

SUMMARY
Neuronal migration leads to a highly organized laminar structure in the mammalian brain and its mis-regulation causes lissencephaly, behavioral and cognitive defects. Reelin signaling, mediated in part by a key adaptor, disabled-1 (Dab1), plays a critical but incompletely understood role in this process. We found that the neuron-specific RNA binding protein Nova2 regulates neuronal migration in late-generated cortical and Purkinje neurons. An unbiased HITS-CLIP and exon junction array search for Nova-dependent RNAs at E14.5 focused on components of the reelin pathway revealed only one candidate—an alternatively spliced isoform of Dab1 (Dab1.7bc). In utero electroporation demonstrated that Dab1.7bc was sufficient to induce neuronal migration defects in wild-type mice and exacerbate defects when Dab1 levels were reduced, while Dab1 overexpression mitigates defects in Nova2-null mice. Thus Nova2 regulates an RNA switch controlling the ability of Dab1 to mediate neuronal responsiveness to reelin signaling and neuronal migration, suggesting new links between splicing regulation, brain disease and development.
INTRODUCTION
The laminar structure of the mammalian brain is generated by waves of neuronal migration that are coordinated in time and space. Human and mouse genetic studies have revealed critical genes such as Reelin-Dab1 pathway, doublecortin (DCX), LIS1 and NUDEL that are implicated in this process (Ayala et al., 2007). Proper lamination of the mammalian neocortex is dependent on an inside-out gradient of reelin protein secreted from Cajal-Retzius cells. This process is unique to mammals, as lower vertebrates do not use radial “inside-out migration” (Molnar et al., 2006; Rakic, 2003), although they do make reelin proteins. Therefore, evolutionary modifications and amplifications of reelin-Dab1 signaling are thought to play a key role in evolution of the mammalian cortex, although their precise nature remains uncertain. These distinctions are of clinical as well as fundamental interest, as defects of these signaling pathways are implicated in many brain disorders, including Lissencephaly, epilepsy and schizophrenia (Ayala et al., 2007; Gleeson and Walsh, 2000; Rakic, 2003).
Brain development, including regional identity, mitotic activity, cell fate determination and differentiation, have been related to regional specification by transcriptional factors, extrinsic factors and cell-cell interactions. However, sequencing of complete genomes, and the realization that both humans and worms both harbor on the order of 20,000 protein coding genes, while human and chimpanzee genomes are 99.7% identical, has led to the belief that species diversity may depend upon the post-transcriptional complexity of RNA processing (Licatalosi and Darnell, 2010; Ponting et al., 2009). Indeed, the fastest evolving difference identified to date between human and chimpanzee is in a gene encoding a noncoding RNA (HAR1F) that is expressed during the critical period for neuronal migration where it co-localizes with reelin in Cajal-Retzius cells (Amadio and Walsh, 2006; Pollard et al., 2006). It may be anticipated that RNA regulation will play roles in establishing brain complexity during development, although how such regulation may be manifest is currently unclear.
Genetic studies have indicated that RNA mis-regulation is involved in a growing list of neurodevelopmental disorders, including fragile X syndrome, spinal muscular atrophy, spinocerebellar ataxias, and others (Cooper et al., 2009; Licatalosi and Darnell, 2006; Lukong et al., 2008). Relatively little is known about specific roles that RNA regulation plays in the developing mammalian brain in vivo. Two different forms of ROBO3, arising by alternative splicing, act tightly in commissural axon midline crossing (Chen et al., 2008), although the factors that regulate ROBO3 splicing are unknown. In neuronal cell lines, a switch between expression of two paralogous RNA binding proteins, Ptbp1 and Ptbp2 (Boutz et al., 2007; Makeyev et al., 2007), as well as expression of the Musashi and Elavl family of RNA binding proteins (Sakakibara et al., 2002; Okano et al., 2005; Akamatsu et al., 1999), regulate RNA transcripts during mammalian neuronal differentiation.
Alternative splicing provides a major means of generating protein diversity from single genes, providing a significant source of variation in cell type, tissue type and species variation (Li et al., 2007; Wang et al., 2008). Alternative splicing is regulated through the action of numerous RNA binding proteins (RBPs). Recent advances have dramatically increased our understanding of this by regulation, through the development of genome wide analyses such as microarray, including exon-junction array and tiling array, RNA Seq and HITS-CLIP to assess protein-RNA interactions in vivo (Blencowe et al., 2009; Chen and Manley, 2009; Licatalosi and Darnell, 2010). These techniques have begun to lead to the discovery of new features of RNA regulation, suggesting for example that the position of protein-RNA interaction determines the outcome of alternative splicing (Chen and Manley, 2009), or that regulatory factors such as Nova control of transcripts encoding biologically coherent sets of proteins (Huang et al., 2005; Ruggiu et al., 2009; Ule et al., 2005).
Here we find that Nova2 deficiency results in aberrant migration of cortical and Purkinje neurons, but not alterations of neural progenitor cell fate, suggesting that alternative splicing mediated by Nova2 regulates migration of post-mitotic neurons and plays an important role in lamination of the brain. Exon junction arrays (Ule et al., 2005) and HITS-CLIP (Licatalosi et al., 2008) were used previously to generate genome-wide maps of functional Nova-RNA interactions (Ule et al., 2006), and here we used these strategies in a more directed manner to refine the search for biologically relevant Nova2 RNA targets in the Reelin signaling pathway. These studies revealed that only one RNA among ~20 candidates assessed, encoding Dab1, an adaptor molecule mediating signaling from reelin receptors, is the responsible transcript mediating Nova2-dependent neuronal migration. These data illustrate the use of high throughput RNA methods to reveal a previously unsuspected alternative splicing switch that controls neuronal migration, demonstrating that Nova2 regulation of alternative splicing plays a critical role in brain development by regulating reelin-Dab1 signaling in a neuron-cell autonomous manner.
