

Abstract
The effects of alcohol on Ca2+ signalling remains poorly understood. Here we have investigated the effects of acute ethanol exposure on Ca2+ influx in mouse pancreatic acinar cells. Cells were loaded with fura-2 and the changes in fluorescence were monitored by spectrofluorimetry and imaging analysis. Stimulation of cells with 20 pM cholecystokinin evoked an oscillatory pattern in [Ca2+]c, both in the presence and in the absence of extracellular Ca2+. Stimulation of cells with cholecystokinin in the presence of 50 mM ethanol led to a transformation of physiological oscillations into a single transient increase in [Ca2+]c. This effect was observed when Ca2+ was present in the extracellular medium, and did not appear in its absence. Addition of 1 mM CaCl2 to the extracellular medium, following release of Ca2+ from intracellular stores by stimulation of cells with 1 nM cholecystokinin or 1 μM thapsigargin in the absence of extracellular Ca2+, was followed by an increase in [Ca2+]c. Ca2+ influx was increased in the presence of 50 mM ethanol. The anti-oxidant cinnamtannin B-1 (10 μM) or inhibition of alcohol dehydrogenase by 4-MP (1 mM), significantly reduced Ca2+ influx evoked by cholecystokinin in the presence of ethanol. In summary, intoxicating concentrations of ethanol may lead to over stimulation of pancreatic acinar cells by cholecystokinin. This might be partially explained by the generation of reactive oxygen species and an increased Ca2+ entry in the presence of ethanol. Potentially ethanol might lead to Ca2+ overload, which is a common pathological precursor that is implicated in pancreatitis.
Keywords: calcium, cholecystokinin, ethanol, pancreas, reactive oxygen species
Cholecystokinin acts in a concentration-dependent fashion on its binding sites in pancreatic acinar cells, and results in the generation of different second messengers in the signal cascades that lead to enzymatic secretion (González et al. 1999). The activation of phospholipase C-linked receptors by cholecystokinin produces an increase in the concentration of inositol 1,4,5-trisphosphate (IP3) in the cytosol. IP3 in turn releases calcium (Ca2+) from cytoplasmic stores leading to an increase in cytosolic free calcium concentration ([Ca2+]c) (Streb et al. 1983). Ca2+ signals are not only a result of release from intracellular stores but also a coordinate influx from the extracellular space (Putney 1988), Ca2+ extrusion across the plasma membrane (Carafoli 1991) as well as Ca2+ uptake into intracellular organelles (González et al. 1997a). A rise in [Ca2+]c is an important early signal by which physiological secretagogues elicit the release of digestive enzymes from pancreatic acinar cells.
In pancreatic acinar cells, cholecystokinin regulates secretion via generation of repetitive local cytosolic Ca2+ signals in the apical pole. In addition, the spatiotemporal pattern of agonist-induced Ca2+ signals is of critical importance for exocytosis of enzymes (Habara & Kanno 1994).
Cytosolic Ca2+ overload may result from excessive loss of Ca2+ from the intracellular stores by its release through specific channels and by the inhibition of Ca2+ pumps, and/or followed by entry of extracellular Ca2+ (Petersen & Sutton 2006; González et al. 2008). Therefore, it is important to know how Ca2+ homeostasis is regulated because, despite being one of the initial steps involved in cellular function, abnormally elevated [Ca2+]c is a shared phenomenon that could induce trypsin premature activation (Ding et al. 2006). This is a previous step that can trigger acute pancreatitis and necrosis (Ward et al. 1995; Petersen & Sutton 2006).
Although cholecystokinin is a major physiological regulator of secretion by the exocrine pancreas, an over stimulation can cause injury to the pancreas which may lead to dysfunction of the gland and even to the activation of death signalling pathways involving caspases (Saluja et al. 1999; Gukovskaya et al. 2002).
It has been long recognized that a significant percentage of patients with pancreatitis often presents a history of excessive alcohol consumption. Alcohol is involved in approximately 40% of cases of acute pancreatitis (Kristiansen et al. 2008; Ellis et al. 2009). Nevertheless, the mechanisms underlying alcohol-derived deleterious effects are not completely understood.
The exocrine pancreas can metabolize ethanol mainly via an oxidative pathway involving the enzymes alcohol dehydrogenase (ADH) and cytochrome P4502E1, although a non-oxidative pathway involving fatty acid ethyl ester synthases has also been proposed (Criddle et al. 2004). Metabolism of ethanol by pancreatic acinar cells and the consequent generation of toxic metabolites are postulated to play an important role in the development of alcohol-related pancreatic injury (Petersen & Sutton 2006; Lawrencia et al. 2009). In these terms, ethanol may induce its deleterious effects in pancreatic acinar cells through the generation of oxidative and non-oxidative metabolites (Criddle et al. 2006; González et al. 2006; Palmieri et al. 2007).
One of the early events leading to alcoholic pancreatitis seems to be the effect of ethanol on stimulus-secretion coupling mechanisms. It has been suggested that ethanol acts to sensitize the pancreas to the deleterious effects of other stimuli such as the physiological agonist cholecystokinin octapeptide (CCK-8), which leads to an inflammatory response and pancreatitis. This effect is in part mediated by augmenting activation of proinflamatory factors (Pandol et al. 2003), although a decrease in the levels of prostaglandin E2 by ethanol could be involved in alcohol-induced injury in the pancreas too (Siegmund et al. 2003). Furthermore, it has been proposed that ethanol induces generation of oxygen radicals in pancreatic acinar cells (Wittel et al. 2003; González et al. 2006; Lawrencia et al. 2009), impairs CCK-8-evoked secretion of digestive enzymes (González et al. 2006) and leads to a delayed or reduced Ca2+ extrusion from the cytosol towards the extracellular space or into the cytosolic stores (González et al. 2008).
Despite the great amount of research carried out to understand the action of ethanol on cell physiology, the involvement of Ca2+ metabolism in ethanol-evoked effects needs to be further studied, because most cellular activity is initiated by changes in [Ca2+]c. In this study, we have investigated the early effects of acute ethanol exposure on CCK-8-evoked Ca2+ influx in mouse pancreatic acinar cells. Our objective is to shed more light on the effects of ethanol on CCK-8 physiological actions. The findings will contribute to a better understanding of the mechanisms by which ethanol causes pancreatic disorders.
