

Abstract
Length-dependent activation (LDA) is a prominent feature of cardiac muscle characterized by decreases in the Ca2+ levels required to generate force (i.e., increases in Ca2+ sensitivity) when muscle is stretched. Previous studies have concluded that LDA originates from the increased ability of (strong) cross-bridges to attach when muscle is lengthened, which in turn enhances Ca2+ binding to the troponin C (TnC) subunit of the troponin complex. However, our results demonstrate that inhibition of strong cross-bridge attachment with blebbistatin had no effect on the length-dependent modulation of Ca2+ sensitivity (i.e., EC50) or Ca2+ cooperativity, suggesting that LDA originates upstream of cross-bridge attachment. To test whether LDA arises from length dependence of thin-filament activation, we replaced native cTnC with a mutant cTnC (DM-TnC) that is incapable of binding Ca2+. Although progressive replacement of native cTnC with DM-TnC caused an expected monotonic decrease in the maximal force (Fmax), DM-TnC incorporation induced much larger increases in EC50 and decreases in Ca2+ cooperativity at short lengths than at long lengths. These findings support the conclusion that LDA arises primarily from the influence of length on the modulation of the Ca2+ cooperativity arising from interaction between adjacent troponin-tropomyosin complexes on the thin filament.
Introduction
Myofilament activation is a highly cooperative phenomenon where force development increases steeply as a function of the Ca2+ concentration (i.e., [Ca2+]) (1–3). Furthermore, as sarcomere length (SL) is increased, the amount of Ca2+ required to produce 50% of the maximal force (EC50) decreases (2,4,5). Since the ATPase rates per myosin cross-bridge do not depend on SL (6,7), it appears that the length dependence of force production originates from alterations in the number of cross-bridges attached to the thin filaments, a phenomenon known as length-dependent activation (LDA). Current models of the thin filament (8) postulate that troponin/tropomyosin (Tn/Tm) complexes exist in three states: 1), a blocked state, wherein myosin cannot effectively bind actin and no Ca2+ is bound to troponin C (TnC); 2), a closed state, wherein Ca2+ is bound to TnC, leading to a shift in Tn/Tm to expose the myosin binding sites, but myosin is still not bound to actin; and 3), an open state, wherein myosin has bound to actin, thereby causing a further shift in the Tn/Tm complex.
Although early studies concluded that LDA arises from the intrinsic Ca2+ binding properties of TnC (9,10), subsequent studies concluded that TnC alone was not responsible for LDA (11), suggesting that other factors are responsible for LDA. The observation that the addition of exogenous myosin fragments (NEM-S1) increased the number of strongly attached cross-bridges while reducing the level of Ca2+ required to generate force (12–15) supports the possibility that LDA arises from intrinsic differences in binding properties of strongly attached cross-bridges (16) with length. It has been postulated that this length-dependent binding of strongly attached cross-bridges arises from reductions in myofilament lattice spacing that occur with increases in sarcomere length, thus making cross-bridge attachment easier at longer lengths (14,17). In this model, it is predicted that the length-dependent changes in the amount of Ca2+ required for myofilament activation are secondary to increased cross-bridge attachment. Yet it is also conceivable that LDA involves the influence of length on the Ca2+-binding properties of the Tn/Tm complexes along the thin filaments. This Ca2+ binding is highly cooperative as a result of the influence of end-to-end nearest-neighbor interactions between adjacent TnC-Tn/Tm complexes on the energetics of Ca2+ binding (18). Cross-bridge attachment to actin within a given TnC-Tn/Tm complex can also participate in the cooperativity of Ca2+ binding between adjacent TnC-Tn/Tm complexes (19,20).
To dissect the mechanism for LDA, we examined the force-Ca2+ relationship as a function of length in the presence of blebbistatin, an agent that inhibits myosin II ATPase activity by preventing strong cross-bridge attachment (21–23), and after replacing native cTnC with mutant cTnC (DM-cTnC) that cannot bind Ca2+. We found that when force was inhibited with blebbistatin, estimates of the Ca2+ sensitivity (i.e., EC50) and Hill coefficient of the force-Ca2+ relationship were shifted by similar amounts at short and long SLs. By contrast, increased incorporation of DM-cTnC caused larger reductions in the Ca2+ sensitivity (i.e., EC50) and Hill coefficients at short SL than at long SL. Our results establish that the length-dependent change in Ca2+-dependent myofilament activation is independent of the number of strong-binding cross-bridges. Furthermore, reductions in the number of TnC binding sites disrupted the cooperativity of myofilament Ca2+ activation more at shorter sarcomere lengths, suggesting a greater dependence on Ca2+-dependent end-to-end (nearest-neighbor) interactions between neighboring TnC-Tn/Tm at short versus long sarcomere lengths.
Materials and Methods
Skinned rat cardiac trabeculae
All experiments were performed according to institutional guidelines concerning the care and use of experimental animals. Male rats (Sprague-Dawley, n = 40) weighing ∼275–350 g received intraperitoneal injections of 50 mg/kg sodium pentobarbital and 1.5 ml heparin (24). Under deep anesthesia, the heart was excised, transferred to a dissection dish, and perfused retrograde with a modified Krebs-Henseleit solution containing, in mM, 118.5 NaCl, 5 KCl, 2 NaH2PO4, 1.2 MgSO4, 10 glucose, 26.4 NaHCO3, and 0.2 CaCl2, as well as 20 2,3-butanedione monoxime (BDM) to inhibit spontaneous contractions. This solution, when aerated at room temperature with a 95% O2/5% CO2 mixture, had a pH of 7.4. Only single, isolated, unbranched trabeculae located along the exterior wall connecting the myocardial valve to the right ventricular wall were removed for further experiments. The muscles were placed in a dish containing standard relaxing solution with 1% (v/v) Triton X-100 added to chemically permeabilize all membranes to allow solutes to flow freely into and out of the myofilament lattice.
Solutions
The compositions of all solutions have been reported previously (24). Activating solution and relaxing solution were mixed to obtain activating solutions containing between 0.64 and 46.8 μM [Ca2+] (pCa 6.2–4.3).
cTnC double-mutant construction
For these experiments, human cDNA for cardiac TnC was used as the starting material. The amino acid sequences for human and rat cTnC are identical except for amino acid 119, which is isoleucine (I) in the human isoform and methionine (M) in the rat isoform. Accordingly, the sequence was altered by inserting a methionine in place of the isoleucine. In addition, glutamates at positions 40 and 76 were mutated to alanines via site-directed mutagenesis. Glu76, the last amino acid in the binding cleft of site II, was chosen as the likely candidate for mutation, since structural studies of TnC have demonstrated that the Glu76 position plays an important role (25) in the N-terminal rearrangement of TnC. Glu40, which is in the defunct site I of cardiac TnC, has been shown to be involved in the rearrangement of the N-terminal portion of cTnC (26–28) in the presence of TnI, and was chosen to inhibit any structural rearrangement of TnC that might be induced by cTnI during cooperative activation of neighboring active cTn units.
