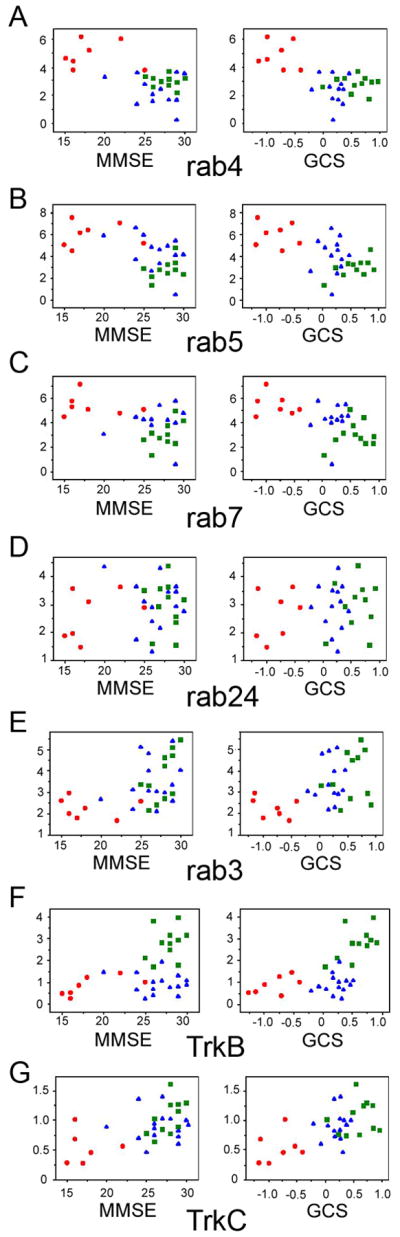
Association between select rab GTPase and neurotrophin receptor gene expression levels within CA1 neurons and antemortem cognitive measures in the same subjects. Scatterplots illustrate the relationship of gene expression levels and MMSE (left panel) and GCS scores (right panel) from individuals classified as NCI (green squares), MCI (blue triangles), and AD (red circles).
A. A highly significant negative association exists with rab4 and MMSE (p < 0.0001) and GCS performance (p < 0.0002).
B. A highly significant negative association exists with rab5 and MMSE (p < 0.001) and GCS performance (p < 0.001).
C. A significant negative association exists with rab7 and MMSE (p < 0.01) and GCS performance (p < 0.001).
D. rab24 expression levels are inversely correlated with MMSE (p < 0.0004) and GCS (p < 0.0002) scores.
E. A significant association between rab3 gene expression down regulation and poor MMSE (p < 0.002) and GCS (p < 0.005) performance was observed.
F. Down regulation of TrkB expression correlated with MMSE (p < 0.02) and GCS (p < 0.0001) performance.
G. Down regulation of TrkC expression correlated with MMSE (p < 0.003) and GCS (p < 0.0004) performance.
qPCR validation
Based upon our custom-designed microarray analysis of CA1 pyramidal neurons, select rab GTPase and neurotrophin receptor gene expression levels were assessed via qPCR using micropunches of hippocampal CA1 tissue harvested from NCI, MCI, and AD cases. The ddCT method was employed to determine relative rab GTPase and neurotrophin receptor gene level differences using Gapdh qPCR products as a control (22–24). No differences in Gapdh expression levels were found between NCI, MCI, and AD subjects, consistent with previously published observations using brain tissues from the Religious Orders Study (17). qPCR experiments using TaqMan primers revealed no cross reactivity between individual rab GTPases or Trk receptors. qPCR analysis independently validated our microarray observations (Figure S1 in the Supplement), including a significant up regulation of rab4, rab5, rab7, and rab24 within the hippocampal CA1 region. Specifically, we observed an ‘early’ alteration (AD & MCI>NCI) in rab4, rab7, and rab24, and an ‘early’ stepwise alteration in rab5 expression (AD>MCI>NCI). A non-significant trend for down regulation of rab3 PCR products was also observed in AD. qPCR analysis for the neurotrophin receptors within the CA1 sector of the hippocampus demonstrated a significant down regulation of TrkB (AD & MCI<NCI, indicative of an ‘early’ alteration) and TrkC (AD<MCI<NCI, suggesting a ‘step down’ alteration; Figure S1 in the Supplement). No differences were found in TrkA (which was expressed at low levels in the CA1 micropunches, making qPCR quantitation difficult), p75NTR, and rab27 qPCR products. In summary, the majority of single population cell microarray findings (e.g., rab4, rab5, rab7, rab24, rab27, TrkB, TrkC, and p75NTR) were validated via qPCR on independent micropunched hippocampal CA1 samples, including the evaluation of ‘early’ and ‘later’ changes in differential regulation (e.g., rab5, rab7, TrkB, and TrkC) across the progression of dementia.