Materials and methods
Animals and chemicals
Adult male Swiss mice were used for this study. Mice were humanely handled and killed in accordance to the institutional Bioethical Committee. Fura-2-AM was obtained from Invitrogen (Spain) and cinnamtannin B-1 from Alexis (Switzerland). All other materials used were obtained from Sigma Chemicals Co. (Spain).
Preparation of isolated pancreatic acinar cells
A suspension of mouse pancreatic single cells and small acini (2–4 cell clusters) was prepared by collagenase treatment following a previously described method (González et al. 1997a). Briefly, the pancreas was incubated in the presence of collagenase for 10 min at 37 °C. This enzymatic digestion of the tissue was followed by gently pipetting the cell suspension through tips of decreasing diameter for mechanical dissociation of the cells. After centrifugation, cells were resuspended in a buffer without collagenase. With this isolation procedure, single cells, as well as small clusters consisting of up to five cells, were obtained.
Throughout the preparation procedure, as well as during dye loading, we employed a physiological solution containing: 130 mM NaCl, 4.7 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 1.2 mM KH2PO4, 10 mM glucose, 10 mM Hepes, 0.01% trypsin inhibitor (soybean) and 0.2% bovine serum albumin (pH = 7.4 adjusted with NaOH). During fluorescence determinations, trypsin inhibitor and bovine serum albumin were not included in the medium. In experiments where Ca2+ free medium is indicated, Ca2+ was omitted from the extracellular solution and 250 μM EGTA was added to the cells in the cuvette.
The viability of cell suspensions, either of cells obtained after the isolation procedure and at the end of the experimental treatments, was assayed by trypan blue exclusion test. Cell viability was not significantly changed and was greater than 95%. After loading with fluorescent dyes, cells were kept at 4 °C until use and the experiments were performed within the next 4 h.
Dye loading and determination of intracellular free Ca2+ concentration ([Ca2+]c)
Freshly isolated mouse pancreatic acinar cells were loaded with fura-2-acetoxymethyl ester (4 μM) at room temperature (23–25 °C) for 40 min as previously established (González et al. 1999).
Monitorization of changes in fluorescence signals was performed by placing aliquots of dye-loaded pancreatic acinar cells into a cuvette in a fluorescence spectrophotometer (Cary Eclipse, mod. EL 0210-6479 from Varian, USA). Cells were continuously stirred and experiments were performed at 37 °C. All stimuli were dissolved in the extracellular Na-Hepes buffer without Ca2+, and were directly added into the cuvette to yield the final concentration required.
For determination of [Ca2+]c fura-2 loaded pancreatic acinar cells were alternatively excited at 340/380 nm and fluorescence emission was detected at 505 nm.
Several values of the Ca2+ response were calculated: (1) the plateau of [Ca2+]c, calculated once [Ca2+]c had reached a stable level after Ca2+ influx; (2) the amount of initial Ca2+ influx was estimated using the integral of the rise in [Ca2+]c for 90 s after addition of CaCl2; (3) to calculate the initial rate of Ca2+ elevation after the addition of Ca2+ to the medium, the traces were fitted to the equation y = A + Kx, where K is the slope. This analysis was performed for the first 30 s of the rising phase of Ca2+ entry process. Results for [Ca2+]c are expressed in nM ± SEM (n), where n is the number of independent experiments, following the calibration method proposed by Grynkiewicz et al. (1985).
In single cell experiments, monitorization of Ca2+-dependent fluorescence signals was carried out by placing small aliquots of cell suspensions into a cover slip mounted on an experimental perfusion chamber, and placed on the stage of an epifluorescence inverted microscope (Nikon Diaphot T200; Melville, NY, USA). The cells were continuously superfused with Na-Hepes buffer. For fluorescence change determination, an image acquisition and analysis system for video microscopy was employed (Hamamatsu Photonics, Hamamatsu, Japan). Cells were alternatively excited with light from a xenon arc lamp passed through a high-speed monochromator (Polychrome IV, Photonics) at 340/380 nm. Fluorescence emission at 505 nm was detected using a cooled digital CCD camera (Hisca CCD C-6790, Hamamatsu) and recorded using dedicated software (Aquacosmos 2.5; Hamamatsu Photonics). All fluorescence measurements were made from areas considered individual cells. In this series of experiments, results are expressed as the ratio of fluorescence emitted at 340 nm and 380 nm excitation wavelengths.
All stimuli were dissolved in the extracellular Na-Hepes buffer and applied directly to the cells in the perfusion chamber. Experiments were performed at room temperature (23–25 °C).
We can discard an effect of ethanol on fura-2-emission fluorescence that could induce artefacts in the determination of [Ca2+]c. In our experimental conditions, the concentration of ethanol employed did not influence fura-2-derived fluorescence. Furthermore, no apparent morphological changes of pancreatic cells that could introduce errors in Ca2+ measurements in our experimental conditions were observed (data not shown).
Statistical analysis
Statistical analysis of data was performed by one-way analysis of variance (anova) followed by Tukey post hoc test, and only P-values <0.05 were considered statistically significant. For individual comparisons and statistics between individual treatments, we employed Student's t test and only P values <0.05 were considered statistically significant.
Results
CCK-8-evoked changes in [Ca2+]c and effect of ethanol in single cell studies
In the presence of extracellular Ca2+, stimulation of fura-2-loaded cells with 20 pM CCK-8 led to an oscillatory pattern in [Ca2+]c. This pattern of Ca2+ mobilization was observed both in the presence (n = 5 experiments/26 cells studied; Figure 1a) and in the absence (n = 4 experiments; 25 cells studied; Figure 1b) of extracellular Ca2+, although the frequency and amplitude of [Ca2+]c spikes were reduced in the absence of external Ca2+. These patterns of changes in [Ca2+]c have been observed previously (González et al. 1997b, 1999).
Figure 1.
