

Abstract
The antimicrobial peptide microcin J25 (MccJ25) is posttranslationally matured from a linear preprotein into its native lasso conformation by two enzymes. One of these enzymes cleaves the preprotein and the second enzyme installs the requisite isopeptide bond to establish the lasso structure. Analysis of a mimic of MccJ25 that can be cyclized without the influence of the maturation enzymes suggests that MccJ25 does not spontaneously adopt a near-lasso structure. In addition, we conducted atomistically detailed replica-exchange molecular dynamics simulations of pro-microcin J25 (pro-MccJ25), the 21-residue uncyclized analog of MccJ25, to determine the conformational ensemble explored in the absence of the leader sequence or maturation enzymes. We applied a nonlinear dimensionality reduction technique known as the diffusion map to the simulation trajectories to extract two global order parameters describing the fundamental dynamical motions of the system, and identify three distinct pathways. One path corresponds to the spontaneous adoption of a left-handed lasso, in which the N-terminus wraps around the C-terminus in the opposite sense to the right-handed topology of native MccJ25. Our computational and experimental results suggest a role for the MccJ25 leader sequence and/or its maturation enzymes in facilitating the adoption of the right-handed topology.
Introduction
The remarkable threaded-lasso structure of microcin J25 (MccJ25), and the exceptional thermal and chaotropic stability and resistance to protease degradation (1–6) that it imparts, are of substantial interest in the field of protein design and engineering. MccJ25 is a β-hairpin with an eight-residue ring formed by an isopeptide bond between the Gly1 N-terminal amino group and the Glu8 γ-carboxyl side chain through which the C-terminus is threaded. The C-terminus is encircled by the N-terminus in a clockwise or right-handed manner, and is held in place by the bulky aromatic groups borne by the Phe19 and Tyr20 steric locks flanking the ring (2–4,7,8).
Only two enzymes, McjB and McjC, are required for the in vitro maturation of the 58-residue pre-peptide McjA (pre-MccJ25) into native MccJ25 (5). McjB is a putative peptidase that likely cleaves the leader peptide from McjA, while McjC is thought to catalyze the ATP-dependent formation of an isopeptide bond between the Gly1 N-terminus and the γ-carboxyl of the Glu8 side chain (5). These two enzymatic activities can be mapped onto McjB and McjC through homology studies, but neither McjB nor McjC have homology to any known chaperones. Furthermore, the β-hairpin portion of mature MccJ25 (2–4) consists of three very strong hydrogen bonds with donor-acceptor distances of 1.8 Å or less. Finally, steric considerations dictate that the C-terminus cannot slip through the Gly1–Glu8 ring after formation of the isopeptide bond (2,3,5,7). Based on these three lines of reasoning, we hypothesize that the segment of the McjA protein corresponding to mature MccJ25 (amino acids 37–57, excluding the N-terminal methionine) likely, at least fleetingly, prefolds into a near-native conformation prior to synthesis of the lasso.
To test our hypothesis, we built a model clickable MccJ25 peptide (cMccJ25) that allowed us to probe the folding of the peptide without the influence of the maturation enzymes McjB and McjC. In this model peptide (Fig. 1), Gly1 and Glu8 are replaced with nearly isosteric azidoacetic acid and propargyl glycine, respectively. The copper-catalyzed click reaction between azides and terminal alkynes (9–13) permits cyclization of cMccJ25 without the influence of the maturation enzymes. In addition, we carried out extensive molecular dynamics (MD) simulations of the linear 21-residue peptide corresponding to the uncyclized form of MccJ25, termed pro-MccJ25, to provide molecular-based understanding of pro-MccJ25 with a level of detail inaccessible by experimentation alone, and assess the degree to which it approached the near-native lasso conformation. For notational clarity, lasso-like structures of pro-MccJ25 which lack the isopeptide bond will henceforth be referred to as coils.
Figure 1.
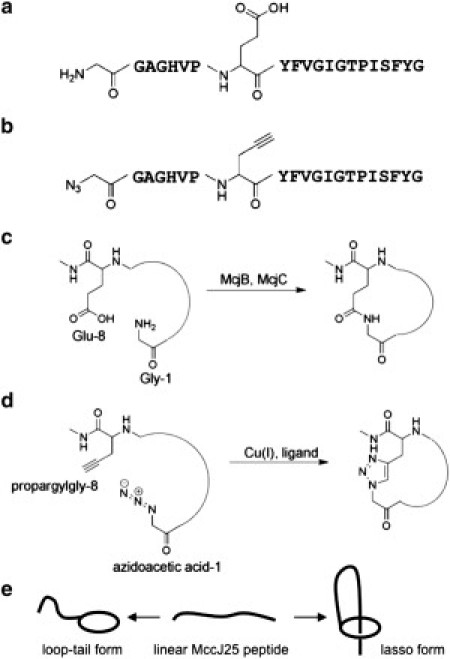
Sequence and cyclization chemistry of native microcin J25 (MccJ25) and clickable microcin J25 (cMccJ25). (a) Sequence of MccJ25 with the structure of Gly1 and Glu8 indicated. (b) Sequence of cMccJ25. Gly1 of the native peptide is replaced with azidoacetic acid, and Glu8 is replaced with propargyl glycine. (c) In native MccJ25, the maturation enzymes McjB and McjC install an isopeptide bond to cyclize the peptide. (d) In cMccJ25, cyclization occurs via Cu-mediated alkyne azide cycloaddition. (e) Schematic of possible topologies upon cyclization of cMccJ25. If the peptide prefolds into a near-native lasso topology, the cyclization reaction results in a lasso topology. Otherwise, cyclization results in a loop-tail form.