RESULTS
Laminar structure defect of neocortex in Nova2 null mice
Nova was identified as a target antigen in a human neurological syndrome characterized by ataxia and tremor (Darnell, 2006), a phenotype also reported in mice harboring defects in neuronal migration (Rice and Curran, 2001). Although biochemical and genetic studies revealed that Nova proteins regulate transcripts encoding synapse related molecules in the postnatal brain (Darnell, 2006; Licatalosi et al., 2008; Ule et al., 2005), developmental actions of Nova proteins have not previously been explored. We found that at mouse embryonic day 14.5 (E14.5) and postnatal day 0 (P0), Nova2 protein is expressed in all cortical layers, including post-mitotic migrating neurons and the cortical plate, whereas Nova1 is not detectable (Yang et al., 1998) (Figure S1A–B). We compared the laminar structure of the neocortex in wild type (WT) and Nova2 null (Nova2 KO; see (Huang et al., 2005)) mice at postnatal 10 days by histological analysis with markers for transcription factors expressed in different cortical layers (Molyneaux et al., 2007). Double staining with Brn2 (Layer II/III and Vb) and Er81 (Layer V) revealed a reduced upper cortical layer and a concomitant increase in mis-localized Brn2 positive cells/Er81 negative cells under layer V neuron in Nova2 KO (Figure 1A and S1C). These ectopic cells expressed the neuronal marker NeuN (Figure S1C). Similarly, Cux1 (Layer II–IV) positive/Brn2 negative cells were also mis-located in the deep cortical layer (Figure S1E). We noted two visible bands of neurons positive for Cux1; one localized in the correct position in the upper layer, and a second mis-localized in the peri-ventricular area under FoxP2 positive (layer V–VI) neurons (Figure 1B). The FoxP2 and ectopic Cux1 positive neurons were distinct (Figure 1B), suggesting that the cell fate for the ectopic layer II–IV and layer V neurons remained distinct. This mutant phenotype was very similar to that seen in mice in which the reelin receptor ApoER2 is disrupted (Hack et al., 2007), and suggested that some late-born neurons in Nova2 KO do not migrate normally to the brain surface.
Figure 1. Loss of Nova2 results in mislocation of late-generated cortical neurons, and, to a lesser degree, disorganization of early generated cortical neurons.

(A) Sagittal sections of postnatal P10 WT and Nova2 KO (N2KO) neocortex at the level of the anterior hippocampus were immnnostained with layer-specific marker proteins, Brn-2 (layer II–III, V in part; green), Er81 (layer V; red) and DAPI (blue). (B) FoxP2 (layerV–VI; green) or Cux1 (layerII–IV; Red). High magnification of deep layers in Nova2 KO (box region in B). These two marker proteins are not completely merged. Scale bar; 100 μm. WT.
Nova2 regulates neuronal migration in late-generated neurons
To clarify when the aberrantly positioned neurons in Nova2 KO were born, we performed BrdU labeling experiments to label cell populations born at E14 or E16, when late-generated neurons are born and analyzed their location at P0. In WT cortex, a single peak of labeled cells was evident in the outer cortex (Figure 2A; fraction 3 and Figure S2A), with cells in a more superficial cortical layer after E16 relative to E14 BrdU injection, as expected given the “inside-out” nature of laminar cortical generation (Cooper, 2008; Rakic, 2003). In contrast, BrdU labeling of cells in Nova2 KO cortex at either E14 or E16 led to formation of a reduced number of outer cortical neurons, and a new ectopic BrdU-positive band of cells in deep cortical layers (Figure 2AB; fraction 6 and Figure S2A). We confirmed that these mis-migrated BrdU positive cells were Cux1 positive neurons in P10 Nova2 KO deep layers (Figure S2B). We also found a small number of mislocalized Er81 positive neurons in P10 Nova2 KO (Figure S1D arrow). These cells, committed to a deep layer, are known to born at early stages of cortical development, around E12–13 (Takahashi et al., 1999). BrdU labeling of cells in Nova2 KO at E12 exhibit no ectopic BrdU positive band and very few mislocalized cells when analyzed at P0 (Figure S2C arrow). These results suggest that Nova2 regulates neuronal migration during cortical development and its action is mainly late-born neurons rather than early-born neurons.
Figure 2. Nova2 controls laminar formation of late generated neurons.

(A,C) BrdU-labeling was performed at E14.0 or E16.0 and cortical sections of WT or Nova2 KO animals were immunostained with BrdU antibody at P0 as indicated. (B,D) Graphs show a quantification of BrdU positive cells percentage in each of 10 bins (along the dorsal/ventral axis) relative to the total number of BrdU positive cells in WT or Nova2 KO. Data represents mean +/– standard deviation (SD) from experiments using three independent animals for each genotype. Results showed statistically significant differences for Nova2 KO relative to WT in both E14 and E16 BrdU injections (F(9,35) = 6.56, P<0.0001, F(9.35) = 2.9348, p=0.0106, respectively by repeated measures ANOVA). Scale bar; 100 μm.
Next, to assess the proliferation and localization of neural stem/progenitor cells in Nova2 KO cortex, we performed BrdU short-term labeling and immunostaining with a marker for M phase (phospho-Histone 3; PH3). WT and Nova2 KO embryos fixed 1hr after BrdU injections at E14.5 showed no difference in BrdU positive cell number or the location of PH3 positive cells, which were located at the ventricular surface with minor populations in sub-ventricular zone in both genetic backgrounds (Figure 3A–C and Figure S3). Taken together, these BrdU labeling experiments indicated that the observed neuronal migration defect in Nova2 KO mice could be due to a failure of postmitotic neurons to reach brain surface, but not to a defect in proliferation of progenitor cells or morphology of radial glia (Figure 3A). Therefore, these data suggest that the migration defect is intrinsic to newborn cortical neurons after their final cell division.
Figure 3. Neural progenitor cells in WT and Nova2 KO cortex.
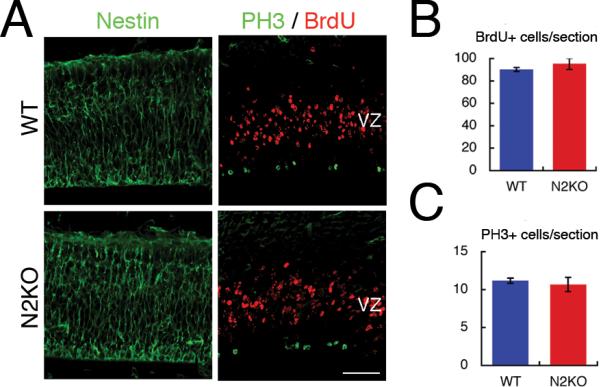
(A) BrdU was injected 1 hr before fixation. Coronal sections of embryonic day 14.5 cortex in wild type and Nova2 KO brains were immunostained with antibodies against Nestin (left panel; green), which is neural progenitor marker protein, BrdU (right panel; red) and PH3 (right panel; green). (B) This graph represents the number of BrdU positive cells per section. (C) This graph represents the number of PH3 positive cells per sections. Error bar indicates SD of three biological replicates. Scale bar; 50 μm. PH3; phospho-Histone3.