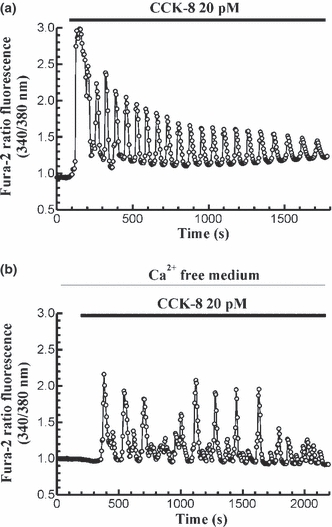
Time-course of changes in [Ca2+]c in response to CCK-8. Fura-2 loaded pancreatic acinar cells were stimulated with 20 pM CCK-8 in the presence (a) and in the absence (b) of Ca2+ in the extracellular medium. The horizontal bars indicate the time during which CCK-8 was applied to the cells. The traces are typical of 4–5 independent experiments.
To study the effects of ethanol on CCK-8-evoked Ca2+ signals, we have chosen a 50 mM ethanol concentration. It is a concentration that falls within the range of alcohol levels found in the blood of intoxicated humans (Lamarche et al. 2004). In addition, this concentration of ethanol induces Ca2+ mobilization and ROS generation, and clearly affects Ca2+ transport mechanisms and amylase secretion in response to CCK-8 in pancreatic acinar cells (González et al. 2006, 2008).
In a set of experiments, we evaluated the effect of ethanol on CCK-8-evoked Ca2+ signals. In the presence of extracellular Ca2+, perfusion of cells with 20 pM CCK-8 induced oscillations in [Ca2+]c as we have shown. The amplitude of oscillations progressively decreased after 15 min in the presence of the agonist in the extracellular solution. Under these conditions, inclusion of 50 mM ethanol in the perfusion medium induced a potentiation of CCK-8-evoked oscillations in [Ca2+]c, which is observed as an increase in the amplitude of Ca2+ spikes in the 55.64% of cells (n = 9 experiments/74 cells of 133 total cells studied; Figure 2a). However, when pancreatic acinar cells were challenged with 20 pM CCK-8 in the absence of Ca2+ in the extracellular medium, ethanol treatment did not induce any change on CCK-8-evoked oscillations in [Ca2+]c (n = 9 experiments, 41 total cells studied; Figure 2b).
Figure 2.
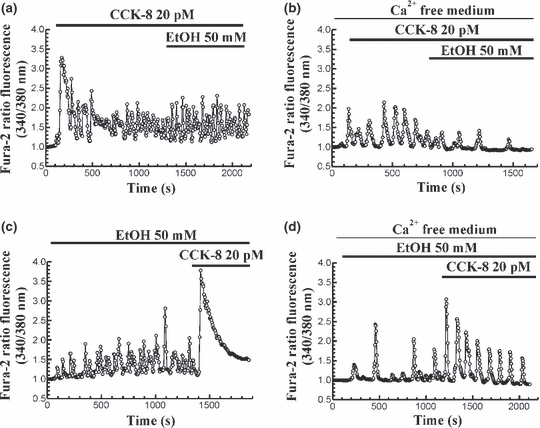
Time-course of CCK-8-evoked changes in [Ca2+]c in the presence of ethanol. (a) Fura-2 loaded pancreatic acinar cells were perfused with 20 pM CCK-8 in the presence of Ca2+ in the extracellular medium. Following CCK-8 stimulation, 50 mM ethanol (EtOH) was included in the perfusion medium. (b) The same protocol was performed in the absence of Ca2+ in the extracellular space (medium containing 250 μM EGTA). (c) Pancreatic acinar cells were perfused with 50 mM ethanol (EtOH) in the presence of Ca2+ in the extracellular medium. In the presence of ethanol, 20 pM CCK-8 was included in the perfusion medium. (d) The same protocol was performed in the absence of Ca2+ in the extracellular medium (containing 250 μM EGTA). The horizontal bars indicate the time during which CCK-8 and ethanol were applied to the cells. The traces are typical of 3–9 independent experiments.
On the other hand, when 50 mM ethanol was first applied to the cells in the presence of Ca2+ in the perfusion medium, an oscillatory mobilization of [Ca2+]c was observed. In the presence of ethanol (50 mM), perfusion of cells with 20 pM CCK-8 evoked a single transient change in [Ca2+]c, similar to that evoked by a supramaximal concentration of CCK-8 in the majority of cells (89.56%) (n = 9 experiments/103 cells of 115 total cells studied; Figure 2c).
However, ethanol failed to increase CCK-8-evoked mobilization of Ca2+ in the absence of external Ca2+, i.e. inclusion of 20 pM CCK-8 in the perfusion medium after stimulation of cells with 50 mM ethanol evoked the typical oscillatory pattern of [Ca2+]c in the majority of cells (86.48% of cells) (n = 3 experiments, 32 cells of 37 total cells studied; Figure 2d).
Changes in [Ca2+]c in response to thapsigargin and effect of ethanol in cell-suspension studies
As the maintenance of secretagogue-evoked oscillations in [Ca2+]c highly depends on the replenishment of cytosolic Ca2+ stores, i.e. endoplasmic reticulum (ER), by Ca2+ entry from the extracellular space, and the potentiation effect of ethanol was only observed when Ca2+ was present in the external medium, it was of interest to analyse the effect of ethanol on Ca2+ influx.
Thapsigargin (Tps), the most potent selective sarcoendoplasmic reticulum Ca2+–ATPase inhibitor, is often used to inhibit this pump (Nielsen et al. 1995). This will release Ca2+ from the ER and consequently will activate Ca2+ influx.
Stimulation of cells with Tps in the absence of extracellular Ca2+, led to a transient change in [Ca2+]c, consisting of an initial increase followed by a decrease of [Ca2+]c towards a value close to the prestimulation level. Subsequent addition of CaCl2 (1 mM) to the external medium resulted in a sustained increase in [Ca2+]c, indicative of Ca2+ entry into the cells. The time-course of changes in [Ca2+]c in response to Tps can be observed in Figure 3a.
Figure 3.