The primary intent of this study is to test the hypothesis that pro-MccJ25 spontaneously prefolds into near-native coil conformations. Recovery of cyclized cMccJ25 product in the click reaction, and/or observation of coil conformations in the molecular simulations, would provide support for this hypothesis and suggest that the absence of active MccJ25 product upon exposure of pro-MccJ25 to the McjB and McjC maturation enzymes may be due to the removal of residues in the leader peptide crucial to enzyme binding (5,14). Conversely, the absence of cyclized cMccJ25 and/or pro-MccJ25 coils would provide evidence for a role of the leader peptide or the maturation enzymes in assisting the formation of the near-native fold. To provide an understanding of the underlying dynamics, we employ a nonlinear dimensionality reduction technique known as the diffusion map (15–17). By generating a low-dimensional embedding of the simulation trajectories, this approach permits the systematic extraction of order parameters with which to describe the important dynamical motions of the peptide.
Experimental Methods
Solid-phase peptide synthesis of clickable MccJ25 (cMccJ25)
A clickable variant of the linear precursor to MccJ25 was assembled using standard manual solid-phase peptide synthesis procedures with Boc-protecting groups. Boc-protected canonical amino acids were purchased from Novabiochem (EMD Biosciences, San Diego, CA). Boc-propargylamine and Boc-His(Dnp), coupling reagents HBTU and DIEA, and ninhydrin test kit were purchased from AnaSpec (San Jose, CA). Azidoacetic acid was synthesized using a published procedure (18). The sequence of the peptide, starting from the N-terminus, is
where X is azidoacetic acid (azide analog of glycine) and Z is propargylglycine (alkyne analog of glutamic acid). The synthesis was performed on a 0.1 mmol scale. Cleavage of the peptide from the resin was accomplished by treating the resin with a mixture of trifluoroacetic acid (TFA) and trifluoromethanesulfonic acid. Commonly added thiol-based scavengers, such as thioanisole, were found to reduce the N-terminal azide to amine, so these scavengers were omitted from the cleavage cocktail. The peptide was purified on an analytical scale (4.6 × 150 mm) Agilent (Santa Clara, CA) Zorbax 300SB-C18 reverse-phase column with eluent A, water (0.1% TFA); eluent B, acetonitrile (0.1% TFA); gradient, 10–50% B over 20 min followed by a linear increase to 90% B in 5 min at 0.75 mL/min. Under these conditions, the peptide eluted at a retention time of 17.7 min. The identity of the peptide was confirmed by electrospray mass spectrometry at the Princeton University Molecular Biology mass spectrometry core facility.
Copper-catalyzed azide-alkyne cycloaddition of cMccJ25
A panel of different reaction conditions was evaluated for cyclization of the cMccJ25 peptide. The conditions evaluated were the solvent for the reaction (water or phosphate-buffered saline), the temperature of the reaction (room temperature or 4°C), and the accelerating ligand for the Cu(I) species. The tris-(benzyltriazolylmethyl)amine ligand (19) is an excellent accelerating ligand for copper-catalyzed azide-alkyne cycloaddition (CuAAC), but it suffers from poor water solubility. The water-soluble ligand bathophenanthrolinedisulfonic acid (20) was evaluated as an alternative. In all cases, the requisite Cu(I) species was generated in situ via reduction of CuSO4 by sodium ascorbate. We converged upon optimum conditions for the cMccJ25 CuAAC reaction as 71 μM peptide, 100 μM CuSO4, 2000 μM sodium ascorbate, and 200 μM bathophenanthrolinedisulfonic acid ligand in water at room temperature. This reaction was carried out on a 100 μL scale (0.0071 μmol of peptide) for test reactions and a 1 mL scale (0.071 μmol of peptide) to generate samples for tandem mass spectrometry (MS/MS).
HPLC and tandem mass spectrometry analysis
The reaction products from CuAAC of the cMccJ25 peptide were (without further purification or concentration) analyzed initially by analytical high-performance liquid chromatography (HPLC) on an Agilent Zorbax 300SB-C18 reverse-phase column using an Agilent 1200 Series instrument. The gradient used for the separation was the same as described above. Peaks desired for mass spectrometry analysis were collected using an automated fraction collector and concentrated via a speed-vac for optimal signal. Liquid chromatography-tandem mass spectrometry was carried out in a NanoLC Ultra system from Eksigent (Dublin, CA) and an LTQ Orbitrap hybrid mass spectrometer from ThermoFisher (Waltham, MA). Eluent A was 0.5% acetic acid and 0.5% formic acid in 95% water/5% acetonitrile; eluent B was 0.5% acetic acid and 0.5% formic acid in 95% acetonitrile/5% water. A gradient was chosen to start with 5% eluent B, then hold for 1 min, raise to 40% eluent B at 39 min, raise to 98% at 40 min, hold at 98% eluent B for 15 min for washing, go back to 5% eluent B at 55.5 min, and hold for 10 min for reequilibration. Flow was 300 nL/min in a 75 μm × 20 cm, C18 column with 5 μm particle size. The MS/MS data was analyzed with Qualbrowser software from ThermoFisher.