Primer design
E76A. 5′ GGCAGCGGCACGGTGGACTTTGATGAGTTCCTGGTCATGATGGTTCGGTGCA 3′ (Parent)
5′ GGCAGCGGCACGGTCGACTTTGATGCGTTCCTGGTCATGATGGTTCGGTGCA 3′ (Primer)
E40A. 5′ GAGGATGGCTGCATCAGCACCAAGGAGCTGGGCAAGGTGATGAGGATGCTG 3′ (Parent)
5′ GAGGATGGCTGCATCAGCACCAAAGCTTTGGGCAAGGTGATGAGGATGCTG 3′ (Primer)
Silent mutations were introduced into both primers to insert enzymatic cut sites in the new DNA that would make identification of successful mutagenesis quicker and easier, eliminating unnecessary sequencing delays. For the E76A primer, the codon for glutamate (GAG) had a point mutation inserted to convert A to C, thus converting the glutamate to an alanine. The second point mutation converted GTG to GTC, allowing for the introduction of a Sal I cleavage site. For the E40A primer, a little more work was done to insert the new cut site and convert the glutamate to an alanine. A HindIII cleavage site (AAGCTT) was inserted encompassing the codon sequence for lysine-glutamate-leucine. The insertion of the HindIII cleavage site allowed us to convert the glutamate to alanine by switching the GAG codon to GCT, with the silent mutations in the lysine (AAA to AAG) and leucine (CTG to TTG) codons.
cTnC exchange protocol
A stock solution of TnC (either wild-type (WT) or double-mutant (DM)) was made by resuspension in relaxing solution with protease inhibitors (protease inhibitor cocktail P8340, Sigma, St. Louis, MO) to an initial concentration of ∼2 mg/mL, and from this stock, working solutions of 200 μg/mL were prepared. To make the final working exchange solutions used in these experiments, appropriate amounts of the DM and WT TnC were mixed to yield double mutant/wild type mixtures of 0/100, 10/90, 25/75, 50/50, and 75/25. Endogenous cTnC was passively exchanged (100 μL working solution) in fibers via mass action at room temperature for 2 h. Preliminary experiments using 100% DM-cTnC were run to measure the approximate amount of time needed. No force-Ca measurements were performed in these studies.
Blebbistatin studies
To further study the effects of strong-binding cross-bridges on LDA, we used various amounts of a myosin II ATPase inhibitor, blebbistatin. Blebbistatin, recently discovered by means of high-throughput drug screening, is an ATPase-inhibiting agent specific for myosin (21,29–31). The drug is stereospecific: the (−) stereo isoform is the active compound, as determined by in vitro motility assay and by its ability to prevent cell division (32). Blebbistatin has a high affinity only for the myosin II isoform, with no appreciable reactivity for the other members of the myosin superfamily (I, V, and X) (29). Recently, we reported results from experiments on both intact and skinned myocardial tissue, which demonstrated that blebbistatin acts to inhibit force production without altering cross-bridge kinetics or excitation-contraction coupling (22). For these experiments, the fibers were chemically skinned for 6 h, incubated as described in Farman et al. (22) in either 0.1, 0.4, or 1.0 μM blebbistatin overnight in dark conditions, and mounted and viewed with red light in a light box with red filter (Kodak, Rochester, NY).
Mechanical experiments
For mechanical studies (except in the case of the blebbistatin experiments), the muscles were incubated in the skinning solution for 14–18 h at 4°C, then transferred to fresh relaxing solution. Trabeculae 1–2 mm long and 150–250 μm thick were attached to aluminum T-clips and then mounted on hooks between a silicon strain gauge (AE801; SenSonor, Horten Norway) and a servomotor (308, Cambridge Technology, Cambridge, MA; ∼1 ms, 90% step response). SL was adjusted and maintained first at 2.0 ± 0.02μm, then at 2.2 ± 0.02 μm, during activation, using the first-order diffraction band from a He-Ne laser (4). If the final maximal calcium activation run generated <90% of the force of the initial full activation (for each SL), the data for that fiber were discarded; all experiments performed at 15°C ± 0.5°C, a temperature known to dampen force production (33) but used here to lower the amount of rundown in each fiber. As previously described (24), force-[Ca2+] relationships were fit individually to a modified Hill equation,
| (1) |
where Frel is the force as a fraction of maximum force at saturating [Ca2+] (Fmax), EC50 is the [Ca2+] at which the Frel is half of Fmax, and n is the Hill coefficient.
Confocal microscopy
To examine whether both our mutant and wild-type cTnC were incorporating into the skinned trabeculae uniformly, ∼1 mg of each expressed protein was labeled with an excess of maleimide fluorophores (Alexa Fluor 546, Molecular Probes, Eugene, OR) via disulfide bonds with one or both of the cysteines on TnC, according to the protocol provided by Molecular Probes. Adding an excess of BME to the reaction solution quenched remaining fluorophores with the unreacted fluorophore removed via dialysis. Fibers were incubated in either pure wild-type or pure double mutant spiked with a fraction of the labeled protein, according to the initial protein-exchange protocol. After incubation in the cTnC, incubated fibers were fixed in a 3.7% formaldehyde/phosphate-buffered saline solution, then labeled with rhodamine phalloidin (Molecular Probes) to stain for actin; to prevent photobleaching, the fibers were then mounted in prolong gold antifade solution (Molecular Probes). Images were obtained using a Radiance 2100 system with a krypton laser (Bio-Rad, Hercules, CA).
Gel analysis
To determine the amount of incorporation of DM cTnC into the myofilament lattice, we labeled both WT cTnC and DM cTnC with Alexa Fluor maleimide dyes (546 for WT and 488 for DM) from Invitrogen (Carlsbad, CA). After the labeling reaction, each protein mixture was diluted to a concentration of ∼200 μg/mL, the same as the experimental protocol, with the appropriate mixtures prepared. Fibers were incubated with the labeled protein(s) for 2 h, at the end of which time the sample was removed and placed in fresh relaxing solution treated with 8 M urea to give a final urea concentration of 6 M to ensure complete dissociation of the proteins from the myofilament lattice. To ensure complete gel separation of WT and DM cTnC, calcium was added to induce, even in the presence of urea, a conformational change in cTnC. Samples were then run on a 15% SDS-PAGE apparatus and imaged on a Bio-Rad molecular imager FX. After imaging for cTnC, the gel was treated with Blue Silver stain overnight, for total protein content, and the resulting gel was imaged on the same system. The ratio of WT and DM cTnC in each fiber was calculated by first normalizing the cTnC protein content in each lane. The relative amounts of WT or DM cTnC were calculated by dividing the relative intensity of the full DM or WT into the relative intensity of the other lanes. The relative amount of DM to WT for each labeled protein for each gel was averaged to give a representative amount of exchange for the fibers.