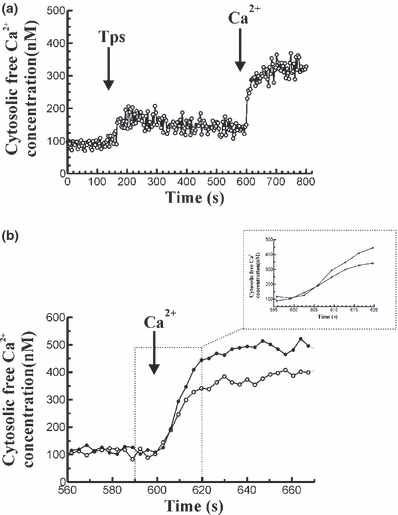
Changes in [Ca2+]c in response to thapsigargin and stimulated Ca2+ influx in mouse pancreatic acinar cells. (a) Cells were stimulated with 1 μM Tps in the absence of Ca2+ in the extracellular medium (250 μM EGTA). At the desired time point 1 mM CaCl2 was added to the cells in the cuvette. The arrow indicates the time point at which Tps and CaCl2 were added. The trace is typical of eight such independent experiments. (b) Following a previous stimulation of mouse pancreatic acinar cells with 1 μM Tps in a Ca2+ free medium (250 μM EGTA) to release of Ca2+ from intracellular stores, 1 mM CaCl2 was added to the cuvette to induce Ca2+ influx. Ca2+ influx was monitorized in the presence of Tps alone (open circles) or in the presence of 50 mM ethanol (full circles). The arrow indicates the time point at which 1 mM CaCl2 was added to the cells. The traces are typical of 7–8 such independent experiments (inset: the time scale has been changed for a better observation of the changes in the rate of Ca2+ influx under the different treatments).
On the other hand, pretreatment of pancreatic acinar cells with 50 mM ethanol for 1 min, prior to Tps (1 μM) stimulation, significantly increased Ca2+ influx following emptying of cytosolic stores by the inhibitor, compared with the values obtained in the presence of CCK-8 alone.
Pretreatment of cells with 50 mM ethanol significantly increased the initial rate of [Ca2+]c elevation (3.10 ± 0.22 Δ nM/s, n = 7 vs. 1.77 ± 0.42 Δ nM/s, n = 8; P < 0.05), the calculated amount of Ca2+ entry (10710 ± 1042 nM, n = 7 vs. 7440 ± 1060, n = 8; P < 0.05) and the plateau of [Ca2+]c (429.60 ± 39.91 nM, n = 7 vs. 310.40 ± 36.81, n = 8; P < 0.05), compared with the effect of Tps alone.
The time course of changes in [Ca2+]c after addition of 1 mM CaCl2 to the cells in the presence of Tps or Tps plus ethanol is shown in Figure 3b.
Changes in [Ca2+]c in response to CCK-8 and effect of ethanol in cell-suspension studies
To better analyse the effects of ethanol on CCK-8 evoked Ca2+ mobilization, we performed a series of experiments in which cells were stimulated with the peptide. These expeiments differ from the former ones in the presence of Tps, in the sense that all Ca2+ pumps are active, and therefore there is no influence of an inhibited pump on the Ca2+ signal observed.
Throughout these studies, pancreatic acinar cells were stimulated with 1 nM CCK-8 alone or in the presence of 50 mM ethanol, in the absence of extracellular Ca2+ (medium containing 250 μM EGTA). This concentration of CCK-8 successfully releases Ca2+ from intracellular stores in a Ca2+ free medium, and therefore decreases the content of Ca2+ in cytosolic stores. As a consequence, store operated Ca2+ entry will take place. Thus, it can be employed to study activation of Ca2+ influx in pancreatic acinar cells (González et al. 1997b, 2002).
Stimulation of cells with 1 nM CCK-8 in the absence of extracellular Ca2+, led to a transient change in [Ca2+]c, consisting of an initial increase followed by a decrease of [Ca2+]c towards a value close to the prestimulation level. Subsequent addition of CaCl2 (1 mM) to the external medium resulted in a sustained increase in [Ca2+]c, indicative of store-operated Ca2+ entry into the cells. The initial rate of [Ca2+]c elevation was 2.36 ± 0.26 Δ nM/s (n = 14), the calculated amount of Ca2+ entry was 4886 ± 298 nM (n = 14) and the plateau of [Ca2+]c was 320.80 ± 19.42 nM (n = 14). The time course of changes in [Ca2+]c following stimulation of cells with 1 nM CCK-8, and after Ca2+ influx, is shown in Figure 4a.
Figure 4.
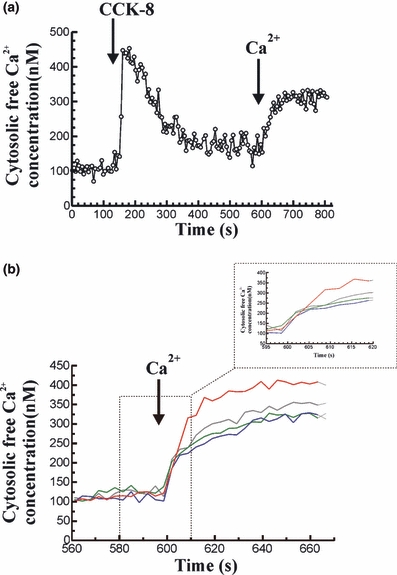
Changes in [Ca2+]c in response to CCK-8 and stimulated Ca2+ influx in mouse pancreatic acinar cells. (a) Cells were stimulated with 1 nM CCK-8 in the absence of Ca2+ in the extracellular medium (250 μM EGTA). At the desired time point, 1 mM CaCl2 was added to the cells in the cuvette. The arrow indicates the time point at which CCK-8 and CaCl2 were added. The trace is typical of 14 such independent experiments. (b) Following a previous stimulation of mouse pancreatic acinar cells with 1 nM CCK-8 in a Ca2+ free medium (250 μM EGTA) to release of Ca2+ from intracellular stores, 1 mM CaCl2 was added to the cuvette to induce Ca2+ influx. Ca2+ influx was monitorized in the presence of CCK-8 alone (blue line), in the presence of 50 mM ethanol (red line), in the presence of 10 μM cinnamtannin B-1 (grey line) or in the presence of the 1 mM of the ADH inhibitor 4-MP (green line). The arrow indicates the time point at which 1 mM CaCl2 was added to the cells. The traces are typical of 7–14 such independent experiments. (Inset: the time scale has been changed for a better observation of the changes in the rate of Ca2+ influx under the different treatments).