Up regulation of rab5 and rab7 protein levels parallel cognitive decline in MCI and AD
Protein-based assessment of rab5 and rab7 expression was performed via immunoblotting using well-established antibodies combined with frozen human hippocampal CA1 punches. Immunoblotting identified an ~27 kDa band and an ~25 kDa band using rab5 and rab7 antibodies, respectively. Quantitative analysis demonstrated a significant up regulation of rab5 (p < 0.0001; AD & MCI>NCI), indicative of an ‘early’ alteration and rab7 expression (p < 0.01; AD>MCI>NCI, indicative of an ‘early’ alteration that was a ‘step-down’ between MCI and AD (Figure S2 in the Supplement). Up regulation of hippocampal rab5 and rab7 expression also correlated with Braak staging in this cohort (rab5, r = 0.70; rab7, r = 0.47), indicating a high correspondence between select rab GTPase gene expression levels found in CA1 neurons and microdissected CA1 tissue, protein levels in the hippocampus, and neuropathologic criteria during the progression of AD.
Elevated rab5 levels down regulate TrkB expression
A potential mechanistic interaction between rab5 up regulation and TrkB down regulation was investigated using fibroblasts infected with rab5 constructs. Human fibroblast preparations have been used previously by our group in concert with the well-established rab5 overexpression vectors to assess endocytic perturbations relevant to cell biological processes underlying AD and Down syndrome (DS) (27, 29). Viral infection of primary neurons using HSV1 vectors is also possible. However, HSV1 infection of neurons has been shown to cause intracellular Ab accumulation as well as phosphorylation of several serine/threonine sites on tau and glycogen synthase kinase 3b/protein kinase A activation (32, 33), and was not pursued in this study.
In the present experiments, lysates obtained from homogenized fibroblasts were immunoblotted for TrkB-FL, TrkB-T1, and EGFR. Infected cells were also collected and RNA extracted for TrkB qPCR. Quantitative analysis revealed a highly significant down regulation of TrkB-FL and TrkB-T1 in fibroblasts infected with HSV1 vectors encoding rab5wt and the constitutively active rab5 construct rab5Q79L compared to mock infected (sucrose) and control (LacZ) infected cells (Fig. 4A). Moreover, TrkB-FL and TrkB-T1 were down regulated in cells infected with HSV expressing rab5wt and rabQ79L compared to the level in cells infected with HSV expressing dominant negative construct rab5S34N (Fig. 4A). No significant differences were observed in TrkB-FL and TrkB-T1 expression between cells infected with rab5S34N, LacZ control virus, or mock infected cells.
Figure 4.

Histograms showing expression of TrkB and EGFR levels in fibroblasts infected with rab5 viral vector constructs and expression of select rab GTPases following TrkB siRNA in cultured human fibroblasts.
A. Upper left panel; down regulation of TrkB-FL is observed in fibroblasts infected with rab5wt (single asterisk denotes p < 0.005) and the constitutively active construct rab5Q79L (asterisk) compared to mock transfected (sucrose) control (LacZ) transfected cells. TrkB-FL down regulation was also found in the rab5wt and rabQ79L constructs compared to the dominant negative construct rab5S34N (double asterisk denotes p < 0.01). Upper right panel; down regulation of TrkB-T1 is seen in fibroblasts infected with rab5wt (single asterisk denotes p < 0.007) and the constitutively active construct rab5Q79L (asterisk) compared to mock transfected (sucrose) control (LacZ) transfected cells. Similar to TrkB-FL, TrkB-T1 down regulation was observed in the rab5wt and rabQ79L constructs compared to rab5S34N (double asterisk denotes p < 0.02). Lower left panel; down regulation of TrkB is observed via qPCR in fibroblasts infected with rab5wt and rab5Q79L (single asterisk denotes p < 0.001) compared to control (LacZ) infected cells and to the dominant negative rab5S34N construct (double asterisk denotes p < 0.04), suggesting that rab5 overexpression regulates TrkB at the transcriptional level. Lower right panel; in contrast to differential TrkB expression levels, no significant differences in related EGFR expression were observed with any of the fibroblasts infected with rab5 constructs or controls. TrkB and EGFR levels were normalized to TUBB ± SD for quantitative immunoblot analysis.