Results
Impact of reduced cross-bridge attachment on the force-Ca2+ relationship
It is conceivable that the length-dependent differences observed with DM-cTnC incorporation are mediated by the influence of length on cross-bridge attachment. To examine this possibility, skinned fibers were treated with blebbistatin, a potent inhibitor of myosin II ATPase and strong cross-bridge attachment. Fig. 1 shows the impact of 1.0 μM blebbistatin on the (normalized) force-Ca2+ relationship and Fmax (Fig. 1, A and B, insets) on fibers at sarcomere lengths of 2.0 μm (Fig. 1 A) and 2.2 μm (Fig. 1 B). The relative reductions in Fmax were similar at the two sarcomere lengths (Fig. 1 C), indicating similar actions of blebbistatin at the two lengths. Moreover, myofilament Ca2+ sensitivity, as indexed by the EC50 parameter, was shifted by similar amounts at the two lengths (Fig. 1 D). For example, at a concentration of 0.4 μM blebbistatin, Fmax was reduced by ∼61% at an SL of 2.0 μm and ∼64% at 2.2 μm, whereas the EC50 was increased by ∼49% and ∼39%, respectively. These results support the conclusion that interference with cross-bridge attachment has minor effects, if any, on length modulation of Ca2+ activation properties of the myofilaments.
Figure 1.
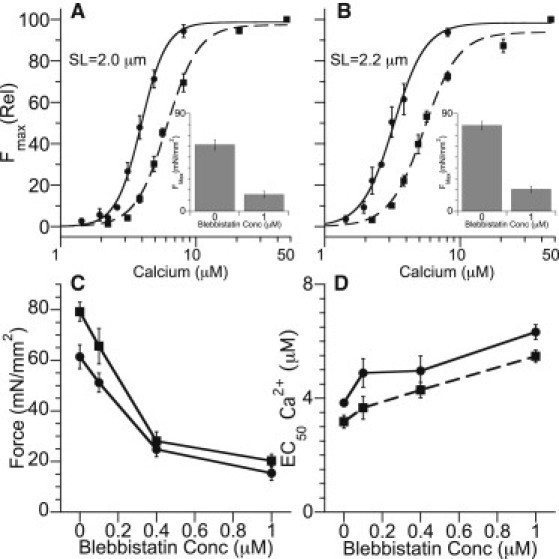
Impact of blebbistatin on strong binding cross-bridges and the Force-Ca relationship. Panels A and B illustrate the impact of increasing blebbistatin concentration (0 mM circle/solid line, 1 μM squares/dashed line) on the force calcium relationship as a function of the maximally calcium activated force. Blebbistatin induced a similar rightward shift in the Force-Ca relationship at both SL's indicating no SL dependent shift, in contrast to the results observed in DM-cTnC exchanged fibers (c.f. Fig. 3). Blebbistatin induced an equal decline in the maximally activated force production (Fmax) at both short (circles) and long (squares) lengths (Panel C) that smoothly declined up to the maximum amount used (1 μM). As with force, the impact of blebbistatin on EC50 (Panel D) was to decrease calcium sensitivity with increasing concentration yet demonstrating no SL dependent shifts in the EC50 as opposed to that observed in DM-cTnC exchanged fibers. Error bars represent the mean ±SE.
Based on the findings above, we speculated that LDA might originate from length-dependence properties of the thin filament. To test this, we replaced native wild-type TnC in isolated skinned cardiac trabeculae with varying ratios of wild-type TnC (WT-TnC) and mutant TnC (DM-TnC) that cannot bind Ca2+ (25). In these studies, DM-TnC and WT-TnC were covalently labeled with Alexa-Fluor 488 and Alexa-Fluor 546, respectively, to allow imaging of the fibers. The total concentration of bacterially expressed WT-cTnC plus DM-cTnC was fixed at 200 μg/mL. Fig. 2 A shows typical western blot results for isolated skinned trabeculae that were incubated for 2 h in solutions containing various ratios of bacterially expressed and fluorescently labeled DM-cTnC and WT-cTnC. Note that the estimated cTnC levels in the fibers include both the endogenous cTnC (remaining in the fiber) and exogenous WT-cTnC. Fig. 2 B establishes that the amount of exogenous DM-TnC relative to exogenous WT-TnC incorporated into the fiber correlated tightly with the relative amount of DM-TnC in the solution. Moreover, the results using solutions with 100% DM-cTnC revealed that the 2-h incubation period resulted in replacement of ∼95% of the endogenous cTnC by exogenous cTnC protein. Furthermore, as illustrated in Fig. 2 C, 2-h exchange with either WT- or DM-cTnC (200 μg/mL), covalently labeled with Alexa-Fluor 546, resulted in a uniform distribution along the fiber (Fig. 2 C, i and iv) and colocalized with rhodamine phalloidin (Fig. 2 C, iii and vi) within the myofilament lattice.
Figure 2.
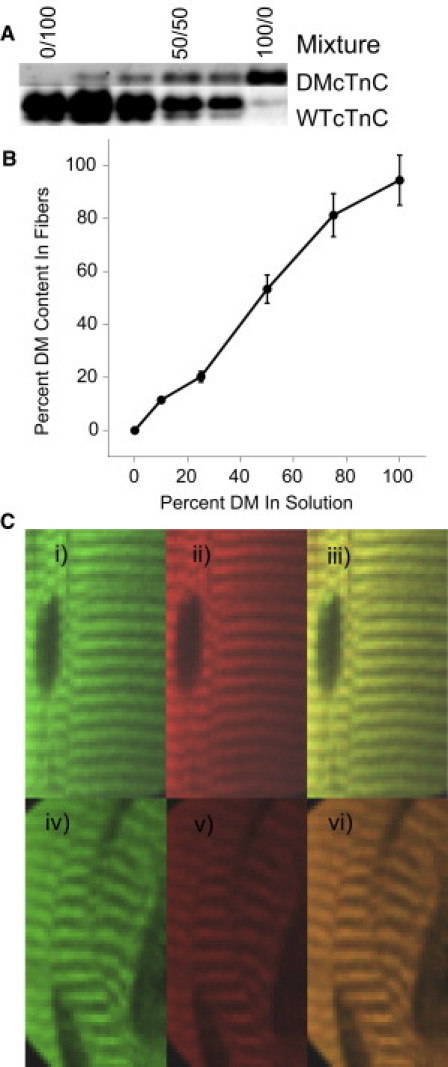
Incorporation of DM-cTnC into the myofilament lattice. Panel A illustrates a typical example of SDS-PAGE gel analysis using Blue Silver staining to visualize TnC protein. Due to the fluorescent label, DM-TnC migrated slower allowing separation from WT-TnC. Note that exchange with 100% DM-TnC revealed a small (∼5%) amount endogenous TnC remaining in the fiber (consistent with the virtual complete elimination of active force development in 100% DM-TnC exchanged fibers, c.f. Fig. 3). Fig. 2B illustrates the percent of DM-cTnC incorporated into the myofilament (y axis) as a function of the percent DM-cTnC used in the exchange solution (x axis). Error bars represent the mean ±SE. As shown, the percent of DM-cTnC incorporated into the myofilament was linearly related to the amount of DM-cTnC in the exchange solution. Fig. 2C illustrates confocal images of two fibers labeled with fluorescent WT-cTnC or fluorescent DM-cTnC (i and iv) or rhodamine phalloidin (ii and v), as well as the merge images (iii and vi). Note the sarcomeric pattern of the TnC labeling in comparison to the rhodamine phalloidin staining. Merging the images demonstrates that both bacterially expressed proteins localize to the thin filament when they are allowed to incubate with skinned fibers.