Nova2 regulates radial migration of Purkinje neuron
Some cases of mutant mice showing cortical migration defects also have ectopic Purkinje neurons in the cerebellum due to radial migration defects of Purkinje neurons (Howell et al., 1997b; Larouche et al., 2008; Sheldon et al., 1997). Therefore, we performed histological analysis of the developing cerebellum in Nova2 KO mice. In the E18.5 cerebellum, calbindin positive young migrating Purkinje neurons express Nova2, but not Nova1 (which is expressed in the deep nuclei), as evident by the absence of Purkinje neuron staining with a pan-Nova antibody at E18.5 (Figure S4A). Calbindin positive Purkinje neurons showed delayed migration in E18.5 Nova2 KO (Figure S4B) and ectopic Purkinje neurons were evident in the cerebellar white matter at P10 (p<0.01; Figure 4A). These ectopic Purkinje cells co-expressed other Purkinje markers such as RORα and IP3R1, but not markers for deep cerebellar nuclei neurons (Figure 4A and Figure S4C–E). Migrating Purkinje neurons were BrdU labeled at E11.5 and the cerebellum fixed at P4, revealing that ~15% of BrdU/calbindin double positive cells were ectopically located in the white matter of Nova2 KO cerebellum (Figure 4BC). These data demonstrate that Nova2 is necessary for correct cellular positioning of both cortical and Purkinje neurons via radial migration.
Figure 4. Nova2 regulates Purkinje neurons migration.

(A) Calbindin staining (green) and counter staining with DAPI (blue) in P10 wild type and Nova2 KO sagittal sections at the level of vermis in cerebellum. High magnification orthogonal picture was enlarged from the boxed region in Nova2 KO cerebellum and double stained with RORα (red). Quantitative result of ectopic calbindin positive neurons in cerebellum white matter in wild type and Nova2 KO using three biological replicates (each ~6 sections in cerebellar vermis). Error bar represents SD using three biological replicates. CaBP; calbindin, Scale bar; 100 μm (B,C) BrdU-labeling at E11.5 and immunostained with BrdU (red) and calbindin (green) antibodies at P4 cerebellum. (B) Whole cerebellum in WT and Nova2 KO at the level of vermis in cerebellum (C) High magnifications of Purkinje cell layers in WT (box region in B in wild type) and Nova2 KO (box region in B in N2KO-1) and white matter in Nova2 KO (box region in B in N2KO-2). Arrow indicates BrdU/calbindin double positive cells. Bar graph represents the percent of BrdU/calbindin double positive cells in white matter per total double positive cells in whole cerebellum. Error bar represents SD using three biological replicates. Asterisk indicates P-value < 0.05 by Student t-test. Scale bar; 200 μm (B), 20 μm (C) EGL; External granule layer, PCL; Purkinje cell layer, GL; granule cell layer N2KO; Nova2 KO
Nova2 modulates reelin signaling via alternative splicing of Dab1 during Neural development
Defects in cortical and Purkinje neuronal migration seen in Nova2 KO mice are similar to those seen with defects in reelin signaling (D'Arcangelo et al., 1995; Ogawa et al., 1995; Larouche et al., 2008). Moreover, we have previously found that Nova regulates RNAs involved in synaptic molecules. Thus we hypothesized that in the developing brain (E14.5), Nova2 might be a modifier of RNAs encoding reelin signaling molecules, leading to the neuronal migration defect we observed in Nova2 KO mice. We did an unbiased search for potential Nova action on such RNAs, using HITS-CLIP, a method (Jensen and Darnell, 2008; Ule et al., 2005) that has been used to map Nova-RNA direct interaction sites in vivo (Licatalosi et al., 2008), and an exon junction array previously shown to be able to identify RNA processing differences in Nova KO and WT brain (Licatalosi et al., 2008; Ule et al., 2005). Nova2 HITS-CLIP in E14.5 cortex identified 27,576 robust clusters Nova2 binding sites in the mouse transcriptome (Figure S5A), including several reelin pathway RNAs. When exon junction arrays were probed with three biologic replicates of E14.5 WT or Nova2 KO cortical RNA, we identified no splicing changes in ~20 transcripts encoding reelin and other migration signaling pathway proteins (Figure S5A and data not shown) other than a very significant change (ΔI ~ 0.8) (Licatalosi et al., 2008; Ule et al., 2005) in the Dab1 transcript. This change was in a previously noted evolutionarily conserved pair of alternative exons with no defined function, termed Dab555 (Howell et al., 1997a) or Dab1 exons 7b and 7c (Jelen et al., 2007; Ule et al., 2006)} (hereafter referred to as Dab1.7bc). We had previously noted Dab1.7bc exons were among ~41 exons mis-regulated in Nova1/Nova2 double KO mice (Ule et al., 2006).
The candidate reelin pathway RNAs identified as harboring Nova binding sites by HITS-CLIP were tested by RT-PCR. We found no evidence for a specific early (E14.5) action of Nova2 to regulate any RNA other than Dab1. For example, alpha-catenin2, which was previously identified as a Nova2 target RNA (Ule et al., 2005), and whose deficiency also leads to ectopic Purkinje neurons (Park et al., 2002), was regulated by Nova2 at P10, but had little Nova regulation evident at E14.5 (Figure S5A). Other transcripts had no significant Nova-dependent changes in splicing or steady state mRNA levels at E14.5 (Figure S5AB), suggesting that these components of the reelin signaling pathway were unlikely to underlie the neuronal migration defect evident in Nova2 null mice.
In contrast, Dab1 regulation correlated well with the observed neuronal migration defect. The Dab1 transcript harbored a large number of Nova2 CLIP tags in intronic sequences upstream of the Dab1 7b/c exons (Figure 5AB), in positions predicted (Licatalosi et al., 2008; Ule et al., 2006) to inhibit exon inclusion. To address the possibility of Nova2-dependent regulation of Dab1 splicing during development, we performed RT-PCR analysis of RNA obtained from the cortex of mice ranging in age from E10.5 to P10. This data confirmed that the Dab1.7bc exons were markedly over represented in E14.5–E16.5 Nova2 KO cortex relative to WT cortex, where they were barely utilized (<20% of total) from E14.5–E16.5 onward; the Dab1.7bc exons were downregulated in Nova2 KO cortex postnatally (Figure 5CD and Figure S5C). These results suggest that Nova2 normally suppresses Dab1.7bc exon inclusion during the time of cortical development, and that other factors further suppress these exons later in postnatal cortex.