Computational Methods
Molecular simulations of pro-MccJ25
Fully atomistic molecular dynamics (MD) simulations of pro-MccJ25 in explicit water were conducted using the GROMACS 3.3.2 simulation suite (21,22). The initial coordinates of pro-MccJ25 were adapted from the Protein DataBank database (23) structure for native MccJ25 (PDB entry 1Q71 (4)). To accelerate sampling of conformational phase space, NPT replica-exchange MD (REMD) simulations (24,25) at 1 bar were conducted on a logarithmically spaced temperature ladder spanning 298–423 K (24,26,27). The data collected at the 298 K and 1 bar state point over the 95 ns production run will henceforward be referred to as the REMD simulation trajectory. Technical details of the simulations are provided in the Supporting Material, Methods: Molecular Simulations of pro-MccJ25.
Free energy surfaces (FES) were constructed to within an arbitrary constant using the relationship (28)
where is the probability with which configurations with order parameter are observed in the simulation trajectory, is the Gibbs free energy of such configurations and β = 1/kBT, where kB is Boltzmann's constant and T the temperature. Suitable order parameters for the parameterization were determined by the application of nonlinear dimensionality reduction to the REMD simulation trajectory.
Nonlinear dimensionality reduction: the diffusion map
Couplings between degrees of freedom in biophysical systems make the effective dimensionality far lower than that in which the Hamiltonian is formulated (28–31). This permits meaningful descriptions of such systems to be constructed in a relatively small number of degrees of freedom describing the most important dynamics of the system, with the other degrees of freedom restrained to this essential subspace (30). It is useful to consider this subspace a low-dimensional hypersurface, termed the intrinsic manifold, upon which the dynamical motions of the system are effectively constrained (28). This manifold may be considered to arise from a separation of timescales in which the fundamental dynamics constitute a low-dimensional slow subspace (32) to which the remaining fast degrees of freedom are slaved.
Principal component analysis (PCA) is a powerful linear dimensionality reduction technique, first applied to biomolecular simulations by García (29) and Amadei et al. (30), and now considered a standard tool for the analysis of simulation data (31,33–36). Details of the method are provided in the Supporting Material, Methods: Principal Component Analysis. For systems exhibiting large conformational changes and which may be expected to possess highly nonlinear intrinsic manifolds (28,33), the inherent linearity of PCA can lead to poor global performance, making it a more appropriate technique for localized regions of phase space. Nonlinear dimensionality reduction techniques provide, in principle, a means to construct low-dimensional descriptions valid over the entire phase space sampled by the system. The diffusion map (15–17) is one such technique which we recently employed to construct low-dimensional descriptions for the collapse dynamics of n-alkane chains in water (28). Similar to other nonlinear methodologies such as local linear embedding (37) and Isomap (38), the diffusion-map approach reconstructs the intrinsic manifold by patching together local structural information within the simulation trajectory into a coherent global construct (28,39). Considering PCA as a process which constructs linear hyperplanes through a data set upon which the data points are projected, the diffusion map approach may be considered its nonlinear analog in which the data points are instead projected onto curved manifolds.
The diffusion map approach has been described in greater depth by ourselves and others (28,39–41), but it may most easily be considered a means to synthesize a low-dimensional embedding of a simulation trajectory in a manner that preserves dynamic distances between the constituent snapshots. Considering the trajectory as a set of n 3R-dimensional snapshots recording R atomic positions, the method proceeds by computing the value of a scalar similarity metric, Δij, i,j = 1…n, between all snapshot pairs. This measure serves as a proxy measure of the kinetic closeness of the simulation snapshots, and need only be meaningful for small distances (i.e., within the immediate phase space locale of a particular configuration). We elect to use the translationally and rotationally minimized root mean-squared deviation between the coordinates of the peptide atoms for this metric, which is a natural choice in biophysical simulations, although others are possible (28,42). The n-by-n real, symmetric matrix L, is constructed by the application of a Gaussian kernel of bandwidth ɛ to the pairwise distances (28),
where the bandwidth specifies the radius within which values of Δij serve as a meaningful measure of kinetic proximity. The range of appropriate ɛ-values is delimited by the linear region of a plot of
twice the gradient of which provides an estimate of the effective dimensionality (39).
Normalizing the L matrix by its row sums yields a right-stochastic Markov matrix W with eigenvectors and associated eigenvalues on the interval (0,1] arranged in nonascending order (28). The top eigenvector is the all-ones vector, , with an associated eigenvalue of unity, μ1 = 1. Dimensionality reduction from 3R to k dimensions proceeds by mapping the jth -dimensional snapshot, into the jth element of each of the top nontrivial k eigenvectors, under the so-called diffusion map (15,17,28,40),
| (1) |
For shorthand, this operation will be referred to as an embedding in the top k eigenvectors (28) and represents the intrinsic or slow manifold of the system, data-mined out of the simulation trajectory. An appropriate number of eigenvectors to include in the embedding may be inferred by the presence of a spectral gap in the eigenvalue spectrum (17,40).