B. qPCR analysis following TrkB siRNA (Silencer 753) indicated significant knockdown of TrkB expression {34.4% ± 0.9 (SEM); p < 0.001; asterisk} compared to vector treated cells. In contrast, no significant regulation of Gapdh (118.8% ± 7.8), rab4 (86.4% ± 7.8), rab5 (98.5% ± 11.5), or rab7 (114.9% ± 9.3) levels was observed following TrkB siRNA. C. Upper panel; immunoblot analysis demonstrated significant knockdown of TrkB expression (56.4% ± 4.1 (SEM); p < 0.005; asterisk), but not rab5 expression (95.0% ± 3.0), following TrkB siRNA. Lower panel; representative immunoblots demonstrating knockdown of TrkB expression with no significant alterations in rab5 or GAPDH expression.
qPCR using RNA harvested from infected cells revealed significant down regulation of TrkB in rab5wt and rabQ79L constructs compared to control (LacZ) infected cells as well as to the dominant negative rab5S34N construct, which tracked with protein expression levels (Fig. 4A). These coordinated immunoblotting and qPCR data suggest that regulation of TrkB via rab5 overexpression occurs at the transcriptional level. qPCR studies also demonstrated down regulation of TrkA and TrkC expression following rab5wt overexpression (data not shown). In contrast to differential TrkB expression levels, no significant differences in related tyrosine kinase receptor EGFR expression were observed with any of the fibroblasts expressing rab5 constructs compared to controls (Fig. 4A), supporting a selective negative drive of rab5 on TrkB expression levels.
TrkB siRNA and rab GTPase regulation
To determine whether regulation of TrkB expression effects rab GTPase levels, siRNA experiments were carried out in human fibroblasts using a TrkB siRNA construct (31). Following transfection of normal human fibroblasts, qPCR and immunoblot analysis for TrkB was performed to verify knockdown at the mRNA and protein levels, respectively. qPCR revealed that TrkB expression was knocked down to approximately 35% of control levels (vector transfected cells) with a concomitant decrease in TrkB protein to approximately 55% of control levels (Fig. 4B, C). qPCR assessment of other select rab GTPases demonstrated no significant difference between fibroblasts treated with TrkB siRNA and vector treated cells (Fig. 4B). Immunoblot analysis using an antibody directed against rab5 confirmed a lack of differential regulation following TrkB siRNA administration (Fig 4C), indicating no reciprocal regulation of TrkB on rab4, rab5, and rab7 expression. These observations suggest that expression of rab5 is as an upstream regulator of TrkB levels.
Discussion
CA1 pyramidal neuron expression profiling combined with hippocampal qPCR and immunoblotting demonstrate significant up regulation of select endosomal-lysosomal markers along with a marked down regulation of Trk receptors within these selectively vulnerable neurons in MCI and early AD. These data suggest a reciprocal change in expression of genes regulating endocytosis and neurotrophin signaling at incipient stages of the disease. Importantly, in vitro experiments demonstrated a functional interelationship between increased endocytic drive and decreased neurotrophin receptor expression. Specifically, wild type and constitutively active rab5 down regulated TrkB at the transcriptional level, but knockdown of TrkB does not feedback onto rab GTPase expression, providing a mechanistic molecular pathologic link between endosomal-lysosomal dysfunction and neurotrophin signaling deficiencies (Fig. 5). These findings suggest that that alterations of select early and late endocytic rab GTPases contribute to CA1 neurodegeneration, in part, by impairing neurotrophin receptor signaling, and that these genes are early molecular markers for the development of AD.
Figure 5.