Figure 5. Nova2 regulates alternative splicing of Dab1 in a context dependent manner.

(A) Scheme of Dab1 protein, which consists of PTB domain and tyrosine phospholylation sites, and amino acids encoded by Dab1 7bc exons. PTB: phospho-tyrosine binding. (B) Nova2 CLIP tags from E14.5 cortex (blue/cyan/purple) and P7 brains (red/orange/pink) by each three individual CLIP experiments, map to Nova2 regulating Dab1 transcripts. The alternatively spliced region is highlighted. Two red boxes in highlight indicate Dab1 7bc exons. (C) Alternative splicing analysis of Dab1 in wild type and Nova2 KO using 5 time points: E10.5 brain, E14.5 cortex (Ctx), E16.5 Ctx, P0 Ctx and P10 Ctx total RNA. Each corresponding band was confirmed by sequencing. (D) Quantitation of Dab1 vs Dab1.7bc mRNA expression data in (C); each point represent the average of three biological replicates (see detailed data in Figure S5C contains the ratio of Dab1.7b/c and steady state level of total Dab1 between embryo and postnatal cortex in Figure S5B). (E–G) Two upstream intronic sequences of Dab1 7b and 7c exons are necessary for the regulation of alternative splicing of Dab1.7bc exons as a pair by Nova2. (E) Schematic representation of pGloDab1.7bc and its derivative minigenes containing the mouse intronic regions surrounding and including exon 7b and 7c between human globin constitutive exon1 and 3. Asterisks indicate site of point mutations in Nova binding YCAY clusters (see Figure S5D, F for details). (F) total RNA was isolated from 293T cells transiently transfected with WT or mutant pGloDab1.7bc minigenes (0.25 μg)and pNova2 (0.5 μg) as indicated, and spliced products analyzed by RT-PCR. Three biological replicates were used in each analysis. (G) Model of Nova2-mediated Dab1.7bc exon repression. .
To assess whether Nova2 might act directly on the Dab1 transcript to inhibit Dab1.7bc exon inclusion, we generated a reporter minigene expressing the Dab1.7bc genomic region. After transfecting this reporter into 293T cells (which do not express Nova2) together with increasing amounts of a Nova-expressing plasmid, we measured Dab1.7bc alternative splicing by RT-PCR. These experiments confirmed that Nova2 inhibits exon inclusion in both 7b/c exons as a pair (Figure S5D–F). We then generated minigenes in which three clusters of Nova binding sites (YCAY elements), identified by HITS-CLIP (Figure 5B) and bioinformatic analysis, were independently mutated. Cotransfection assays revealed that the Nova2 binding sites upstream of both 7b and 7c exons were necessary for Dab1.7bc exon exclusion by Nova2; each binding site was independently necessary for Nova-inhibition of the immediate downstream exon (Dab1.7b or c), and mutation of both sites abrogated Nova-inhibited of the Dab1.7bc exons as a pair (Figure 5E–G). Interestingly, these two regions were highly conserved element among mammalian genome compared with the third Nova2 binding site downstream of the Dab1 7bc exons, and mutation of this third site had no effect on Nova-dependent splicing (mt3; Figure 5F). Therefore Nova acts directly on the Dab1 transcript to inhibit inclusion of the paired 7b/c exons in a manner consistent with the rules of the positional Nova-dependent splicing map (Licatalosi and Darnell, 2010; Licatalosi et al., 2008; Ule et al., 2006).
The aberrant Dab1.7bc exons encode a 33 amino acid peptide of unknown function. We confirmed that a larger Dab1 protein isoform was the predominant isoform detectable by Western blot of E14.5 mouse cortex in the absence of Nova2. This band co-migrated with Dab1.7bc expressed in transfected tissue culture cells (Figure S6AB), and was downregulated postnatally in WT or Nova2 KO cortex (data not shown). Dab1 protein levels are known to be tightly controlled by phosphorylation following reelin-induction of tyrosine kinases and subsequent protein degradation, and Dab1 levels are increased in reelin deficient mice (Hiesberger et al., 1999; Park and Curran, 2008; Rice and Curran, 2001). Consistent with these prior studies, c-src over-expression induced equivalent levels of Dab1 or Dab1.7bc phosphorylation and interaction with Crk; however, in contrast to Dab1, the majority of Dab1.7bc is stable by Western blot after c-src over-expression, perhaps by escaping ubiquitination (Feng et al., 2007) (Figure S6C). Taken together, our observations prompted us to explore whether aberrant production of the Dab1.7bc isoform could account for the Nova2 cortical migration phenotype.
The balance between Dab1 and the Dab1.7bc isoform regulates neuronal migration
To test whether Dab1.7bc was sufficient to induce the neuronal migration defect evident in Nova2 KO mice, we performed in utero electroporation experiments, an extension of an approach originally used to study the effects of neuronal Elavl proteins on neuronal development (Akamatsu et al., 1999). In these studies, we introduced plasmid DNA encoding WT Dab1 or the Dab1.7bc isoform into lateral ventricle in the developing neocortex of WT or Nova2 KO mice at E14.5. Control pcDNA3 vector or Dab1 expressing plasmids were electroporated together with a plasmid encoding mRFP to allow tracing of electroporated cells in vivo. Following electroporation, pregnant mothers were allowed to continue gestation, and 4 days later pups were sacrificed and the effects of plasmid transfection assessed by monitoring mRFP labeled cells. In WT mice, 80% of mRFP labeled cells were clearly located at upper cortical plates 4 days after electroporation. Remarkably, transfection of plasmid encoding Dab1.7bc in WT mice caused cortical plate neurons to migrate to aberrant positions (Figure 6AB), recapitulating the migration defect seen in Nova2 KO mice (Figure 1–2). Although we cannot determine the degree of Dab1.7bc overexpression in the small number of neurons transfected in these experiments, we anticipate that they are high relative to the very small amount of residual Dab1 (Figure S6), given the robust expression of Dab1.7bc from this construct in tissue culture cells (Figure S6A), and evidence that this promoter expresses well in early postmitotic cortical neurons (Hagino-Yamagishi et al., 1997). These results indicate that the Dab1.7bc isoform itself is able to induce a neuronal migration defect.