Unlike PCA, where the transformation of the simulation snapshots into the basis of principal components is explicitly known, the mapping of the diffusion map embedding to physical variables is unknown a priori. Established procedures to assign physical meaning to diffusion map order parameters (i.e., the components of the eigenvectors ) do not yet exist, with physical insight and interpretation typically provided by correlation with prospective physical variables and visualization of representative system configurations (28,41).
Biophysical systems with low effective dimensionalities (28–31) are describable by a set of coupled stochastic differential equations, and their dynamics are therefore expected to be well modeled as diffusion processes (28). The microscopic diffusive motions of such systems arise from thermal fluctuations in the constituent molecules, which are expected to be captured by the rotationally and translationally minimized root mean-squared deviation employed as the pairwise similarity metric in this work (28). When both of these conditions are satisfied, the diffusion map embedding is kinetically relevant in the sense that routes over the intrinsic manifold defined by the diffusion map embedding correspond to the slow, fundamental dynamical motions of the system. When these conditions are not met, the routes over the diffusion map embedding do not correspond to the fundamental, underlying motions, but the embedding still provides a good parameterization of the metastable states of the system and the transitions between them (28).
In this work, we apply the diffusion map approach to construct a low-dimensional embedding of the pro-MccJ25 REMD simulation trajectory, and identify a small number of variables with which to describe the fundamental dynamical motions of the peptide. Full details of the procedure, including the subtleties introduced by the application of the approach to a REMD data set, are provided in the Supporting Material, Methods: Application of the Diffusion Map to the REMD Simulation Trajectory.
Results and Discussion
Cyclization of cMccJ25 via CuAAC
There are two distinct cyclized topologies possible upon azide-alkyne ligation of the model cMccJ25 peptide (Fig. 1 e). Spontaneous folding into a near-native structure in which the β-turn is formed and residues 1–8 of the peptide begin to wrap around the peptide in a right-handed manner would lead to a lasso conformation similar to authentic MccJ25. If prefolding into lasso-like coils does not occur, the peptide can cyclize instead into a simpler loop-tail form. Analysis of the products of an intramolecular azide-alkyne ligation reaction on the cMccJ25 peptide therefore allows us to determine whether cMccJ25 adopts a near-native state. As described in the Experimental Methods, a panel of azide-alkyne ligation conditions was tested to evaluate which conditions led to high extents of intramolecular cyclization, and the products from several different reaction conditions were tested for antimicrobial activity using a Salmonella newport killing assay (see the Supporting Material, Methods: Salmonella Newport Killing Assays). No growth inhibition was observed for any of the reaction conditions (see Fig. S1 in the Supporting Material).
We have previously developed an HPLC gradient for the analysis of MccJ25 (14,43), and authentic MccJ25 has a retention time of 16.1 min under these conditions. HPLC analysis of linear cMccJ25 revealed that this peptide had a longer retention time of 17.7 min, consistent with its larger radius of gyration relative to authentic MccJ25. HPLC analysis of the cyclization product of cMccJ25 revealed a new peak intermediate between these two extremes, with a retention time of 16.9 min (Fig. 2 a). We hypothesize that this peak corresponds to the loop-tail cyclized form of cMccJ25, with further support provided by MS/MS. The fragmentation of lasso peptides in MSn experiments has been previously reported as being inefficient (44), whereas the C-terminal portion of the loop-tail form of the peptide is expected to be readily fragmented. As a control experiment, the linear cMccJ25 peptide was analyzed by MS/MS and yielded peaks corresponding to residues 5–20 of the peptide (Fig. 2 b). In contrast, fragmentation of click-cyclized cMccJ25 resulted only in peaks corresponding to residues 9–20, consistent with the difficulty in fragmenting the macrocycle composed of residues 1–8 (Fig. 2 c). The appearance of a clean fragmentation pattern corresponding to residues 9–20 is consistent with the loop-tail form of cyclized cMccJ25 rather than the lasso form. Taken together with the HPLC data presented above, this MS/MS data provides compelling evidence that the loop-tail form of cMccJ25 is the predominant species formed upon CuAAC-induced cyclization. From these experiments, we conclude that cMccJ25 does not adopt a near-native lasso structure in appreciable amounts.
Figure 2.