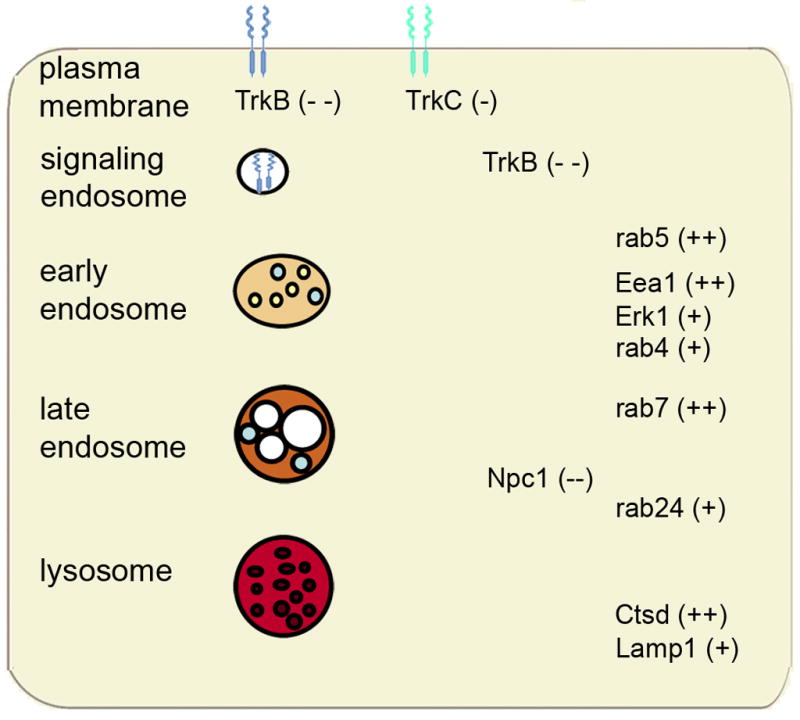
Schematic diagram illustrating gene expression level changes within components of the endosomal-lysosomal system and neurotrophins at the cell surface and within signaling endosomes of vulnerable CA1 pyramidal neurons in MCI and AD. Genes demonstrating up regulation are denoted with a plus sign (one plus sign for ‘late’ changes observed only in AD and two plus signs indicating ‘early’ up regulation seen in MCI and AD). Genes demonstrating down regulation are denoted with a minus sign (one minus sign for ‘late’ changes observed only in AD and two minus signs indicating ‘early’ up regulation seen in MCI and AD).
Prior to the present findings, there was virtually no data of multiple rab GTPase expression at the genomic level in postmortem human brain during the progression of AD. CA1 gene expression assessment demonstrates simultaneous dysregulation of several rab GTPases within discrete endosomal, lysosomal, synaptic, and trafficking compartments as dementia occurs in humans. These observations extend the concept that endosomal abnormalities reflect an over activation of the endocytic pathway in sporadic AD and in DS (29). In the latter, accelerated endocytosis is mediated, in part, by elevated rab5 protein levels (29). Microarray analysis also identified dysregulation of several genes associated with increased endocytic drive and lysosomal activation. Up regulation of Ctsd, a lysosomal protease, is consistent with our previous assessments within NFT-bearing CA1 neurons and cholinergic basal forebrain (CBF) neurons in AD (16, 34, 35). Although our data support an up regulation of endocytosis, this does not exclude the possibility that downstream impairments of endosomal maturation may also contribute to endocytic pathway dysfunction (36, 37). Collectively, the present data highlights the importance of endosomal transcripts in the pathogenesis of AD, and is supported by emerging evidence implicating genes influencing endocytosis as risk factors for AD (38–41).
The mechanistic link between rab5 overexpression and TrkB down regulation is highly significant as it potentially uncovers a regulatory pathway between a well-established endosomal effector and a neurotrophin signal transduction receptor pathway, which is enriched in vulnerable forebrain neurons. An interaction between TrkB and rab GTPases is intriguing in light of the findings that BDNF levels are significantly reduced in the brains of people with MCI (42) and TrkB polymorphisms have been implicated as susceptibility genes contributing to AD pathology (43–45). rab5 overexpression appears to be relatively specific for TrkB, as EGFR expression levels were not perturbed. This may indicate that EGFR and TrkB are being processed, sequestered, and/or degraded through different pathways (46–48). Intriguingly, accelerated rab5-mediated endocytosis has been associated with increased production of neurotoxic A (49, 50). Ab has been shown to down regulate TrkB signaling and BDNF retrograde trafficking (51, 52), whereas BDNF is neuroprotective against Ab-induced cellular damage (53, 54). This relationship between Ab and neurotrophin receptor levels suggests that rab5-positive endosomal trafficking acts as a critical factor regulating cell survival. Taken together, the present results strongly implicate deregulation of early endosomal function as a causative factor for TrkB down regulation, suggesting a novel site for pharmacotherapeutic interventions via small molecule activators and/or TrkB transactivation (55–59).