Having established that we can tightly control the amount of DM-cTnC incorporated into the myofilament lattice, we next examined the effects of DM-TnC on LDA. Fig. 3 (A and B) summarizes the force-Ca2+ relationships measured in fibers at SLs of 2.0 μm and 2.2μm after incubation (for 2 h) with solutions containing several DM-TnC/WT-TnC ratios (0/100, 10/90, and 50/50). The force in these figures is normalized to the maximal force (i.e., Fmax) which is the force generated at saturating Ca2+ levels (i.e., ∼43 μM). Consistent with previous reports on LDA (2,34), the Ca2+ concentration required for generating a force that is 50% of Fmax (i.e., the EC50) is lower (P < 0.01) and the Fmax is greater (P < 0.01) at the longer SL compared to the shorter SL when fibers are bathed in solutions without DM-TnC. Fig. 3 shows Fmax and EC50 as a function of the DM-cTnC/cTnC ratio as measured in fibers. It should be noted that the estimates of cTnC include both the exogenous cTnC and the residual endogenous cTnC remaining after the incubation period (i.e., ∼5%). Fig. 3 C demonstrates that increasing DM-cTnC incorporation causes progressive and similar reductions in Fmax at both lengths, as expected from the loss of TnC binding sites on the thin filament. Moreover, exchange with 100% Dm-TnC virtually eliminated Ca2+-activated force development (c.f. Fig. 3). Despite similar relative reductions in Fmax at the two lengths, DM-cTnC incorporation caused far greater (P < 0.01) increases in EC50 (i.e., greater rightward shifts in the force-Ca2+ relationship) at short SLs than at longer SLs (Fig. 3 D). For example, when fibers were bathed in solutions with a DM-TnC/WT-TnC of 50/50, reduction of Fmax at short length (∼45%) was similar to that at long length (∼38%) and the EC50 increased (P < 0.001) from 7.40 ± 0.58 to 14.94 ± 1.81 μM at SL = 2.0 μm, compared with a relatively small shift (P = 0.17) from 5.70 ± 0.48 to 7.25 ± 1.86 μM at SL = 2.2 μm. It should be noted that when the DM-cTnC/cTnC ratios exceeded ∼0.5, we observed a large variability in EC50, as well as in Fmax, which prevented reliable estimation of these parameters. Yet even with these data points we can see the impact of SL changes on the activation levels of the myofilament lattice. Comparing the impacts of different amounts of DM-cTnC, it can be seen that when FMax levels (e.g., with ∼10% DM-cTnC at short SL and 25% DM- cTnC at long SL in Fig. 3 C) were similar, the EC50 values (Fig. 3 D) were still different (p < 0.05), providing further evidence for the importance of SL changes on the submaximal activation of the thin filament. As discussed further below, these observations have important implications on the length dependence of Ca2+ cooperative sarcomere activation.
Figure 3.
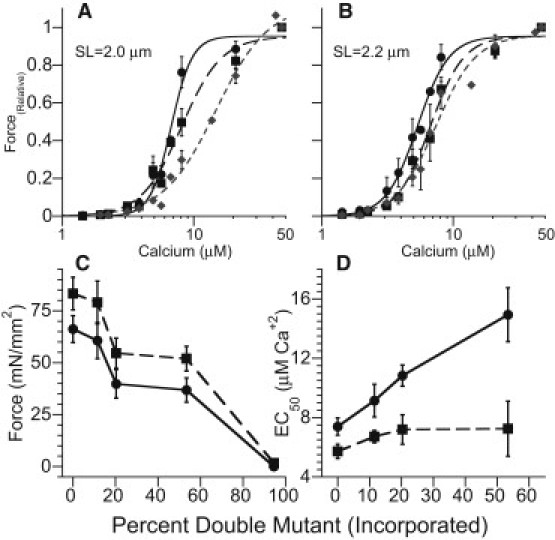
Impact of DM-cTnC incorporation on the force-Ca relationship. (A and B) Impact of increasing DM-cTnC (circles, 0%; squares, 10%; diamonds, 50%) on the force-Ca relationship as a function of the maximally calcium-activated force. The impact of DM-cTnC is greater at the short SL (2.0 μm) (A) than at the long SL (2.2 μm) (B), with a larger shift in the relationship to the right (lower Ca2+ sensitivity) at the short SL with a concomitant shift in the slope (Hill coefficient). (C) The impact of DM-cTnC incorporation into the myofilament caused an equal shift in the maximally activated force production (Fmax) at both short (circles) and long (squares) SLs that reached an approximate maximum at 25% DM-cTnC incorporation. (D) The impact of DM-cTnC incorporation on the EC50 illustrates that the reduction of force had a greater impact at short SLs (circles) than at long SLs, indicating a large calcium dependence for activation at short SL. Error bars represent the mean ±SE.
Interplay between thick- and thin-filament regulation on LDA
The results above establish that force inhibition by DM-cTnC incorporation has more distinct effects on the length dependence of myofilament activation than does that by blebbistatin. To further illustrate these differences, we plotted the relationships between EC50 and the Hill coefficient (cooperative activation) with Fmax for both blebbistatin-treated and DM-cTnC-exchanged fibers. The rationale for this analysis is that Fmax provides an estimate of the extent of interference with myofilament activation, thereby allowing comparisons between the DM-TnC interventions at the level of the thin filament and the blebbistatin interventions at the level of the thick filament. Fig. 4 shows that the EC50 correlated inversely with Fmax at both sarcomere lengths for either DM-TnC or blebbistatin treatment. This inverse relationship between EC50 and Fmax is predicted from current models of Ca2+-dependent myofilament activation that arises from the interaction between Ca2+ binding and cross-bridge attachment (13,35). It is clear that the slope of the EC50-Fmax relationship in the DM-cTnC-exchanged fibers at short length (Fig. 4, blue) was much greater (P < 0.05) than for the long sarcomere length or for the blebbistatin-treated fibers at either length. On the other hand, the slopes of the Fmax-EC50 relationship did not differ (P = 0.20) between the two lengths in the blebbistatin experiments, nor did these slopes differ (P = 0.24) from the DM-TnC-exchange group at long length. The absence of difference between the long and short lengths in the blebbistatin group is somewhat surprising, since previous studies had concluded that LDA results from length-dependent difference in strong cross-bridge attachment (36). It is interesting to note that the same relationship is observed in the plot of Fmax with the Hill parameter, suggesting a possible relationship between cooperativity and calcium sensitivity. These results support the notion that length-dependent differences in Ca2+ sensitivity arise from the effects of length on thin-filament properties.
Figure 4.