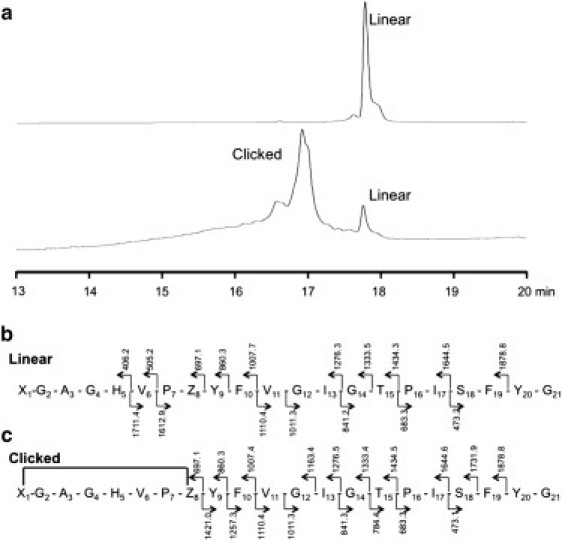
HPLC and MS/MS analysis of linear and cyclized cMccJ25. (a) The linear peptide exhibits an HPLC retention time of 17.7 min. After cyclization, much of the linear peptide was consumed and a new peak at 16.9 min emerged; the putative loop-tail form. As a point of reference, authentic MccJ25 has a retention time of 16.1 min (data not shown). (b) Mass of MS/MS fragments found in the linear peptide. (c) No MS/MS peaks corresponding to the macrocycle (amino acids 1–8) were found in the cyclized form due to the difficulty in fragmenting the ring.
Systematic determination of order parameters for pro-MccJ25
Application of the diffusion map approach to the pro-MccJ25 REMD simulation trajectory at 298 K and 1 bar resulted in a three-dimensional diffusion map embedding into eigenvectors 2–4 (evecs2–4). The physical interpretation of the order parameters associated with these three eigenvectors was facilitated by correlating evecs2–4 with physical intermediary variables (28). Our analysis revealed that evec2 was well correlated with the average length of the seven vectors linking the Cα atoms of adjacent residues in the native MccJ25 β-hairpin (Fig. 3). This order parameter, which we denote Λ, is similar to that employed by Yang et al. (45) in the study of a β-hairpin fragment of protein G. Small values of Λ correspond to Ile13 occupying the position at the top of the β-turn, with larger values corresponding to a ratcheting-down of the C-terminal with respect to the N-terminal strand such that residues closer to the N-terminus (Gly12, Val11, Phe10) occupy the turn position. Because ratcheting in the opposite direction was never observed during the course of the simulation, a scalar valued metric was sufficient to characterize the identity of the residue at the turn. Fig. 4 a presents a heat map of the [evec2, evec3, evec4] embedding in which the points are colored according to Λ, illustrating good correlation with evec2. Similarly, Fig. 4 b presents a heat map in the Glu8 Ψ angle, which demonstrates good correlation with evec4. The correlation coefficient between evec2 and Λ is 0.90, and between evec4 and the Glu8 Ψ angle—accounting for 360° periodicity—is 0.84. The strong, but not bijective, correlation in both cases indicates that although Λ and Glu8 Ψ are good descriptors of the order parameters extracted by the diffusion map, they are not identically the order parameters themselves. Physical variables showing good correlation with evec3 remained elusive.
Figure 3.
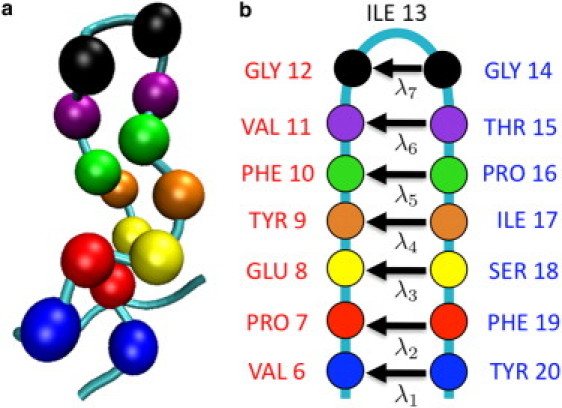
The physical order parameter Λ for pro-MccJ25 is defined as the mean length of the vectors connecting the Cα atoms of the seven pairs of adjacent residues in the native MccJ25 β-hairpin. The residue pairs are illustrated in (a) a visualization of pro-MccJ25 in a conformation formed by hydrolyzing the MccJ25 Gly1–Glu8 amide bond, in which the backbone is represented as a tube and the Cα atoms of the residue pairs as van der Waals spheres, and (b) a schematic representation of pro-MccJ25 showing the seven vectors , where . Reading down from the β-turn, the residue pairs are: Gly12–Gly14 (black), Val11–Thr15 (purple), Phe10–Pro16 (green), Tyr9–Ile17 (orange), Glu8–Ser18 (yellow), Pro7–Phe19 (red), and Val6–Tyr20 (blue). Increases in Λ correspond to lengthening of the λi vectors, and ratcheting residues closer to the N-terminus into the turn position; ratcheting in the opposite direction was never observed.
Figure 4.