Figure 6. WT Dab1 mitigates and Dab1.7bc exacerbates neuronal migration defects.

Expression plasmids of mRFP together with control or Dab1 or Dab1.7bc were injected into lateral ventricle of E14.5 embryo brain and electroporated into the region of neocortex. (A) WT (C) Nova2 KO brains were fixed at 4 days later and examined. High magnifications were double stained with Cux-1 and mRFP. (B and D) Graphs quantifying mRFP positive cells in each of 4 regions (marginal zone (MZ), and upper, middle and lower cortical plates (CP), as illustrated in (A) and (C)) as a percentage of total mRFP positive cells. Scale bar; 100 μm, 20 μm (high magnifications). Data represent the mean +/− SD of three WT or Nova2 KO brains. Results were compared to WT (B) or Nova2 KO (D) control vector and showed no significant difference in WT brain transfected with Dab1 (p=0.2), but significant differences with WT brain transfected with Dab1.7bc (p<0.001) or Nova2 KO brain transfected with Dab1 (p<0.01) by repeated measures ANOVA. (E–F) Control and Dab1shRNA expression vectors were injected together with Dab1 or Dab1.7bc expression vectors into the lateral ventricle of E14.5 embryo brain, electroporated and analyzed 4 days later. (E) indicates representative results and (F) shows the ratio of the mRFP positive cells in upper cortical layer per total mRFP positive cells in cortical plate as indicated in (E). Error bar represents SD using three biological replicates. Asterisk indicates P-value < 0.01 by Student t-test.
In addition to the de novo production of Dab1.7bc, Nova2 KO mice are deficient in production of the WT Dab1 signaling molecule; Dab1 levels are decreased to 35% WT levels in E14.5 Nova2 KO cortex (Figure S6AB). Therefore, we next addressed whether reintroduction of the WT Dab1 isoform can mitigate the migration defect in Nova2 KO neocortex. When control plasmid together with mRFP was introduced into Nova2 KO neocortex, mRFP positive cells were separated in upper cortical plate and aberrant cortical positions (Figure 6CD), reflecting the neuronal migration defect. In the rescue experiment, in which plasmid expressing the WT Dab1 isoform was electroporated into Nova2 KO cells, we found that the migration of the mRFP positive cells population was significantly shifted toward its normal position in the upper cortical plate. Importantly both the mis-positioned and normal positioned mRFP positive cells in Nova2 KO are Cux1 positive, which marks layer II–IV neurons (Figure 6CD). These results demonstrate that the neuronal splicing factor Nova2 regulates neuronal migration during embryonic brain development through reelin-Dab1 signaling pathway, and that maintenance of an appropriate balance between Dab1 and Dab1.7bc isoforms may be required for normal migration.
We next assessed whether Dab1.7bc is able to dysregulate neuronal migration in the setting of reduced Dab levels. We performed Dab1 knock-down (KD) with Dab1 specific shRNAs introduced by in utero electroporation. As expected, this led to a neuronal migration defect in WT mice (a 43 % reduction in the number of neurons successfully migrating to the upper cortical plate compared with control (Figure 6EF). Again, we were unable to measure the degree of Dab1 knockdown in the small number of neurons transfected in vivo, but this shRNA construct led to ~80–90% reduction of Dab1 expression in tissue culture cells (Figure S6D); the small residual Dab1 expression may explain the failure to completely block migration in this experiment. Importantly, this phenotype by Dab1 KD could be rescued by co-electroporation of the wild-type Dab1 expression construct (Figure 6DE). Remarkably, co-transfection of the Dab1.7bc expression construct not only failed to rescue the migration defect in Dab1 KD neurons, but exacerbated the migration defect (Figure 6DE). These results suggest that Dab1.7bc itself may have a toxic or dominant negative action to antagonize wild-type Dab1 action in mediating neuronal migration.
DISCUSSION
In the present work we link Nova2 function to a switch in alternative splicing of Dab1 isoforms and in neuronal migration. Genome wide mapping of Nova:RNA interactions in the brain with HITS-CLIP and exon junction arrays was previously used to identify and predict functional interaction sites (Licatalosi et al., 2008; Ule et al., 2005; Racca et al., 2010). The present study extends these findings to identify a new role for Nova2 in the developing brain by combining histologic analysis of cortical development with a directed analysis of HITS-CLIP and exon array data to examine regulation of the reelin pathway. Biologic experiments in Nova2 null mice demonstrate that Nova2 is a novel reelin signal modifier and regulates radial migration of cortical neuron and Purkinje neurons. To our knowledge, this provides the first in vivo evidence that alternative splicing regulation controls neuronal migration, as well as providing a mechanism for how it does so.
Histological analysis showed that Nova2 deficiency results in ectopic cortical neurons and Purkinje neurons (Figures 1 and 4). In the neocortex, cux1 (layer II–IV) and brn-2 (Layer II/III and Va) positive late-generated neurons are more severely mis-localized in Nova2 null mice in deep layers than early-generated neurons (Er81 (LayerV) and FoxP2 (layerVI) positive cells; Figure 1 and S1). These late generated neurons generally use radial glia processes as a scaffold to move long distances from the proliferative zone to the superficial cortical layer when the layer of cortex formed in an inside-out manner, while early generated neurons mainly use somal translocation mode that is unnecessary in a radial fiber early in development when the radial glial scaffolds has not formed yet (Rakic, 2003; Rakic and Caviness, 1995; Nadarajah et al., 2001; Nadarajah and Parnavelas, 2002). Importantly, the radial glia forms normally in Nova2 KO; histone H3 staining and short term BrdU labeling showed that the proliferation and mitotic polarity of radial glia, which give rise to new born neurons, were normal in Nova2 null mice (Figure 3). This suggests that the mis-location of neurons in Nova2 null mice is due to a defect of neuronal migration after their terminal cell division, a conclusion supported by the BrdU birth-date studies and electroporation analysis of mRFP positive cells in Nova2 KO animals demonstrating neuronal migration defects (Figure 2, 4 and 6).