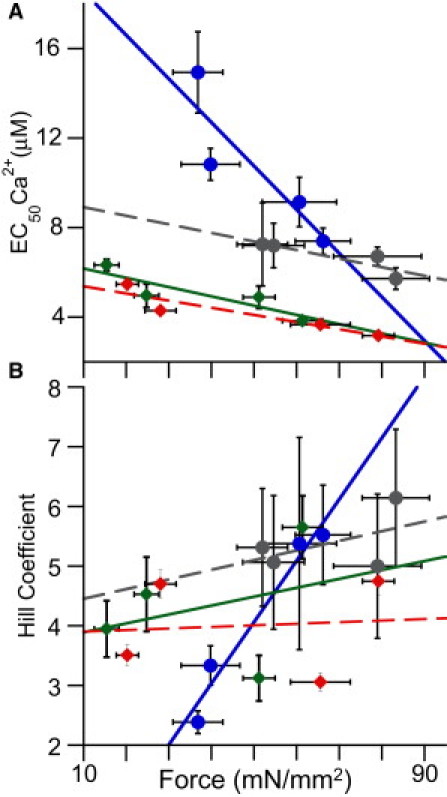
(A and B) Impact of reducing Fmax on the calcium sensitivity (EC50) (A) and cooperative activation (n) (B) of isolated myocardial tissue. As demonstrated, the use of blebbistatin at short (green) and long (red) SLs reduced Fmax while having no significant impact on either the EC50 (A) or the Hill coefficient (B), as illustrated by the shallow slopes. The use of DM-cTnC at long sarcomere lengths (gray) mimics the impact of blebbistatin, indicating that the loss of thin-filament activity at long SLs did not have a great impact on either the calcium sensitivity or the cooperativity; meanwhile, at short SLs (blue), the impact of DM-cTnC exchange on calcium sensitivity (A) and cooperative activation (Hill coefficient) (B) was severe, causing a significantly larger slope (p < 0.05) in both cases than that observed in any of the other three groups. Error bars represent the mean ±SE.
Discussion
We compared the contribution of thin-filament activation via the troponin-tropomyosin complex to that of strong cross-bridge attachment on the LDA in cardiac muscle. Strong cross-bridge attachment was altered in our studies using blebbistatin, an agent that has previously been shown to block myosin attachment to actin and myosin ATPase activity (21,22,29). To alter thin filament activation, we exchanged native TnC in the skinned fibers with various mixtures of wild-type TnC and a mutant form of TnC, which is incapable of binding Ca2+ but which is otherwise structurally normal (25). Under the experimental conditions used in our replacement studies, we found a ∼95% replacement of endogenous TnC. Moreover, the exogenous TnC (WT-TnC and DM-TnC) was uniformly incorporated into the fibers, as expected (37,38). Importantly, increasing the ratios of mutant DM-TnC to WT-TnC in the bathing solution proportionally increased the amount of DM-TnC incorporation into the fiber leading to an progressive decreases in Fmax as a result of reduced numbers of functional troponin/tropomyosin (Tn/Tm) complexes. Consistent with our ability to displacement most of the endogenous TnC with exogenous TnC, we found that exchange with 100% DM-TnC abolished force development.
Although the sarcomere length (SL) of cardiac muscle during contraction can vary between ∼1.5-2.3 μm, we purposely focused our LDA studies on lengths of 2.0 and 2.2μm in order to avoid complications associated with the effects internal loads or restoring forces that are observed at SL's below ∼1.9μm. The presence of these internal forces would interfere with estimations of strongly attached cross-bridge numbers from the measurement of developed tension (1). Our force-Ca2+ results are consistent with previous studies showing that the ability of Ca2+ to activate contraction (i.e., the Ca2+ affinity) improves with increasing sarcomere length (2,4,39,40).
As summarized in the Introduction, the basis for LDA remains controversial, and several cooperative mechanisms have been proposed (13,20,39,41–43). Since increased Ca2+ sensitivity is observed with enhanced cross-bridge attachment via increased rigor cross-bridges or the addition of cross-bridge fragments, it was concluded that LDA originated from the reduced ability of strong cross-bridges to form as SL decreases (14,36). We found that when strong cross-bridge formation was blocked with blebbistatin, only minor changes occurred in the EC50 and Ca2+ cooperativity (Hill coefficients), even with >60% force inhibition. More important, the effect of blebbistatin was not length-dependent. Indeed, shifts in EC50 and Hill coefficients induced by SL changes were unaffected by blebbistatin at all levels of force inhibition. Thus, although strong-binding cross-bridges can participate in the cooperative activation of the thin filament by pushing the troponin/tropomyosin complex into the open conformation (16,44), our investigations demonstrate that blocking strong cross-bridge attachment using blebbistatin does not differentially affect the contractile system's responsiveness to Ca2+ when SL is varied. This suggests, as discussed below, that Ca2+ cooperativity and LDA arise from the properties of thin filaments. Moreover, also as discussed below, these results suggest that feedback of cross-bridges on Ca2+ binding and Ca2+ cooperativity might primarily involve the weakly attached cross-bridges, as opposed to strongly attached (force-generating) cross-bridges as previously suggested (45). If correct, our conclusion (that Ca2+ cooperativity arises from weak cross-bridge attachment) would predict that the effects of increased rigor cross-bridges and cross-bridge fragments on Ca2+ cooperativity are mediated primarily by simultaneous alterations in weak cross-bridge attachment.
An alternative mechanism for LDA focuses on thin filament properties. Specifically, it has been hypothesized that movements of TnC/Tm complexes combined with end-to-end interactions between tropomyosin leads to cooperativity in the Ca2+ binding between nearest-neighbor Tn/Tm complexes (3,46). In this model, the free energy of Ca2+ binding to TnC is coupled to the energy required for the net movement of a Tn/Tm complex, which is coupled to neighboring Tn/Tm complexes via end-to-end interactions. Thus, Ca2+ binding and Tn/Tm movement in one Tn/Tm complex is coupled to that in neighboring complexes (47,48). We anticipated that incorporation of exogenous mutant cTnC (DM-TnC), which is unable to bind Ca2+, would interfere with transmission of the cooperativity arising from end-to-end interactions between Tn/Tm and thereby disrupt Ca2+ cooperativity. Consistent with this prediction, increased DM-TnC incorporation caused progressive increases in EC50 and simultaneous reductions in the Hill coefficient and Fmax at both lengths. However, the impact of DM-TnC incorporation on the EC50 and the Hill coefficient were far more pronounced at the short sarcomere length. This is best appreciated by plots of the EC50 or Hill coefficients as a function of the maximal force (Fmax) generated, where Fmax accounts for the differences in force generation between the fibers (c.f. Fig. 4). It is clear that for the ranges of DM-TnC incorporation studied, which were limited to solutions containing >∼50% DM-TnC because of fiber instability (i.e., large variability between fibers), EC50 and Hill coefficient depended much more steeply on Fmax for the short than for the long sarcomere length. Regardless, for the range of DM-TnC incorporation used in our studies, we observed a twofold increase in the EC50 (i.e., Ca2+ sensitivity) at the short sarcomere length compared with a 40% change in EC50 at the long sarcomere length when Fmax was reduced by ∼40%. In a similar way, the Hill coefficient (i.e., Ca2+ cooperativity) decreased far more at the short length than at the long length. It is interesting that the dependence of EC50 and Hill coefficient on Fmax in the blebbistatin-treated fibers at either length was similar to that observed at the long length in the DM-TnC group.