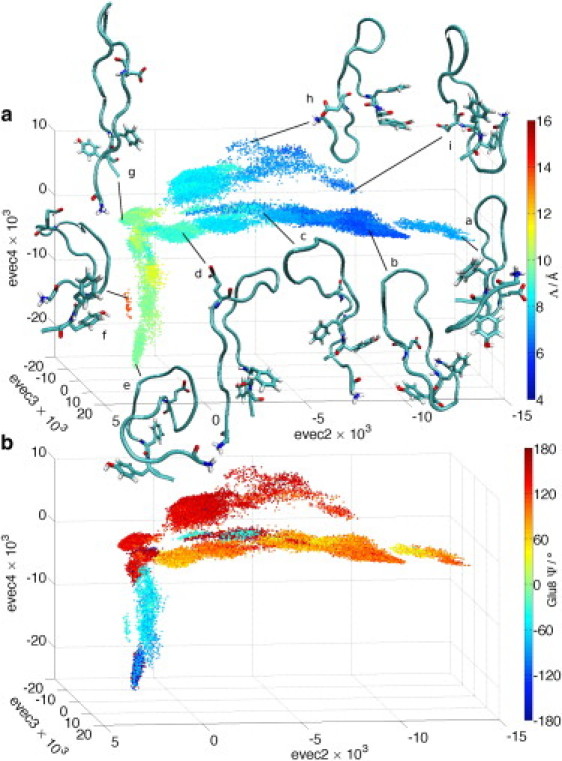
Three-dimensional embeddings of the REMD trajectory in [evec2, evec3, evec4] in which the data points are colored according to (a) Λ (see Fig. 3) and (b) the Glu8 Ψ angle. Representative peptide structures are visualized over the point cloud in panel a. In this and subsequent figures, the peptide backbone is represented as a tube and solvent has been removed for clarity. In this particular figure Gly1, Glu8, Phe19, and Tyr20 residues are of most relevance to the discussion, and are visualized in atomic detail.
The details of the structural transitions over the diffusion map embedding were resolved by visualization of peptide conformations at selected data points, using the molecular visualization package VMD (46). Superposition of representative structures onto the point cloud in Fig. 4 a illustrate the conformational transitions corresponding to each of the three distinct branches. For clarity of viewing in this figure and those that follow, the peptide backbone is represented as a tube, with only a subset of the residues visualized in atomic detail. The solvent is removed for clarity.
Central branch
Transitions along the central branch from g→d→c correspond to a ratcheting-down and concomitant lengthening of the N-terminal strand with respect to the C-terminal strand, shifting the shared occupancy of the β-turn position by Val11/Gly12 in structure g, to Gly12 in structure d, to Gly12/Ile13 in structure c. The N- and C-terminal strands remain essentially parallel and uncrossed. The transition from c→b is associated with Ile13 moving to occupy the turn position, and the N-terminus wrapping around the C-terminus between the Phe19 and Tyr20 steric locks in a left-handed or counterclockwise manner. The Gly1 N-Glu8 Cδ approach distance closes to as little as 6 Å. The N-terminal wrapping is in the opposite sense to the right-handed topology of native MccJ25. Finally, structure a at the farthest extent of the central branch corresponds to a left-handed topology in which the N-terminus is threaded through the cleft formed by the C-terminus and Tyr20 rather than between the steric locks.
Upper branch
As with the central branch, traversing the upper branch from g→h is associated with a lengthening of the N-terminal strand with respect to the C-terminal as the occupancy of the β-turn position by Val11/Gly12 in structure g yields to Ile13 in structure h. This is accompanied by a doubling-back of the N-terminus upon itself whereby the positively charged N-terminal amino and negatively charged Glu8 γ-carboxylate groups are held in close proximity by mutual electrostatic attraction. Phe19-Tyr20 ring stacking is observed at the C-terminus. The transition from from h→i corresponds to a threading of the N-terminus through the C-terminal/Tyr20 cleft, wrapping under rather than around the C-terminus as in structure a. Hydrogen bonding between the N-terminal amino, Ser18 hydroxyl and Tyr9 hydroxyphenyl groups stabilize this conformation.
Lower branch
This branch in which evec2 remains essentially constant as evec4 decreases corresponds to global collapse of the peptide. The N- and C-terminal strands remain essentially parallel along this branch, with the occupancy of the turn position by Val11/Gly12 in the extended conformation in structure g yielding to Phe10 in the collapsed C-shaped conformation in e. The island of points containing structure f corresponds to partially collapsed C-shaped conformations with Tyr9 at the turn.
We now consider the distribution of points over the manifold, by constructing free energy surfaces over the point cloud in Fig. 4 projected into evecs2 and 4 for ease of viewing (Fig. 5). The structure of the point cloud is such that the topology of the three-dimensional FES in evecs2–4 (Fig. 5, upper inset) is preserved under this projection. Five distinct local free energy minima are apparent, within which representative peptide conformations have been projected to illustrate the metastable states of the system. These structures are distinct from those projected in Fig. 4, where the intent was to elucidate the structural transitions along the three branches, and overlap between the two sets of images is purely coincidental. Basin D is the global free energy minimum, associated with peptide conformations with extended, parallel N- and C-terminal strands with Val11/Gly12 in the turn position, stabilized by nine backbone hydrogen bonds. Basins B and E consist of similar β-hairpin structures with Gly12 and Gly12/Ile13 occupying the turn position, respectively. Basin A contains the left-handed coil conformations which possess seven backbone hydrogen bonds, and the docking of the N-terminus behind the Phe19 steric lock. Basin C at the bottom of the lower branch contains C-shaped structures possessing six backbone hydrogen bonds, with the aromatic side chains of Tyr9, Phe10, Ile17, and Phe19 forming a solvent-excluded hydrophobic pocket. The representation of C in the lower inset of Fig. 5 renders these atoms of these residues as van der Waals spheres, illustrating that such conformations may be considered hydrophobically collapsed.