We pursued the mechanism underlying the neuronal migration defect in Nova2 KO mice after recognizing its similarity with previously reported migration defects. Many previous genetics approaches have shown that reelin-Dab1 signaling shows neuronal migration defects in both cortical and Purkinje neurons, as noted in Nova2 null mice. In fact, mis-location of cortical and Purkinje neurons in Nova2 KO is very similar to defects seen in Apoer2 KO mice, which is one of two reelin receptors. Apoer2 KO mice displayed migration defects of late-generated cortical neuron and ectopic Purkinje cells. Interestingly loss of function of another reelin receptor, Vldlr, leads to a different phenotype, showing milder phenotypes for cortical migration and more severe Purkinje neurons migration than Apoer2 KO (Hack et al., 2007; Larouche et al., 2008). Apoer2/Vldlr double mutants, or mutations in their adaptor molecule, Dab1, show defects as severe as reeler phenotypes (Trommsdorff et al., 1999). In this study, we could not find any differences in the steady state RNA levels, alternative splicing or proteins level of reelin or its receptors in Nova2 KO embryo and postnatal brain (Figure 5 and S5).
We focused on alternative splicing changes of Dab1 exon7b/c, previously reported as one of many Nova targets encoding synapse-related proteins (Ule et al., 2006). Interestingly, a previous study had shown that the Dab1.7bc/Dab1 ratio is decreased during differentiations of P19 cells induced by retinoic acid (Bar et al., 2003). Moreover, in chick, where only the Dab1.7c exon is present prior to a likely genome duplication event in mammals (Figure 5; (Ule and Darnell, 2007; Jelen et al., 2007), Dab1.7c was found to be regulated during chick retina development and was again correlated with post-mitotic differentiation (Katyal and Godbout, 2004). These studies are consistent with the results found here for a role for Nova2 in regulating Dab1.7bc expression and post-mitotic neuronal migration. HITS-CLIP, exon array/RT-PCR and in vitro minigene analysis studies demonstrate that Nova2 directly binds predicted YCAY clusters to block inclusion of the Dab1.7bc exons (Figure 5). Although it was previously suggested that Dab1.7bc was expressed and therefore might act earlier in development (Bar et al., 2003), we find that its action is restricted to migrating neurons versus proliferative neural progenitors due to the restricted expression of Nova2 (Figure S1). In summary, our data demonstrate not only a function for Dab1.7bc, but the mechanism by which it is regulated, by direct Nova2 action on upstream YCAY clusters to block exon inclusion in a manner consistent with the predicted map of Nova functional exon regulation (Figure 5 and (Licatalosi et al., 2008; Ule et al., 2006)).
Overexpression of Dab1.7bc induced neuronal migration defects in WT animals and exacerbated this defect in Dab1 KD brain after in utero electroporation analysis, and conversely wild type Dab1 can rescue the neuronal migration defect in Nova2 KO mice (Figure 6). Interestingly this alternative splicing change is regulated in context/age dependent manner. Nova-dependent alternative splicing suppression of Dab1.7bc is highest in the E14.5–E16.5 cortex, the critical time window in neuronal migration for late-born neurons in neocortex, and is gradually diminished up to P10 (Figure 5). Presumably, Dab1.7bc has a transient but, given its evolutionary conservation, important role early in development that is regulated by Nova2 during neuronal migration, and other RNA binding proteins take in the postnatal brain. Recent studies revealed that Reelin signaling is involved in not only early development, including neuronal migration but also dendrite genesis and synapse formation at the postnatal (Niu et al., 2008), and it seems reasonable to assume that Dab1.7bc and/or alternative switching between the two Dab1 isoforms also has a physiological role in the adult brain.
Previous studies have shown that Dab1 is necessary for two critical and quite different events in radial migration, (i) to migrate past early generated neurons and (ii) to allow detachment from radial glia to cause migrating neurons to stay put at a correct layer position (Sanada et al., 2004). Detailed single mutant analysis of the two reelin receptors revealed that Apoer2 signaling plays an important role in proper migration of late generated neurons (i), while Vldlr signaling mediates a “stop signal” to prevent migrating neurons into marginal zones (ii) (Hack et al., 2007). Additionally, these two receptors show completely different cellular localization by a fractionation analysis of lipid rafts—ApoER2 but not VLDLR is mainly localized to lipid rafts, which is caveolin1-enriched fractions (Mayer et al., 2006). Nova2 KO phenotypes are very similar to Apoer2 KO, but not Vldlr KO mice, showing two visible late-generated neuronal bands and not invasion of migrated neurons into the marginal zone (Figures 1–2). Our data are therefore consistent with the Dab1/Dab1.7bc balance acting downstream of ApoER2 but not VLDLR, perhaps through localization of receptor trafficking and protein degradation (Hack et al., 2007; Larouche et al., 2008; Mayer et al., 2006).
Dab1 can orchestrate many downstream signaling pathway including PI3K-Akt, crk, LIS1, integrin and notch signaling (Ayala et al., 2007; Hashimoto-Torii et al., 2008). We do not know whether the Dab1.7bc protein domain might affect the ability of the Dab1 PTB domain to interact with crk. But the Dab1.7bc does appear to escape the degradation pathway after src induction (Figure S6). Improved reagents will be required to undertake more detailed localization and biochemical analysis of Dab1 isoforms.
Our observations suggest that Nova-dependent regulation of Dab1/Dab1.7bc splicing is able to switch neurons from a reelin-responsive to a reelin-unresponsive state, thereby mediating inside-out cortical lamination. Our results add Nova2 dependent control of neuronal migration to the list of factors that contribute to the complexity of cortical development, and suggest that alternative splicing switches may provide a general means of regulating key developmental events in mammalian biology and developmental neurologic disorders.
EXPERIMENTAL PROCEDURES
Histology and BrdU analysis
Postnatal Mice were perfused transcardially with 4% paraformaldehyde phosphate buffered saline and brains removed and postfixed in same fixative followed by cryo-protection with 30% sucrose/PBS for cryostat (16 μm) (CM3050S, LEICA) or sliced with a vibratome (50 μm) (VT1000S, LEICA). For 5-bromodeoxyuridine (BrdU) analysis, the pregnant mothers were injected with BrdU (Sigma-Aldrich) dissolved in 0.9% NaCl (50 mg/kg, i.p.) at indicated periods. The embryos or P0 pups were then processed for Immunohistochemistry (IHC). Sections were incubated in 2N HCl for 30 min at room temperature for anti-BrdU staining or boiled in 10 mM citric acid buffer (pH6.0) for 5min × 3 times for Nova2, PH3, calbindin, IP3R1, GFP, NeuN and RORα before IHC. Sections were incubated overnight at 4°C with primary antibodies (see supplemental information) followed by incubation with Alexa-dye conjugated secondary antibodies (Invitrogen 1:1000) or by the combination of biotinylated secondary antibodies (Jackson laboratory 1:500), the VECTASTAIN Elite ABC kit (Vector Laboratories) and the visualization by using the TSA Fluorescence system (PerkinElmer Life). The images of immunostained specimens were collected by either a universal fluorescence microscope (Axiophot 2) or a confocal laser scan microscope (LSM510) (Carl Zeiss) at the Bio-Imaging Resource Center at The Rockefeller University.