How can we explain our findings that it is interference with the number of TnC/Tm complexes able to bind Ca2+, and not strong cross-bridge attachment, that most affects Ca2+ cooperativity and LDA? In principle, Ca2+ cooperativity of force generation must ultimately arise from interactions between neighboring Tn/Tm complexes (44,49). Previous two-state biochemical models for Tn/Tm (50,51) have linked this cooperativity to feedback of cross-bridge attachment on Ca2+ binding in adjacent Tn/Tm complexes wherein strong cross-bridge attachment drives Tn/Tm into the open state, which has a high Ca2+-binding affinity. However, the modest effect of blebbistatin on the Ca2+ activation properties (i.e., sensitivity and cooperativity) and LDA, though initially surprising given findings in previous studies (13,14,52), cannot be easily explained using a two-state model. On the other hand, our results are consistent with a three-state biochemical model wherein the states have distinct Ca2+ binding properties and are topologically connected in a linear manner (blocked ⇆ closed ⇆ open) (44,47,48). Specifically, because strong cross-bridge attachment did not affect cooperativity, and because strong cross-bridge attachment in this model is associated with closed-to-open-state transitions, this model predicts that there are larger differences in Ca2+ affinity between the blocked and closed states than between the closed and open states. Thus, since closed states are stabilized by weak cross-bridge attachment, weak-binding cross-bridges, which are involved in blocked-to-closed-state transitions, are predicted to be the primary mediators of Ca2+ cooperativity, as proposed previously (53,54). By contrast, closed-to-open-state transitions are not expected to enhance the Ca2+-binding affinity and cooperativity. This conclusion is consistent with those of studies showing that Ca2+ cooperativity for myofilament activation is mediated by increased probabilities of closed-state occupancy of a Tn/Tm complex after Ca2+ binding to the adjacent Tn/Tm complex (55,56). This mechanism also readily explains the effects of exogenous myosin and rigor bridges on Ca2+ sensitivity without invoking the need for strong cross-bridge binding. Specifically, it is predicted that both exogenous myosin and low ATP conditions to promote rigor bridge formation force Tn/Tm complexes in the process of binding to force transitions initially through the closed state either by stoichiometric mass action or by distortion of the cross-bridge binding equilibria with actin (due to reduced off rates of rigor bridges when ATP is low). Consistent with this suggestion, it has been shown that the structural impact of rigor cross-bridges on Tn/Tm complexes is different from that induced by cycling cross-bridges (57).
The remarkable ability of DM-TnC incorporation to strongly influence Ca2+ sensitivity and Ca2+ cooperativity at short, compared to long, lengths suggests that SL affects interactions between Tn/Tm complexes. In other words, Ca2+ cooperativity appears to depend more strongly on the number of TnC Ca2+ binding sites at shorter SLs than at longer SLs. These findings suggest that coupling between adjacent Tn/Tm complexes increases with length, thereby making activation less dependent on the number of TnC sites with Ca2+ bound. Thus, when Ca2+ binds to a given Tn/Tm complex, the probability of the neighboring complexes (and beyond) to transition to the closed state is enhanced at longer lengths due to stronger coupling (Fig. 5). In an equivalent manner, within the framework of the flexible Tn/Tm model (49), increases of SL increase the persistence length by reducing Tn/Tm flexibility, thereby allowing the influence of Ca2+ binding to propagate further along the thin filament. Regardless of the model, the propagation of activation is, of course, aided by weak cross-bridge attachment, which stabilizes the closed state, leading ultimately to open-state transitions of Tn/Tm and strong cross-bridge binding. If propagation of activation is indeed reduced at short SLs, we predict, incorporation of DM-TnC molecules will cause greater disruptions of activation at short lengths than at longer lengths, with the effects being more pronounced at submaximal Ca2+ levels (i.e., affecting EC50 and Hill coefficients) than at maximal Ca2+ levels (i.e., affecting Fmax), as shown by our results. The mechanism for the length dependence of the propagation of Ca2+ activation is unclear but could involve the interactions observed between cTnT and regulatory light chains (58,59) or titin (60) binding to the thin filaments at the Z-disc (61) and other proteins (62,63). Clearly, further studies will be needed to assess the molecular basis for the enhanced propagation of Ca2+ with increased SL.
Figure 5.
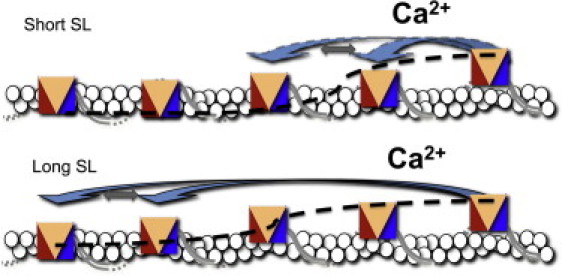
Schematic drawing of the proposed mechanism of LDA, showing tropomyosin (gray lines), actin (ovals), and cardiac troponin units (tricolored rectangles). In our version of the three-state model, the impact of calcium on the activation of the neighboring units (dashed black line) at short SL works to activate the neighboring cTn unit, and thus, calcium needs to bind to more Tn units (arrows), whereas at longer SL, the activation of one cTn unit, due to the proposed stiffening of the tropomyosin, is able to activate more cTn units, thus requiring less calcium to activate the same number of cTn units (arrows). In other words, stretching the SL theoretically shifts the requirement for calcium binding from every 1–2 troponins to approximately every 3–4 troponins, allowing for more binding sites to be activated with the same levels of calcium activation.
Acknowledgments
We thank Dr. Kate Sheehan for her assistance in gathering and analysis of the confocal images.
These studies were supported in part by the National Institutes of Health (grant HL-75494 to P.P.d.T.) and the Canadian Institutes for Health Research (grant MOP-79460 to P.H.B.).