Treatment of pancreatic acinar cells with 50 mM ethanol for 1 min, prior to CCK-8 (1 nM) stimulation, significantly increased Ca2+ influx following emptying of cytosolic stores by the agonist, compared with the values obtained in the presence of CCK-8 alone. In the presence of ethanol, the initial rate of [Ca2+]c elevation was 3.26 ± 0.14 Δ nM/s (n = 12), the calculated amount of Ca2+ entry was 7822 ± 733 nM (n = 12) and the plateau of [Ca2+]c was 378.60 ± 13.22 nM (n = 12), which were significantly increased compared with the effect of CCK-8 alone (P < 0.05, P < 0.01 and P < 0.05, respectively). The time course of changes in [Ca2+]c after addition of 1 mM CaCl2 to the cells in the presence of ethanol is shown in Figure 4b.
Effect of alcohol dehydrogenase inhibition on ethanol-induced changes in Ca2+ influx in response to CCK-8
Alcohol dehydrogenase plays an important role in the metabolism of alcohol. The following set of experiments was carried out to evaluate whether the effects of ethanol on Ca2+ influx are direct or not. To characterize the differences in Ca2+ responses between the control experiments with CCK-8 alone and those performed in the presence of ethanol, it was of interest to investigate the changes in Ca2+ influx after inhibition of ADH. These experiments were performed on cell suspensions.
Under these experimental conditions, cells were preincubated for 30 min in the presence of the ADH inhibitor 4-methylpyrazole (4-MP) (Cheung et al. 2003; Dudka et al. 2005; Lawrencia et al. 2009).
The increase evoked by 50 mM ethanol on the initial rate of [Ca2+]c elevation, the calculated amount of Ca2+ entry and the plateau of [Ca2+]c, induced by emptying of cytosolic stores by 1 nM CCK-8, were significantly inhibited by preincubation of pancreatic acinar cells with 1 mM 4-MP (Table 1). These differences were not statistically significant compared with the values obtained following emptying of cytosolic stores by stimulation of pancreatic acinar cells with 1 nM CCK-8 alone. The time course of changes in [Ca2+]c after addition of 1 mM CaCl2 to the cells in the presence of 4-MP is shown in Figure 4b.
Table 1.
Changes in Ca2+ influx in response to CCK-8 alone and in the presence of ethanol, and effect of alcohol dehydrogenase inhibition by 4-MP and cinnamtannin B-1
| CCK-8 | CCK-8/EtOH | CCK-8/EtOH/4-MP | CCK-8/EtOH/CinB | |
|---|---|---|---|---|
| Rate of [Ca2+]c elevation (Δ nM/s) | 2.36 ± 0.26 | 3.25 ± 0.14 n = 12* | 2.47 ± 0.11 n = 7†† | 2.58 ± 0.19 n = 10† |
| Ca2+ entry (nM/90 s) | 4886 ± 298 | 7822 ± 733 n = 12** | 5102 ± 1064 n = 7† | 4954 ± 1094 n = 10† |
| Plateau [Ca2+]c (nM) | 320.80 ± 19.42 | 378.60 ± 13.22 n = 12* | 319.10 ± 15.95 n = 7† | 340.80 ± 12.44 n = 10† |
Pancreatic acinar cells were stimulated, in the absence of extracellular Ca2+, with 1 nM CCK-8 alone, in the presence of 50 mM ethanol (EtOH), after preincubation in the presence of 1 mM of the alcohol dehydrogenase inhibitor 4-MP or following preincubation in the presence of 10 μM of the antioxidant cinnamtannin B-1 (CinB). Following CCK-8 stimulation, 1 mM CaCl2 was added to the extracellular medium and the initial rate of Ca2+ elevation, the calculated amount of Ca2+ entry and the plateau of [Ca2+]c induced by emptying of cytosolic stores were calculated (*P < 0.05; **P < 0.01 vs. CCK-8; †P < 0.05; ††P < 0.01 vs. CCK-8 plus EtOH).
Effect of ethanol-induced ROS generation on CCK-8-evoked Ca2+
To evaluate whether the effects of ethanol are mediated by a secondary ROS generation, we investigated the changes in Ca2+ influx in the presence of the antioxidant cinnamtannin B-1. This substance is a naturally occurring A-type proanthocyanidin, which belongs to a class of polyphenols that is widely distributed throughout the plant kingdom. These compounds have long been investigated as a result of the antioxidant functions (Yamakoshi et al. 1999; Bagchi et al. 2003; Ariga 2004; Bouaziz et al. 2007). These experiments were performed on cell suspensions too.
When Ca2+ influx was analysed after stimulation of pancreatic acinar cells with 1 nM CCK-8 in the presence of 10 μM cinnamtannin B-1 (30 min preincubation), and after 1 min preincubation with 50 mM ethanol, we observed a statistically significant inhibition of the increase evoked by 50 mM ethanol on the initial rate of [Ca2+]c elevation, the calculated amount of Ca2+ entry and the plateau of [Ca2+]c induced by emptying of cytosolic stores by 1 nM CCK-8 (Table 1). These differences were not statistically significant compared with the values obtained following emptying of cytosolic stores by stimulation of pancreatic acinar cells with 1 nM CCK-8 alone. The time course of changes in [Ca2+]c after addition of 1 mM CaCl2 to the cells in the presence of cinnamtannin B-1 is shown in Figure 4b.
Discussion
Alcohol consumption has long been associated with cell damage. Many studies on ethanol toxicity have been conducted following middle or long-term exposition to ethanol (Siegmund et al. 2003; Bhopale et al. 2006; Fortunato et al. 2006; McVicker et al. 2007; Wu et al. 2008). However, attention on the early steps initiated by ethanol is missing. Furthermore, although relevant studies have been performed on the effect of ethanol on the oxidative state of cellular components and/or apoptosis (Saluja et al. 1999; Gukovskaya et al. 2002; Fortunato et al. 2006; Palmieri et al. 2007), its effects on Ca2+ homeostasis have not been dealt with in depth.