Western, RT-PCR and qRT-PCR
Western blotting and RT-PCR analyses were done as described previously. Quantitative PCR were used at least three biological replicates with a MyiQ single-color real-time PCR detection system, using mouse β-actin or GAPDH as an internal control, and calculated with ΔΔ Ct methods.
Exon junction microarray
Nova-dependent alternate splicing was assessed using exon junction arrays as described (Licatalosi et al., 2008).
HITS-CLIP
CLIP was performed on E14.5 wild mouse cortex using three biological replicates as described (Licatalosi et al., 2008) and anti-goat IgG was used as a negative control. In PCR amplification, high-throughput sequencing was performed using Solexa-primers at The Rockefeller University Genome Resource Center. Sequence tags were aligned to the mouse genome (mm9) by BOWTIE. Unique tags were collected by eliminating duplicates of those tags that had the same sequence start on the same strand.
| total read | Annotated to genome | unique tags | |
|---|---|---|---|
| E14.5Ctxl-N2 | 8,782,833 | 1,758,473 | 71,152 |
| E14.5Ctx2-N2 | 9,581,435 | 2,497,832 | 60,406 |
| E14.5Ctx3-N2 | 9,112,841 | 1,840,583 | 37,589 |
In utero electroporation
DNA solution (1 μl) in PBS containing 0.01% fast green was injected into the lateral ventricle of E14.5 embryos as previously described (Tabata and Nakajima, 2001). After injection, electronic pulses of 30 V were charged six times at 950-ms intervals using a square-pulse electroporator (CUY21EDIT; Protech). The plasmid mRFP-pcXN2 (0.5 μg/μl), an expression vector under the control of the CAG promoter, was electroporated together with pcDNA3-Dab1 or Dab1.7bc (1.0 μg/μl). Four days after electroporation, the embryos were then processed for IHC (see above).
Minigene analysis
A Dab1 7bc chimeric minigene (termed pGloDab1.7bc) was derived from the pGLO γ (Dredge and Darnell, 2003) by the insertion of 822 bp of mouse genomic sequence around Dab1 7b and 7c exons by PCR amplification. All mutant pGloDab1.7bc constructs were generated by site-direct PCR (detailed mutant sequences were described in Figure S5D, F). All fragments generated by PCR were confirmed by complete DNA sequencing. 293T cells were transfected with 0.25 μg of the appropriated reporter minigenes together with 0.5 μg of pNova2 (Licatalosi et al., 2008) or EGFP control vector, using Fugene6HD (Roche) as described by the manufacturer. After 40hr, cells were rinsed with ice-cold PBS and collected by TRIZOLE for RNA extraction. Reverse transcription-PCR was performed using PCR primers to human β-globin were E1F and E3R as described previously (Dredge and Darnell, 2003).
HIGHLIGHTS.
Alternative splicing regulates cortical neuronal migration
Nova2 null mice have cortical and Purkinje neuronal migration defects
HITS-CLIP effectively screened Nova splicing targets for a role in neuronal migration
Nova2 regulates the balance of Dab1 spliced isoforms and thereby reelin signaling
Email alert paragraph.
Defects in neuronal migration lead to lissencephaly, behavioral and cognitive defects. Reelin signaling, mediated in part disabled-1 (Dab1), plays a critical role in this process. HITS-CLIP was used to identify RNA targets of the neuron-specific RNA binding protein Nova2 mediating a neuronal migration defect in KO mice. One candidate, an alternatively spliced isoform of Dab1, was sufficient to induce neuronal migration defects, and Dab1 overexpression mitigated defects in Nova2-KO brain. Thus Nova2 regulates an RNA switch controlling neuronal migration.
Supplementary Material
Acknowledgements
We are grateful to Dr. J. Kohyama for helpful advice with in utero electroporation, Dr. J. Ule for help with ASPIRE analysis of exon junction arrays, Dr. N. Heintz for L7–L10a-GFP mice, Dr. Y. Okada and Dr. H. Okano for mRFP-pcXN2, which is mRFP1 cDNA originally from Dr. R. Tsien and Dr. S Arber for Er81 antibody, Dr. H.J. Okano for generating the HuC expression vector construct, Dr. A. Sali and U Pieper for peptide predictions. Drs. J. Darnell, G. Dunn, M. Frias, M. Ruggiu, and C. Zhang for thoughtful comments, J. Fak and N. Rehman for technical support and all the members of Dr. Darnell's laboratory for encouragement and kind support. This work was supported by National Institutes of Health Grants to R.B.D, and the Rockefeller University Hospital CTSA Grant (UL1 RR024143) from the National Center for Research Resources at the National Institutes of Health. M.Y was supported by a JSPS postdoctoral fellowship for research abroad, KANAE Foundation for the promotion of medical science and Mochida Memorial Foundation for medical and pharmaceutical research. R.B.D is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akamatsu W, Okano HJ, Osumi N, Inoue T, Nakamura S, Sakakibara SI, Miura M, Matsuo N, Darnell RB, Okano H. Mammalian ELAV-like neuronal RNA-binding proteins HuB and HuC promote neuronal development in both the central and the peripheral nervous systems. Proc Natl Acad Sci U S A. 1999;96:9885–9890. doi: 10.1073/pnas.96.17.9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadio JP, Walsh CA. Brain evolution and uniqueness in the human genome. Cell. 2006;126:1033–1035. doi: 10.1016/j.cell.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Bar I, Tissir F, Lambert de Rouvroit C, De Backer O, Goffinet AM. The gene encoding disabled-1 (DAB1), the intracellular adaptor of the Reelin pathway, reveals unusual complexity in human and mouse. J Biol Chem. 2003;278:5802–5812. doi: 10.1074/jbc.M207178200. [DOI] [PubMed] [Google Scholar]
- Blencowe BJ, Ahmad S, Lee LJ. Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev. 2009;23:1379–1386. doi: 10.1101/gad.1788009. [DOI] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares MJ, Black DL. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009 doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Gore BB, Long H, Ma L, Tessier-Lavigne M. Alternative splicing of the Robo3 axon guidance receptor governs the midline switch from attraction to repulsion. Neuron. 2008;58:325–332. doi: 10.1016/j.neuron.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Cooper JA. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 2008;31:113–119. doi: 10.1016/j.tins.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Darnell RB. Developing global insight into RNA regulation. Cold Spring Harb Symp Quant Biol. 2006;71:321–327. doi: 10.1101/sqb.2006.71.002. [DOI] [PubMed] [Google Scholar]