Figure 5.
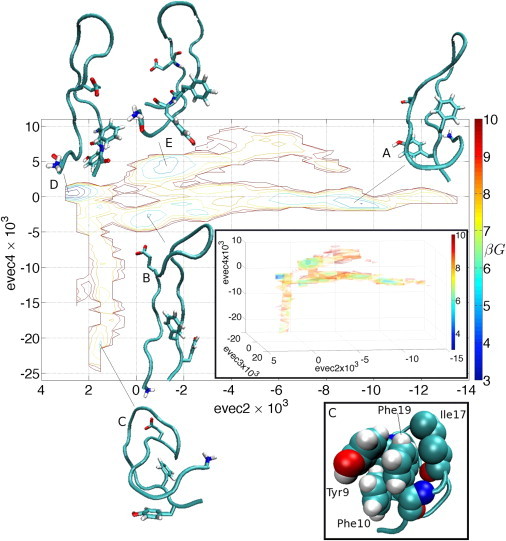
Two-dimensional FES constructed over the embedding of the REMD trajectory in [evec2, evec4]. Gly1, Glu8, Phe19, and Tyr20 residues are visualized explicitly in the representative peptide structures projected into the free energy minima. G is the Gibbs free energy, and β = 1/kBT. The range of βG is 2.5–10.7, and contours are drawn at unit steps of βG from 3 to 10. (Upper inset) The three-branched topology of the three-dimensional FES constructed over the embedding of the REMD trajectory in [evec2, evec3, evec4] is preserved upon projecting out evec3. The range of βG is 2.3–10.7, and isosurfaces are plotted at unit steps of βG from 3 to 10. (Lower inset) A rerepresentation of structure C with the atoms of Tyr9, Phe10, Ile17, and Phe19 rendered as van der Waals spheres to illustrate the hydrophobic pocket formed by these side chains.
The left-handed coils within basin A are entropically disfavored due to the loss of conformational entropy associated with the docking of the N-terminus behind the Phe19 aromatic ring. Such conformations are, however, energetically stabilized by water-mediated Coulombic interactions among the positively charged N-terminal amino, Ser18 hydroxyl, and negatively charged Glu8 carboxylate groups in an electrostatic bridge. Two water molecules are frequently observed in the interstices among the N-terminal amino, Ser18 hydroxyl, and Glu8 carboxylate, forming a linear hydrogen-bonded chain bridging the positively charged N-terminus and negatively charged Glu8 side chain, and stabilizing the low-entropy coil conformation (Fig. 6). It is also likely that the Phe19 steric lock offers a measure of kinetic stabilization of this free energy basin by restricting the motion of the N-terminal strand.
Figure 6.
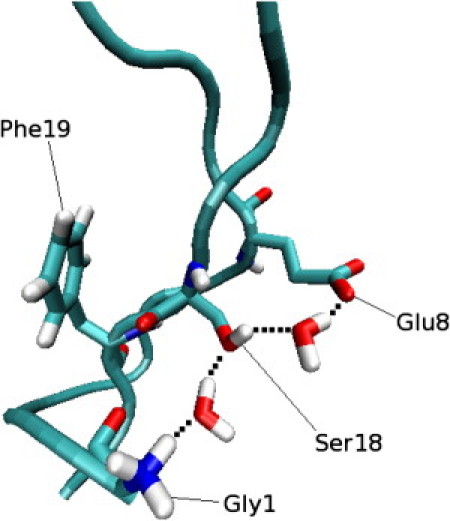
Snapshot from the 30 ns MD trajectory initialized in the vicinity of the left-handed coil at ∼40 ps after the docking of the N-terminus into the left-handed coil conformation. Details of this simulation are provided in the Supporting Material, Results & Discussion: Local Analysis of Spontaneously Adopted Left-Handed Coils in pro-MccJ25. Gly1, Glu8, Ser18, and Phe19 are explicitly visualized on the peptide backbone, along with the two interstitial water molecules. Together with the Ser18 hydroxyl, these water molecules form a linear hydrogen-bonded network between the positively charged N-terminal amino and negatively charged Glu8 carboxylate groups. Hydrogen bonds are indicated by broken lines.
In coarse terms, the three distinct pathways leading away from the global free energy minimum (basin D) may be considered as global hydrophobic collapse (lower branch), adoption of a left-handed coil (central branch), and adoption of an unwrapped left-handed coil (upper branch).