References
- 1.Allen D.G., Kentish J.C. The cellular basis of the length-tension relation in cardiac muscle. J. Mol. Cell. Cardiol. 1985;17:821–840. doi: 10.1016/s0022-2828(85)80097-3. [DOI] [PubMed] [Google Scholar]
- 2.Dobesh D.P., Konhilas J.P., de Tombe P.P. Cooperative activation in cardiac muscle: impact of sarcomere length. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H1055–H1062. doi: 10.1152/ajpheart.00667.2001. [DOI] [PubMed] [Google Scholar]
- 3.Gordon A.M., Homsher E., Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 4.Kentish J.C., ter Keurs H.E., Noble M.I. Comparison between the sarcomere length-force relations of intact and skinned trabeculae from rat right ventricle. Influence of calcium concentrations on these relations. Circ. Res. 1986;58:755–768. doi: 10.1161/01.res.58.6.755. [DOI] [PubMed] [Google Scholar]
- 5.de Beer E.L., Grundeman R.L., Schiereck P. Effect of sarcomere length and filament lattice spacing on force development in skinned cardiac and skeletal muscle preparations from the rabbit. Basic Res. Cardiol. 1988;83:410–423. doi: 10.1007/BF02005827. [DOI] [PubMed] [Google Scholar]
- 6.Palmer S., Kentish J.C. The role of troponin C in modulating the Ca2+ sensitivity of mammalian skinned cardiac and skeletal muscle fibres. J. Physiol. 1994;480:45–60. doi: 10.1113/jphysiol.1994.sp020339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wannenburg T., Janssen P.M., de Tombe P.P. The Frank-Starling mechanism is not mediated by changes in rate of cross-bridge detachment. Am. J. Physiol. 1997;273:H2428–H2435. doi: 10.1152/ajpheart.1997.273.5.H2428. [DOI] [PubMed] [Google Scholar]
- 8.Maytum R., Lehrer S.S., Geeves M.A. Cooperativity and switching within the three-state model of muscle regulation. Biochemistry. 1999;38:1102–1110. doi: 10.1021/bi981603e. [DOI] [PubMed] [Google Scholar]
- 9.Babu A., Scordilis S.P., Gulati J. The control of myocardial contraction with skeletal fast muscle troponin C. J. Biol. Chem. 1987;262:5815–5822. [PubMed] [Google Scholar]
- 10.Gulati J., Sonnenblick E., Babu A. The role of troponin C in the length dependence of Ca2+-sensitive force of mammalian skeletal and cardiac muscles. J. Physiol. 1991;441:305–324. doi: 10.1113/jphysiol.1991.sp018753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moss R.L., Nwoye L.O., Greaser M.L. Substitution of cardiac troponin C into rabbit muscle does not alter the length dependence of Ca2+ sensitivity of tension. J. Physiol. 1991;440:273–289. doi: 10.1113/jphysiol.1991.sp018708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzsimons D.P., Patel J.R., Moss R.L. Cooperative mechanisms in the activation dependence of the rate of force development in rabbit skinned skeletal muscle fibers. J. Gen. Physiol. 2001;117:133–148. doi: 10.1085/jgp.117.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzsimons D.P., Patel J.R., Moss R.L. Cross-bridge interaction kinetics in rat myocardium are accelerated by strong binding of myosin to the thin filament. J. Physiol. 2001;530:263–272. doi: 10.1111/j.1469-7793.2001.0263l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitzsimons D.P., Moss R.L. Strong binding of myosin modulates length-dependent Ca2+ activation of rat ventricular myocytes. Circ. Res. 1998;83:602–607. doi: 10.1161/01.res.83.6.602. [DOI] [PubMed] [Google Scholar]
- 15.McDonald K.S., Moss R.L. Strongly binding myosin crossbridges regulate loaded shortening and power output in cardiac myocytes. Circ. Res. 2000;87:768–773. doi: 10.1161/01.res.87.9.768. [DOI] [PubMed] [Google Scholar]
- 16.Vibert P., Craig R., Lehman W. Steric-model for activation of muscle thin filaments. J. Mol. Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Y., Kawai M. The effect of the lattice spacing change on cross-bridge kinetics in chemically skinned rabbit psoas muscle fibers. II. Elementary steps affected by the spacing change. Biophys. J. 1993;64:197–210. doi: 10.1016/S0006-3495(93)81357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butters C.A., Tobacman J.B., Tobacman L.S. Cooperative effect of calcium binding to adjacent troponin molecules on the thin filament-myosin subfragment 1 MgATPase rate. J. Biol. Chem. 1997;272:13196–13202. doi: 10.1074/jbc.272.20.13196. [DOI] [PubMed] [Google Scholar]
- 19.Campbell K.B., Razumova M.V., Slinker B.K. Nonlinear myofilament regulatory processes affect frequency-dependent muscle fiber stiffness. Biophys. J. 2001;81:2278–2296. doi: 10.1016/S0006-3495(01)75875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Razumova M.V., Bukatina A.E., Campbell K.B. Different myofilament nearest-neighbor interactions have distinctive effects on contractile behavior. Biophys. J. 2000;78:3120–3137. doi: 10.1016/S0006-3495(00)76849-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allingham J.S., Smith R., Rayment I. The structural basis of blebbistatin inhibition and specificity for myosin II. Nat. Struct. Mol. Biol. 2005;12:378–379. doi: 10.1038/nsmb908. [DOI] [PubMed] [Google Scholar]
- 22.Farman G.P., Tachampa K., de Tombe P.P. Blebbistatin: use as inhibitor of muscle contraction. Pflugers Arch. 2008;455:995–1005. doi: 10.1007/s00424-007-0375-3. [DOI] [PubMed] [Google Scholar]
- 23.Dou Y., Arlock P., Arner A. Blebbistatin specifically inhibits actin-myosin interaction in mouse cardiac muscle. Am. J. Physiol. Cell. Physiol. 2007;293:C1148–C1153. doi: 10.1152/ajpcell.00551.2006. [DOI] [PubMed] [Google Scholar]
- 24.Farman G.P., Walker J.S., Irving T.C. Impact of osmotic compression on sarcomere structure and myofilament calcium sensitivity of isolated rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H1847–H1855. doi: 10.1152/ajpheart.01237.2005. [DOI] [PubMed] [Google Scholar]
- 25.Strynadka N.C., Cherney M., James M.N. Structural details of a calcium-induced molecular switch: x-ray crystallographic analysis of the calcium-saturated N-terminal domain of troponin C at 1.75 Å resolution. J. Mol. Biol. 1997;273:238–255. doi: 10.1006/jmbi.1997.1257. [DOI] [PubMed] [Google Scholar]
- 26.Li M.X., Gagné S.M., Sykes B.D. NMR studies of Ca2+ binding to the regulatory domains of cardiac and E41A skeletal muscle troponin C reveal the importance of site I to energetics of the induced structural changes. Biochemistry. 1997;36:12519–12525. doi: 10.1021/bi971222l. [DOI] [PubMed] [Google Scholar]
- 27.Li M.X., Spyracopoulos L., Sykes B.D. Binding of cardiac troponin-I147–163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 28.Spyracopoulos L., Li M.X., Sykes B.D. Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry. 1997;36:12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 29.Limouze J., Straight A.F., Sellers J.R. Specificity of blebbistatin, an inhibitor of myosin II. J. Muscle Res. Cell Motil. 2004;25:337–341. doi: 10.1007/s10974-004-6060-7. [DOI] [PubMed] [Google Scholar]
- 30.Kovács M., Tóth J., Sellers J.R. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 31.Straight A.F., Cheung A., Mitchison T.J. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- 32.Sakamoto T., Limouze J., Sellers J.R. Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry. 2005;44:584–588. doi: 10.1021/bi0483357. [DOI] [PubMed] [Google Scholar]