To date, it is not clear whether the deleterious actions of ethanol on cellular physiology could result from the production of toxic derivatives from alcohol metabolism or from a direct action of alcohol on cellular structures, which would be morphologically and functionally altered (Bhopale et al. 2006; González et al. 2006; Petersen & Sutton 2006; Palmieri et al. 2007; Szabo et al. 2007; Prosser et al. 2008; Wu et al. 2008).
In this study, we have further investigated the early effects of an acute ethanol exposure on CCK-8-evoked Ca2+ signals in mouse pancreatic acinar cells, because Ca2+ signalling is of critical importance for CCK-8-evoked responses in the exocrine pancreas. We show that stimulation of pancreatic acinar cells with a physiological concentration of CCK-8 (20 pM) evoked an oscillatory pattern of [Ca2+]c, and that ethanol increases the amplitude of CCK-8-evoked oscillations in [Ca2+]c. As the potentiation effect was only observed in the presence of external Ca2+, a stimulated Ca2+ influx in the presence of ethanol might underlie the increased Ca2+ signalling. Our results support previously reported works, which propose that ethanol-induces a sensitization of pancreatic acinar cell to low doses of CCK-8, that by itself does not cause pancreatitis (Pandol et al. 2003).
Our observations cannot be explained by a decrease of the viability of cell suspensions as a result of the experimental treatments. If ethanol should cause a high cell death ratio, a higher Ca2+ influx should therefore be observed. In our hands, the viability of cells was not affected by ethanol treatment. In addition, we can exclude an alteration of cell membrane fluidity and permeability that could potentially affect ion channels and other transporters. Under our experimental conditions, the shape of cells was not affected by the treatments (tested under microscopy observation) and, furthermore, the cells maintained their ability to respond to agonists and to effectively transport Ca2+, as we have previously reported (González et al. 2006; Salazar et al. 2008).
We have further studied the mechanisms by which ethanol evokes its deleterious effects in pancreatic acinar cells, to clarify whether the effects of ethanol are direct or mediated through its metabolization, and if ROS are somehow involved. The inhibition of alcohol metabolization would increase those effects that are resulting from a direct action of ethanol, whereas it would block those effects of ethanol that would be resulting from its metabolites.
Through our study, we have employed the ADH inhibitor 4-MP. This compound substantially decreased ethanol effects on connective tissue growth factor mRNA expression in mouse pancreatic stellate cells (Lawrencia et al. 2009). Inhibition of ADH by 4-MP is concentration-dependent, with a range of action from 1 μM up to 1000 μM in different tissues (Xie & Hurley 1999; Cheung et al. 2003; Dudka et al. 2004, 2005). Thus, the concentration of the ADH inhibitor, we have employed falls within the concentration range successfully employed to inhibit ethanol metabolization by this enzyme. Our results show that the effect of ethanol on Ca2+ influx was significantly reduced in the presence of ADH inhibition. Therefore, alcohol metabolization is necessary for ethanol-evoked stimulation of Ca2+ influx. However, the exact mechanisms by which ethanol evokes its effects need to be further clarified.
Ethanol has been proposed to induce its deleterious effects in pancreatic acinar cells through the generation of ROS (González et al. 2006; Palmieri et al. 2007). Therefore, treatment of cells with the antioxidant cinnamtannin B-1 would block those effects of ethanol because of the generation of ROS. As expected, preincubation of pancreatic acinar cells in the presence of this antioxidant inhibited ethanol-stimulated Ca2+ influx. This supports the hypothesis that ethanol metabolization could lead to ROS generation which, in turn, would affect Ca2+ transport mechanisms, and is in agreement with our previous findings.
Therefore, we can hypothesize that ethanol has an indirect action, mediated through ROS generation, on CCK-8-evoked changes in [Ca2+]c, which consists of a stimulated Ca2+ influx from the extracellular space. This will in turn present its consequences on cellular function. According to our observations, we have shown in a previous work that ethanol induces ROS production in mouse pancreatic acinar cells (González et al. 2006). Our findings are in agreement with others showing that ethanol can induce several cellular reactions which result in a modification of cellular red-ox status, and lead to overproduction of ROS (Wittel et al. 2003; Gonthier et al. 2004; Palmieri et al. 2007).
The close relationship between impairment in Ca2+ homeostasis (i.e. abnormal elevated [Ca2+]c) and pancreatitis has been previously reported (Ward et al. 1995; Criddle et al. 2006; Petersen & Sutton 2006). The factors that precipitate pancreatitis, for example hyperstimulation of cells or excessive ROS generation, cause excessive release of Ca2+ or damage to the integrity of the mechanisms that restore low resting levels of [Ca2+]c. Our results show clear effects of ethanol on Ca2+ mobilization, which are minimized by inhibition of oxidative metabolization of ethanol by alcohol dehydrogenase, and by an antioxidant, suggesting ROS generation. Nevertheless, ethanol can be metabolized by non-oxidative pathways in pancreatic acinar cells too, and thus we cannot exclude that non-oxidative metabolites of ethanol exert an action on Ca2+ metabolism (Criddle et al. 2004, 2006). On the other hand, the relationship between ROS generation and Ca2+-overload is so close that it is difficult to assess which one leads to the other, because either high levels of [Ca2+]c lead to ROS generation (Granados et al. 2004) and/or excessive ROS production induces Ca2+-overload (González et al. 2002, 2005).
In summary, our findings show that ethanol, at intoxicating concentrations, leads to a greater Ca2+ mobilization by a physiological concentration of CCK-8. This behaviour depends on a stimulated Ca2+ influx in the presence of ethanol. Furthermore, inhibition of ethanol metabolization by alcohol dehydrogenase, or preincubation of cells in the presence of the antioxidant cinnamtannin B-1, reverted ethanol-evoked effects on CCK-8-induced Ca2+ influx, which shows that ROS generation by ethanol metabolization is involved in the process. Altogether, this leads to higher levels of [Ca2+]c following stimulation of cells with CCK-8. Ethanol will consequently lead to Ca2+ accumulation within the cytosol, creating a situation potentially leading to cytosolic Ca2+ overload, which is a common pathological precursor that mediates pancreatitis.