In summary, the present work defines significant changes within vulnerable CA1 neurons and brings to light potential mechanistic interactions between endosomal-lysosomal trafficking components and neurotrophin signaling cascades early in the progression of AD (Fig. 5). Select rab GTPase overexpression supports evidence that APP-dependent endosome pathology developing early in AD involves abnormal acceleration of endocytosis and endosome recycling, which may promote neurodegeneration at least in part by impairing neurotrophin receptor signaling. Our data suggest that strategies to improve TrkB signaling (possibly via small molecule activators or transactivating compounds) as well as modulating rab5 (or possibly rab4, rab7, and rab24) levels may prove beneficial to deter the progression of AD.
Supplementary Material
Acknowledgments
Support for this project comes from NIH grants AG17617, AG14449, AG10161, AG09466 and the Alzheimer’s Association. We thank Dr. Freddy Jeanneteau and Dr. Katrin Deinhardt for their assistance with the Trk antibodies and critical review of the manuscript. We thank Irina Elarova, Anne Boyer Boiteau, and Shaona Fang for expert technical assistance. We are indebted to the altruism and support of the participants in the Religious Orders Study and the Center for Neurodegenerative Research at UPENN. A list of participating groups can be found at the website: http://www.rush.edu/rumc/page-R12394.html. Sadly, Dr. Anne M. Cataldo passed away prior to the publication of this manuscript, and we dedicate this work to her memory.
Footnotes
Financial Disclosures
The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 2.Kordower JH, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennett D, et al. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49:202–213. [PubMed] [Google Scholar]
- 3.Hyman BT, Van Hoesen GW, Damasio AR. Memory-related neural systems in Alzheimer’s disease: an anatomic study. Neurology. 1990;40:1721–1730. doi: 10.1212/wnl.40.11.1721. [DOI] [PubMed] [Google Scholar]
- 4.Ginsberg SD, Schmidt ML, Crino PB, Eberwine JH, Lee VM-Y, Trojanowski JQ. Molecular pathology of Alzheimer’s disease and related disorders. In: Peters A, Morrison JH, editors. Cerebral Cortex, vol 14 Neurodegenerative and Age-related Changes in Structure and Function of Cerebral Cortex. New York: Kluwer Academic/Plenum; 1999. pp. 603–653. [Google Scholar]
- 5.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cataldo A, Rebeck GW, Ghetri B, Hulette C, Lippa C, Van Broeckhoven C, et al. Endocytic disturbances distinguish among subtypes of Alzheimer’s disease and related disorders. Ann Neurol. 2001;50:661–665. [PubMed] [Google Scholar]
- 7.Howe CL, Mobley WC. Signaling endosome hypothesis: A cellular mechanism for long distance communication. J Neurobiol. 2004;58:207–216. doi: 10.1002/neu.10323. [DOI] [PubMed] [Google Scholar]
- 8.Delcroix JD, Valletta J, Wu C, Howe CL, Lai CF, Cooper JD, et al. Trafficking the NGF signal: implications for normal and degenerating neurons. Prog Brain Res. 2004;146:3–23. doi: 10.1016/s0079-6123(03)46001-9. [DOI] [PubMed] [Google Scholar]
- 9.Valdez G, Philippidou P, Rosenbaum J, Akmentin W, Shao Y, Halegoua S. Trk-signaling endosomes are generated by Rac-dependent macroendocytosis. Proc Natl Acad Sci USA. 2007;104:12270–12275. doi: 10.1073/pnas.0702819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bronfman FC, Escudero CA, Weis J, Kruttgen A. Endosomal transport of neurotrophins: roles in signaling and neurodegenerative diseases. Dev Neurobiol. 2007;67:1183–1203. doi: 10.1002/dneu.20513. [DOI] [PubMed] [Google Scholar]
- 11.Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, et al. Rab5 and rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Lamb D, Chou MM, Liu YJ, Li G. Nerve growth factor-mediated neurite outgrowth via regulation of Rab5. Mol Biol Cell. 2007;18:1375–1384. doi: 10.1091/mbc.E06-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saxena S, Bucci C, Weis J, Kruttgen A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J Neurosci. 2005;25:10930–10940. doi: 10.1523/JNEUROSCI.2029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mufson EJ, Counts SE, Che S, Ginsberg SD. Neuronal gene expression profiling: uncovering the molecular biology of neurodegenerative disease. Prog Brain Res. 2006;158:197–222. doi: 10.1016/S0079-6123(06)58010-0. [DOI] [PubMed] [Google Scholar]