- Dredge BK, Darnell RB. Nova regulates GABA(A) receptor gamma2 alternative splicing via a distal downstream UCAU-rich intronic splicing enhancer. Molecular & Cellular Biology. 2003;23:4687–4700. doi: 10.1128/MCB.23.13.4687-4700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21:2717–2730. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352–359. doi: 10.1016/s0166-2236(00)01607-6. [DOI] [PubMed] [Google Scholar]
- Hack I, Hellwig S, Junghans D, Brunne B, Bock HH, Zhao S, Frotscher M. Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development. 2007;134:3883–3891. doi: 10.1242/dev.005447. [DOI] [PubMed] [Google Scholar]
- Hagino-Yamagishi K, Saijoh Y, Ikeda M, Ichikawa M, Minamikawa-Tachino R, Hamada H. Predominant expression of Brn-2 in the postmitotic neurons of the developing mouse neocortex. Brain Res. 1997;752:261–268. doi: 10.1016/s0006-8993(96)01472-2. [DOI] [PubMed] [Google Scholar]
- Hashimoto-Torii K, Torii M, Sarkisian MR, Bartley CM, Shen J, Radtke F, Gridley T, Sestan N, Rakic P. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron. 2008;60:273–284. doi: 10.1016/j.neuron.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Howell BW, Gertler FB, Cooper JA. Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J. 1997a;16:121–132. doi: 10.1093/emboj/16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997b;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Huang CS, Shi SH, Ule J, Ruggiu M, Barker LA, Darnell RB, Jan YN, Jan LY. Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell. 2005;123:105–118. doi: 10.1016/j.cell.2005.07.033. [DOI] [PubMed] [Google Scholar]
- Jelen N, Ule J, Zivin M, Darnell RB. Evolution of Nova-dependent splicing regulation in the brain. PLoS Genet. 2007;3:1838–1847. doi: 10.1371/journal.pgen.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KB, Darnell RB. CLIP: crosslinking and immunoprecipitation of in vivo RNA targets of RNA-binding proteins. Methods Mol Biol. 2008;488:85–98. doi: 10.1007/978-1-60327-475-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyal S, Godbout R. Alternative splicing modulates Disabled-1 (Dab1) function in the developing chick retina. EMBO J. 2004;23:1878–1888. doi: 10.1038/sj.emboj.7600185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larouche M, Beffert U, Herz J, Hawkes R. The Reelin receptors Apoer2 and Vldlr coordinate the patterning of Purkinje cell topography in the developing mouse cerebellum. PLoS One. 2008;3:e1653. doi: 10.1371/journal.pone.0001653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. 2006;52:93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24:416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 2007;27:435–448. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer H, Duit S, Hauser C, Schneider WJ, Nimpf J. Reconstitution of the Reelin signaling pathway in fibroblasts demonstrates that Dab1 phosphorylation is independent of receptor localization in lipid rafts. Mol Cell Biol. 2006;26:19–27. doi: 10.1128/MCB.26.1.19-27.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar Z, Metin C, Stoykova A, Tarabykin V, Price DJ, Francis F, Meyer G, Dehay C, Kennedy H. Comparative aspects of cerebral cortical development. Eur J Neurosci. 2006;23:921–934. doi: 10.1111/j.1460-9568.2006.04611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Parnavelas JG. Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci. 2002;3:423–432. doi: 10.1038/nrn845. [DOI] [PubMed] [Google Scholar]
- Niu S, Yabut O, D'Arcangelo G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J Neurosci. 2008;28:10339–10348. doi: 10.1523/JNEUROSCI.1917-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Okano H, Kawahara H, Toriya M, Nakao K, Shibata S, Imai T. Function of RNA-binding protein Musashi-1 in stem cells. Exp Cell Res. 2005;306:349–356. doi: 10.1016/j.yexcr.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Park C, Falls W, Finger JH, Longo-Guess CM, Ackerman SL. Deletion in Catna2, encoding alpha N-catenin, causes cerebellar and hippocampal lamination defects and impaired startle modulation. Nat Genet. 2002;31:279–284. doi: 10.1038/ng908. [DOI] [PubMed] [Google Scholar]
- Park TJ, Curran T. Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J Neurosci. 2008;28:13551–13562. doi: 10.1523/JNEUROSCI.4323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, Kern AD, Dehay C, Igel H, Ares MJ, Vanderhaeghen P, Haussler D. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Racca C, Gardiol A, Eom T, Ule J, Triller A, Darnell RB. The neuronal splicing factor Nova co-localizes with target RNAs in the dendrite. Front Neural Circuits. 2010 doi: 10.3389/neuro.04.005.2010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex. 2003;13:541–549. doi: 10.1093/cercor/13.6.541. [DOI] [PubMed] [Google Scholar]
- Rakic P, Caviness VSJ. Cortical development: view from neurological mutants two decades later. Neuron. 1995;14:1101–1104. doi: 10.1016/0896-6273(95)90258-9. [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Ruggiu M, Herbst R, Kim N, Jevsek M, Fak JJ, Mann MA, Fischbach G, Burden SJ, Darnell RB. Rescuing Z+ agrin splicing in Nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc Natl Acad Sci U S A. 2009;106:3513–3518. doi: 10.1073/pnas.0813112106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara S, Nakamura Y, Yoshida T, Shibata S, Koike M, Takano H, Ueda S, Uchiyama Y, Noda T, Okano H. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci U S A. 2002;99:15194–15199. doi: 10.1073/pnas.232087499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D'Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VSJ. Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci. 1999;19:10357–10371. doi: 10.1523/JNEUROSCI.19-23-10357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Ule J, Darnell RB. Functional and mechanistic insights from genome-wide studies of splicing regulation in the brain. Adv Exp Med Biol. 2007;623:148–160. doi: 10.1007/978-0-387-77374-2_9. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen K, Mele A, Darnell RB. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37:376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, Darnell RB. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YY, Yin GL, Darnell RB. The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc Natl Acad Sci U S A. 1998;95:13254–13259. doi: 10.1073/pnas.95.22.13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.