- 33.Ranatunga K.W. Endothermic force generation in skinned cardiac muscle from rat. J. Muscle Res. Cell Motil. 1999;20:489–496. doi: 10.1023/a:1005509731881. [DOI] [PubMed] [Google Scholar]
- 34.Konhilas J.P., Irving T.C., de Tombe P.P. Myofilament calcium sensitivity in skinned rat cardiac trabeculae: role of interfilament spacing. Circ. Res. 2002;90:59–65. doi: 10.1161/hh0102.102269. [DOI] [PubMed] [Google Scholar]
- 35.Lehrer S.S., Geeves M.A. The muscle thin filament as a classical cooperative/allosteric regulatory system. J. Mol. Biol. 1998;277:1081–1089. doi: 10.1006/jmbi.1998.1654. [DOI] [PubMed] [Google Scholar]
- 36.Swartz D.R., Moss R.L. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J. Biol. Chem. 1992;267:20497–20506. [PubMed] [Google Scholar]
- 37.Bell M.G., Lankford E.B., Barsotti R.J. Kinetics of cardiac thin-filament activation probed by fluorescence polarization of rhodamine-labeled troponin C in skinned guinea pig trabeculae. Biophys. J. 2006;90:531–543. doi: 10.1529/biophysj.105.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brenner B., Kraft T., Chalovich J.M. Thin filament activation probed by fluorescence of N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole-labeled troponin I incorporated into skinned fibers of rabbit psoas muscle. Biophys. J. 1999;77:2677–2691. doi: 10.1016/S0006-3495(99)77102-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith S.H., Fuchs F. Length-dependence of cross-bridge mediated activation of the cardiac thin filament. J. Mol. Cell. Cardiol. 2000;32:831–838. doi: 10.1006/jmcc.2000.1124. [DOI] [PubMed] [Google Scholar]
- 40.Konhilas J.P., Irving T.C., de Tombe P.P. Length-dependent activation in three striated muscle types of the rat. J. Physiol. 2002;544:225–236. doi: 10.1113/jphysiol.2002.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martyn D.A., Chase P.B., Gordon A.M. A simple model with myofilament compliance predicts activation-dependent crossbridge kinetics in skinned skeletal fibers. Biophys. J. 2002;83:3425–3434. doi: 10.1016/S0006-3495(02)75342-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDonald K.S., Wolff M.R., Moss R.L. Sarcomere length dependence of the rate of tension redevelopment and submaximal tension in rat and rabbit skinned skeletal muscle fibres. J. Physiol. 1997;501:607–621. doi: 10.1111/j.1469-7793.1997.607bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Tombe P.P., Mateja R.D., Irving T.C. Myofilament length dependent activation. J. Mol. Cell. Cardiol. 2010;48:851–858. doi: 10.1016/j.yjmcc.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKillop D.F., Geeves M.A. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith L., Tainter C., Martyn D.A. Cooperative cross-bridge activation of thin filaments contributes to the Frank-Starling mechanism in cardiac muscle. Biophys. J. 2009;96:3692–3702. doi: 10.1016/j.bpj.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gillis T.E., Martyn D.A., Rivera A.J., Regnier M. Investigation of thin filament near-neighbor regulatory unit interactions during skinned rat cardiac muscle force development. J. Physiol. 2007;580:561–576. doi: 10.1113/jphysiol.2007.128975. comment 580:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisenberg E., Hill T.L. A cross-bridge model of muscle contraction. Prog. Biophys. Mol. Biol. 1978;33:55–82. doi: 10.1016/0079-6107(79)90025-7. [DOI] [PubMed] [Google Scholar]
- 48.Hill T.L., Eisenberg E., Podolsky R.J. Some self-consistent two-state sliding filament models of muscle contraction. Biophys. J. 1975;15:335–372. doi: 10.1016/S0006-3495(75)85823-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boussouf S.E., Geeves M.A. Tropomyosin and troponin cooperativity on the thin filament. Adv. Exp. Med. Biol. 2007;592:99–109. doi: 10.1007/978-4-431-38453-3_10. [DOI] [PubMed] [Google Scholar]
- 50.Geeves M.A., Halsall D.J. Two-step ligand binding and cooperativity. A model to describe the cooperative binding of myosin subfragment 1 to regulated actin. Biophys. J. 1987;52:215–220. doi: 10.1016/S0006-3495(87)83208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKillop D.F., Geeves M.A. Regulation of the acto.myosin subfragment 1 interaction by troponin/tropomyosin. Evidence for control of a specific isomerization between two acto.myosin subfragment 1 states. Biochem. J. 1991;279:711–718. doi: 10.1042/bj2790711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuda N., Kajiwara H., Kurihara S. Effects of MgADP on length dependence of tension generation in skinned rat cardiac muscle. Circ. Res. 2000;86:E1–E6. doi: 10.1161/01.res.86.1.e1. [DOI] [PubMed] [Google Scholar]
- 53.Rice J.J., Wang F., de Tombe P.P. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys. J. 2008;95:2368–2390. doi: 10.1529/biophysj.107.119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rice J.J., Tu Y., De Tombe P.P. Spatially-compressed cardiac myofilament models generate hysteresis that is not found in real muscle. Pac. Symp. Biocomput. 2008;2008:366–377. [PubMed] [Google Scholar]
- 55.Gong H., Hatch V., Tobacman L.S. Mini-thin filaments regulated by troponin-tropomyosin. Proc. Natl. Acad. Sci. USA. 2005;102:656–661. doi: 10.1073/pnas.0407225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gafurov B., Chen Y.D., Chalovich J.M. Ca2+ and ionic strength dependencies of S1-ADP binding to actin-tropomyosin-troponin: regulatory implications. Biophys. J. 2004;87:1825–1835. doi: 10.1529/biophysj.104.043364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun Y.B., Lou F., Irving M. Calcium- and myosin-dependent changes in troponin structure during activation of heart muscle. J. Physiol. 2009;587:155–163. doi: 10.1113/jphysiol.2008.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farman G.P., Miller M.S., Irving T.C. Phosphorylation and the N-terminal extension of the regulatory light chain help orient and align the myosin heads in Drosophila flight muscle. J. Struct. Biol. 2009;168:240–249. doi: 10.1016/j.jsb.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cazorla O., Szilagyi S., Lacampagne A. Transmural stretch-dependent regulation of contractile properties in rat heart and its alteration after myocardial infarction. FASEB J. 2005;19:88–90. doi: 10.1096/fj.04-2066fje. [DOI] [PubMed] [Google Scholar]
- 60.Granzier H., Labeit S. Cardiac titin: an adjustable multi-functional spring. J. Physiol. 2002;541:335–342. doi: 10.1113/jphysiol.2001.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trombitás K., Granzier H. Actin removal from cardiac myocytes shows that near Z line titin attaches to actin while under tension. Am. J. Physiol. 1997;273:C662–C670. doi: 10.1152/ajpcell.1997.273.2.C662. [DOI] [PubMed] [Google Scholar]
- 62.Winegrad S. Cardiac myosin binding protein C. Circ. Res. 1999;84:1117–1126. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]
- 63.Trombitás K., Jin J.P., Granzier H. The mechanically active domain of titin in cardiac muscle. Circ. Res. 1995;77:856–861. doi: 10.1161/01.res.77.4.856. [DOI] [PubMed] [Google Scholar]