- 15.Ginsberg SD, Che S, Counts SE, Mufson EJ. Single cell gene expression profiling in Alzheimer’s disease. NeuroRx. 2006;3:302–318. doi: 10.1016/j.nurx.2006.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginsberg SD, Hemby SE, Lee VM-Y, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48:77–87. [PubMed] [Google Scholar]
- 17.Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75 gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- 18.Ginsberg SD, Che S, Counts SE, Mufson EJ. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Neurochem. 2006;96:1401–1408. doi: 10.1111/j.1471-4159.2005.03641.x. [DOI] [PubMed] [Google Scholar]
- 19.Counts SE, He B, Che S, Ikonomovic MD, Dekosky ST, Ginsberg SD, et al. {alpha}7 Nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol. 2007;64:1771–1776. doi: 10.1001/archneur.64.12.1771. [DOI] [PubMed] [Google Scholar]
- 20.Ginsberg SD, Che S. Expression profile analysis within the human hippocampus: Comparison of CA1 and CA3 pyramidal neurons. J Comp Neurol. 2005;487:107–118. doi: 10.1002/cne.20535. [DOI] [PubMed] [Google Scholar]
- 21.Che S, Ginsberg SD. Amplification of transcripts using terminal continuation. Lab Invest. 2004;84:131–137. doi: 10.1038/labinvest.3700005. [DOI] [PubMed] [Google Scholar]
- 22.Ginsberg SD. Transcriptional profiling of small samples in the central nervous system. Methods Mol Biol. 2008;439:147–158. doi: 10.1007/978-1-59745-188-8_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alldred MJ, Che S, Ginsberg SD. Terminal continuation (TC) RNA amplification enables expression profiling using minute RNA input obtained from mouse brain. Int J Mol Sci. 2008;9:2091–2104. doi: 10.3390/ijms9112091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alldred MJ, Che S, Ginsberg SD. Terminal continuation (TC) RNA amplification without second strand synthesis. J Neurosci Methods. 2009;177:381–385. doi: 10.1016/j.jneumeth.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.SAS Institute Inc. SAS/STAT 9.1 User’s Guide. Cary, NC: SAS Publishing; 2004. [Google Scholar]
- 26.ABI. Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR. Applied Biosystems Product Guide. 2004:1–60. [Google Scholar]
- 27.Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, et al. Alzheimer’s-related endosome dysfunction in Down syndrome is A{beta}-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci USA. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann Neurol. 2004;56:520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- 29.Cataldo AM, Mathews PM, Boiteau AB, Hassinger LC, Peterhoff CM, Jiang Y, et al. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol. 2008;173:370–384. doi: 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coopersmith R, Neve RL. Expression of multiple proteins within single primary cortical neurons using a replication deficient HSV vector. BioTechniques. 1999;27:1156–1160. doi: 10.2144/99276st01. [DOI] [PubMed] [Google Scholar]
- 31.Kajiya M, Shiba H, Fujita T, Ouhara K, Takeda K, Mizuno N, et al. Brain-derived neurotrophic factor stimulates bone/cementum-related protein gene expression in cementoblasts. J Biol Chem. 2008;283:16259–16267. doi: 10.1074/jbc.M800668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wozniak MA, Frost AL, Itzhaki RF. Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimers Dis. 2009;16:341–350. doi: 10.3233/JAD-2009-0963. [DOI] [PubMed] [Google Scholar]
- 33.Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007;429:95–100. doi: 10.1016/j.neulet.2007.09.077. [DOI] [PubMed] [Google Scholar]
- 34.Mufson EJ, Counts SE, Ginsberg SD. Single cell gene expression profiles of nucleus basalis cholinergic neurons in Alzheimer’s disease. Neurochem Res. 2002;27:1035–1048. doi: 10.1023/a:1020952704398. [DOI] [PubMed] [Google Scholar]