Acknowledgments
This work was supported by Junta de Extremadura-FEDER (PRI08A018). Angel Del Castillo-Vaquero was granted a fellowship by the University of Extremadura. The authors would like to thank Mrs. Mercedes Gómez Blázquez for her excellent technical support.
Conflict of Interest
The authors declare that there are no conflicts of interest with other people or organizations that could inappropriately influence (bias) the present work.
References
- Ariga T. The antioxidative function, preventive action on disease and utilization of proanthocyanidins. Biofactors. 2004;21:197–201. doi: 10.1002/biof.552210140. [DOI] [PubMed] [Google Scholar]
- Bagchi D, Sen CK, Ray SD, et al. Molecular mechanisms of cardioprotection by a novel grape seed proanthocyanidin extract. Mutat. Res. 2003;523–524:87–97. doi: 10.1016/s0027-5107(02)00324-x. [DOI] [PubMed] [Google Scholar]
- Bhopale KK, Wu H, Boor PJ, Popov VL, Ansari GA, Kaphalia BS. Metabolic basis of ethanol-induced hepatic and pancreatic injury in hepatic alcohol dehydrogenase deficient deer mice. Alcohol. 2006;39:179–188. doi: 10.1016/j.alcohol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Bouaziz A, Salido S, Linares-Palomino PJ, et al. Cinnamtannin B-1 from bay wood reduces abnormal intracellular Ca2+ homeostasis and platelet hyperaggregability in type 2 diabetes mellitus patients. Arch. Biochem. Biophys. 2007;457:235–242. doi: 10.1016/j.abb.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Carafoli E. Calcium pump of the plasma membrane. Physiol. Rev. 1991;71:129–153. doi: 10.1152/physrev.1991.71.1.129. [DOI] [PubMed] [Google Scholar]
- Cheung C, Davies NG, Hoog JO, Hotchkiss SAM, Smith Pease CK. Species variations in cutaneous alcohol dehydrogenases and aldehyde dehydrogenases may impact on toxicological assessments of alcohols and aldehydes. Toxicology. 2003;184:97–112. doi: 10.1016/s0300-483x(02)00552-8. [DOI] [PubMed] [Google Scholar]
- Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc. Natl Acad. Sci. U.S.A. 2004;101:10738–10743. doi: 10.1073/pnas.0403431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criddle DN, Sutton R, Petersen OH. Role of Ca2+ in pancreatic cell death induced by alcohol metabolites. J. Gastroenterol. Hepatol. 2006;21(Suppl. 3):S14–7. doi: 10.1111/j.1440-1746.2006.04577.x. [DOI] [PubMed] [Google Scholar]
- Ding YX, Yang K, Chin WC. Ethanol augments elevated-[Ca2+]c induced trypsin activation in pancreatic acinar zymogen granules. Biochem. Biophys. Res. Commun. 2006;350:593–597. doi: 10.1016/j.bbrc.2006.09.086. [DOI] [PubMed] [Google Scholar]
- Dudka J, Burdan F, Madej B, et al. Effect of selected alcohol dehydrogenase inhibitors on the human heart lactate dehydrogenase activity – an in vitro study. Acta Physiol. Hung. 2004;91:235–241. doi: 10.1556/APhysiol.91.2004.3-4.7. [DOI] [PubMed] [Google Scholar]
- Dudka J, Burdan F, Szumillo J, et al. Effect of selected alcohol dehydrogenase inhibitors on human hepatic lactate dehydrogenase activity – an in vitro study. J. Appl. Toxicol. 2005;25:549–553. doi: 10.1002/jat.1094. [DOI] [PubMed] [Google Scholar]
- Ellis MP, French JJ, Charnley RM. Acute pancreatitis and the influence of socioeconomic deprivation. Br. J. Surg. 2009;96:74–80. doi: 10.1002/bjs.6414. [DOI] [PubMed] [Google Scholar]
- Fortunato F, Deng X, Gates LK, et al. Pancreatic response to endotoxin after chronic alcohol exposure: switch from apoptosis to necrosis? Am. J. Physiol. 2006;290:G232–G241. doi: 10.1152/ajpgi.00040.2005. [DOI] [PubMed] [Google Scholar]
- Gonthier B, Signorini-Allibe N, Soubeyran A, Eysseric H, Lamarche F, Barret L. Ethanol can modify the effects of certains free radical-generating systems on astrocytes. Alcoholism. 2004;28:526–533. doi: 10.1097/01.alc.0000122271.32522.a7. [DOI] [PubMed] [Google Scholar]
- González A, Camello PJ, Pariente JA, Salido GM. Free cytosolic calcium levels modify intracellular pH in rat pancreatic acini. Biochem. Biophys. Res. Commun. 1997a;230:652–656. doi: 10.1006/bbrc.1996.6026. [DOI] [PubMed] [Google Scholar]
- González A, Pariente JA, Salido GM, Camello PJ. Intracellular pH and calcium signalling in rat pancreatic acinar cells. Pflügers Arch. – Eur. J. Physiol. 1997b;434:609–614. doi: 10.1007/s004240050443. [DOI] [PubMed] [Google Scholar]
- González A, Schmid A, Sternfeld L, Krause E, Salido GM, Schulz I. Cholecystokinin-evoked Ca2+ waves in isolated mouse pancreatic acinar cells are modulated by activation of cytosolic phospholipase A2, phospholipase D, and protein kinase C. Biochem. Biophys. Res. Commun. 1999;261:726–733. doi: 10.1006/bbrc.1999.1106. [DOI] [PubMed] [Google Scholar]
- González A, Schmid A, Salido GM, Camello PJ, Pariente JA. XOD-catalyzed ROS generation mobilizes calcium from intracellular stores in mouse pancreatic acinar cells. Cell. Signal. 2002;14:153–159. doi: 10.1016/s0898-6568(01)00247-9. [DOI] [PubMed] [Google Scholar]
- González A, Granados MP, Salido GM, Pariente JA. H2O2-induced changes in mitochondrial activity in isolated mouse pancreatic acinar cells. Mol. Cell. Biochem. 2005;269:165–173. doi: 10.1007/s11010-005-3457-6. [DOI] [PubMed] [Google Scholar]
- González A, Núñez AM, Granados MP, Pariente JA, Salido GM. Ethanol impairs CCK-8-evoked amylase secretion through Ca2+-mediated ROS generation in mouse pancreatic acinar cells. Alcohol. 2006;38:51–57. doi: 10.1016/j.alcohol.2006.03.002. [DOI] [PubMed] [Google Scholar]