- 35.Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, et al. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuyama K, Yanagisawa K. Late endocytic dysfunction as a putative cause of amyloid fibril formation in Alzheimer’s disease. J Neurochem. 2009;109:1250–1260. doi: 10.1111/j.1471-4159.2009.06046.x. [DOI] [PubMed] [Google Scholar]
- 38.Nixon RA. Niemann-Pick Type C disease and Alzheimer’s disease: the APP-endosome connection fattens up. Am J Pathol. 2004;164:757–761. doi: 10.1016/S0002-9440(10)63163-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 2008;4:590–599. doi: 10.4161/auto.6259. [DOI] [PubMed] [Google Scholar]
- 40.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 41.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol. 2004;63:641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- 43.Vepsalainen S, Castren E, Helisalmi S, Iivonen S, Mannermaa A, Lehtovirta M, et al. Genetic analysis of BDNF and TrkB gene polymorphisms in Alzheimer’s disease. J Neurol. 2005;252:423–428. doi: 10.1007/s00415-005-0667-5. [DOI] [PubMed] [Google Scholar]
- 44.Chen Z, Simmons MS, Perry RT, Wiener HW, Harrell LE, Go RC. Genetic association of neurotrophic tyrosine kinase receptor type 2 (NTRK2) With Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147:363–369. doi: 10.1002/ajmg.b.30607. [DOI] [PubMed] [Google Scholar]
- 45.Cozza A, Melissari E, Iacopetti P, Mariotti V, Tedde A, Nacmias B, et al. SNPs in neurotrophin system genes and Alzheimer’s disease in an Italian population. J Alzheimers Dis. 2008;15:61–70. doi: 10.3233/jad-2008-15105. [DOI] [PubMed] [Google Scholar]
- 46.Bao J, Alroy I, Waterman H, Schejter ED, Brodie C, Gruenberg J, et al. Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J Biol Chem. 2000;275:26178–26186. doi: 10.1074/jbc.M002367200. [DOI] [PubMed] [Google Scholar]
- 47.Madshus IH, Stang E. Internalization and intracellular sorting of the EGF receptor: a model for understanding the mechanisms of receptor trafficking. J Cell Sci. 2009;122:3433–3439. doi: 10.1242/jcs.050260. [DOI] [PubMed] [Google Scholar]
- 48.Stern KA, Visser Smit GD, Place TL, Winistorfer S, Piper RC, Lill NL. Epidermal growth factor receptor fate is controlled by Hrs tyrosine phosphorylation sites that regulate Hrs degradation. Mol Cell Biol. 2007;27:888–898. doi: 10.1128/MCB.02356-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, et al. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- 50.Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poon WW, Blurton-Jones M, Tu CH, Feinberg LM, Chabrier MA, Harris JW, et al. beta-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.05.012. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arancibia S, Silhol M, Mouliere F, Meffre J, Hollinger I, Maurice T, et al. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31:316–326. doi: 10.1016/j.nbd.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 54.Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin B, Pirrung MC, Deng L, Li Z, Liu Y, Webster NJ. Neuroprotection by small molecule activators of the nerve growth factor receptor. J Pharmacol Exp Ther. 2007;322:59–69. doi: 10.1124/jpet.106.118034. [DOI] [PubMed] [Google Scholar]
- 56.Longo FM, Yang T, Knowles JK, Xie Y, Moore LA, Massa SM. Small molecule neurotrophin receptor ligands: novel strategies for targeting Alzheimer’s disease mechanisms. Curr Alzheimer Res. 2007;4:503–506. doi: 10.2174/156720507783018316. [DOI] [PubMed] [Google Scholar]
- 57.Skaper SD. The biology of neurotrophins, signalling pathways, and functional peptide mimetics of neurotrophins and their receptors. CNS Neurol Disord Drug Targets. 2008;7:46–62. doi: 10.2174/187152708783885174. [DOI] [PubMed] [Google Scholar]
- 58.Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang YZ, Pan E, Xiong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57:546–558. doi: 10.1016/j.neuron.2007.11.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.