- González A, Pariente JA, Salido GM. Ethanol impairs calcium homeostasis following CCK-8 stimulation in mouse pancreatic acinar cells. Alcohol. 2008;42:565–573. doi: 10.1016/j.alcohol.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Granados MP, Salido GM, Pariente JA, González A. Generation of ROS in response to CCK-8 stimulation in mouse pancreatic acinar cells. Mitochondrion. 2004;3:285–296. doi: 10.1016/j.mito.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gukovskaya AS, Gukovsky I, Jung Y, Mouria M, Pandol S. Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells. J. Biol. Chem. 2002;277:22595–22604. doi: 10.1074/jbc.M202929200. [DOI] [PubMed] [Google Scholar]
- Habara Y, Kanno T. Stimulus-secretion coupling and Ca2+ dynamics in pancreatic acinar cells. Gen. Pharmacol. 1994;25:843–850. doi: 10.1016/0306-3623(94)90085-x. [DOI] [PubMed] [Google Scholar]
- Kristiansen L, Grønbaek M, Becker U, Tolstrup JS. Risk of pancreatitis according to alcohol drinking habits: a population-based cohort study. Am. J. Epidemiol. 2008;168:932–937. doi: 10.1093/aje/kwn222. [DOI] [PubMed] [Google Scholar]
- Lamarche F, Gonthier B, Signorini N, Eysseric H, Barret L. Impact of ethanol and acetaldehyde on DNA and cell viability of cultured neurons. Cell Biol. Toxicol. 2004;20:361–374. doi: 10.1007/s10565-004-0087-9. [DOI] [PubMed] [Google Scholar]
- Lawrencia C, Charrier A, Huang G, Brigstock DR. Ethanol-mediated expression of connective tissue growth factor (CCN2) in mouse pancreatic satellite cells. Growth Factors. 2009;27:91–99. doi: 10.1080/08977190902786319. [DOI] [PubMed] [Google Scholar]
- McVicker BL, Tuma DJ, Casey CA. Effect of ethanol on pro-apoptotic mechanisms in polarized hepatic cells. World J. Gastroenterol. 2007;13:4960–4966. doi: 10.3748/wjg.v13.i37.4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SF, Thastrup O, Pedersen R, Olsen CE, Christensen SB. Structure-activity relationships of analogues of thapsigargin modified at O-11 and O-12. J. Med. Chem. 1995;38:272–276. doi: 10.1021/jm00002a009. [DOI] [PubMed] [Google Scholar]
- Palmieri VO, Grattagliano I, Palasciano G. Ethanol induces secretion of oxidized proteins by pancreatic acinar cells. Cell Biol. Toxicol. 2007;23:459–464. doi: 10.1007/s10565-007-9007-0. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Gukovski I, Satoh A, Lugea A, Gukovskaya AS. Emerging concepts from the mechanism of alcoholic pancreatitis from experimental models. J. Gastroenterol. 2003;38:623–628. doi: 10.1007/s00535-003-1134-7. [DOI] [PubMed] [Google Scholar]
- Petersen OH, Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol. Sci. 2006;27:113–120. doi: 10.1016/j.tips.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Prosser RA, Mangruma CA, Glassb JD. Acute ethanol modulates glutamatergic and serotonergic phase shifts of the mouse circadian clock in vitro. Neuroscience. 2008;152:837–848. doi: 10.1016/j.neuroscience.2007.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1988;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Salazar M, Pariente JA, Salido GM, Gonzalez A. Ethanol induces glutamate secretion by Ca2+ mobilization and ROS generation in rat hippocampal astrocytes. Neurochem. Int. 2008;52:1061–1067. doi: 10.1016/j.neuint.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer M. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am. J. Physiol. 1999;276:G835–G842. doi: 10.1152/ajpgi.1999.276.4.G835. [DOI] [PubMed] [Google Scholar]
- Siegmund E, Weber H, Kasper M, Jonas L. Role of PGE2 in the development of pancreatic injury induced by chronic alcohol feeding in rats. Pancreatology. 2003;3:26–35. doi: 10.1159/000069141. [DOI] [PubMed] [Google Scholar]
- Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol 1,4,5-trisphosphate. Nature. 1983;306:67–69. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P, Oak S, Mayerle J. Effect of ethanol on inflammatory responses. Implications for pancreatitis. Pancreatology. 2007;7:115–123. doi: 10.1159/000104236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JB, Petersen OH, Jenkins SA, Sutton R. Is an elevated concentration of acinar cytosolic free ionised calcium the trigger for acute pancreatitis? Lancet. 1995;346:1016–1019. doi: 10.1016/s0140-6736(95)91695-4. [DOI] [PubMed] [Google Scholar]
- Wittel UA, Bachem M, Siech M. Oxygen radical production precedes alcohol-induced acute pancreatitis in rats. Pancreas. 2003;26:74–80. doi: 10.1097/00006676-200305000-00018. [DOI] [PubMed] [Google Scholar]
- Wu H, Bhopale KK, Ansari GA, Kaphalia BS. Ethanol-induced cytotoxicity in rat pancreatic acinar AR42J cells: role of fatty acid ethyl esters. Alcohol Alcohol. 2008;43:1–8. doi: 10.1093/alcalc/agm044. [DOI] [PubMed] [Google Scholar]
- Xie PT, Hurley TD. Methionine-141 directly influences the binding of 4-methylpyrazole in human sigma sigma alcohol dehydrogenase. Protein Sci. 1999;8:2639–2644. doi: 10.1110/ps.8.12.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakoshi J, Kataoka S, Koga T, Ariga T. Proanthocyanidin-rich extract from grape seeds attenuates the development of aortic atherosclerosis in cholesterol-fed rabbits. Atherosclerosis. 1999;142:139–149. doi: 10.1016/s0021-9150(98)00230-5. [DOI] [PubMed] [Google Scholar]