Conclusions
The HPLC and MS/MS data collectively provide compelling evidence that the cyclized cMccJ25 peptide is in a loop-tail topology and not a lasso topology, a result for which the MD study of pro-MccJ25 offers a molecular-level interpretation. The simulations suggest that near-native right-handed coils are not spontaneously adopted by pro-MccJ25 in isolation, although left-handed coils in which the N-terminus wraps around the C-terminus in a nonnative, counterclockwise manner were observed. In such left-handed coils, the entropic cost associated with docking the N-terminus behind the Phe19 steric lock is offset by a water-mediated electrostatic interaction between the Gly1 amino and Glu8 carboxylate. Crucially, this interaction is absent in cMccJ25 in which these groups are replaced by azidoacetic acid and propargylglycine, respectively (Fig. 1). Accordingly, while 15% of the pro-MccJ25 conformational ensemble exists as left-handed coils (basin A in Fig. 5), we expect this fraction to be substantially reduced for cMccJ25 due to the absence of the stabilizing electrostatic interaction, with the majority of the population existing in noncoil conformations. The experimentally observed loop-tail topology of cyclized cMccJ25 may be interpreted as arising from a higher probability of the click-reaction occurring with the peptide in an unwrapped conformation.
We have reported the first application, to the best of our knowledge, of the diffusion map approach to a molecular simulation of a protein. This technique allowed us to systematically extract three global order parameters characterizing the underlying dynamical motions of the peptide. Two of these variables were well correlated with the Glu8 Ψ angle and a variable Λ describing the identity of the residue occupying the β-turn position (Fig. 4). We were unsuccessful in assigning a physical interpretation to the third order parameter, highlighting a well-known deficiency of the diffusion map approach in that no systematic means yet exists to relate physical and diffusion map order parameters. We believe that concepts from control theory such as the single input effectiveness (47), and techniques to systematically screen pools of candidate variables such as those proposed by Ma and Dinner (48) and Trout and co-workers (49,50), may provide means to make inroads into this important problem. Nevertheless, a dynamically meaningful free energy surface parameterized by Glu8 Ψ and Λ (Fig. 5) permitted the identification of three distinct pathways corresponding to global hydrophobic collapse, adoption of a left-handed coil conformation, and formation of an unwrapped left-handed coil. As we report in the Supporting Material, Results & Discussion: Application of PCA to the REMD Simulation Trajectory, despite its inherent linearity and absence of a dynamic interpretation, PCA performed remarkably well for this system, extracting similar global order parameters and pathways to those furnished by the diffusion map approach.
Analysis of the important dynamical motions in the vicinity of the left-handed coil conformations spontaneously adopted by pro-MccJ25 (see the Supporting Material, Results & Discussion: Local Analysis of Spontaneously Adopted Left-Handed Coils in pro-MccJ25) show that the coil conformation is adopted by the lateral wrapping of the N-terminus around the C-terminal strand (Fig. S2). Furthermore, this analysis suggests that the Phe19 and Tyr20 steric locks may be pinned-back by hydrophobic and electrostatic interactions, respectively, permitting the docking of the N-terminus. Although the REMD simulations were initialized from a right-handed coil conformation, the absence of right-handed coil conformations during the 95 ns REMD production run suggests that this conformation may be inaccessible to pro-MccJ25. In an analysis of the local dynamics in the region of conformational space in which the closest approach to right-handed coils was observed (see the Supporting Material, Results & Discussion: Local Analysis of Closest Approach to Right-Handed Coils in pro-MccJ25), we find evidence for the kinetic inaccessibility of such conformations in the concerted motion of the Phe19 χ1 angle upon approach of the N-terminal strand in a right-handed manner, causing the Phe19 aromatic ring to sterically block the N-terminus from forming a right-handed coil (Fig. S3).
Although native MccJ25 is a right-handed lasso, the left-handed lasso topology is sterically feasible and compatible with experimental NMR distance restraints (2). By decomposing the various contributions to the interaction energy for the left and right-handed lassos (see the Supporting Material, Results & Discussion: Energetic Comparison of Left and Right-Handed MccJ25 Lassos), we find the remarkable result that the 21-residue MccJ25 peptide supports both left- and right-handed lasso topologies, with nearly identical overall interaction energies (Fig. S4). The rigidity imparted by the lasso implies that the two topologies also possess similar conformational entropies, suggesting that the observed absence of right-handed coils in our pro-MccJ25 MD simulations using the replica exchange methodology—a technique which facilitates the crossing of barriers on the order of several kBT—is likely due to the presence of substantial free energy barriers, possibly associated with an entropic bottleneck involving the motion of the Phe19 side chain.
Because steric considerations dictate that MccJ25 must—at least transiently—adopt a near-native right-handed coil before the formation of the Gly1–Glu8 isopeptide bond, our results suggest a putative role for some portion of the 37-residue McjA leader sequence (14), and/or the maturation enzymes McjB and McjC, in establishing the correct handedness of the lasso. While we are hesitant to speculate on a possible mechanism, we observe that a threonine residue in the penultimate amino acid (P2) position of the leader peptide is critical for the maturation of both MccJ25 (14) and another lasso peptide, capistruin (51). We advance the possibility that the hydrogen-bonding capability of this conserved residue in close proximity to the N-terminus of the mature peptide may be involved in correctly orienting the Glu8 side chain.
Acknowledgments
We thank Prof. Jennifer J. Ottesen of The Ohio State University for providing 75 structures of MccJ25 determined from NMR distance restraints.
A.Z.P. acknowledges financial support from the Department of Energy, Office of Basic Energy Sciences (No. DE-SC0002128) and the Princeton Center for Complex Materials (No. DMR-0819860). P.G.D. acknowledges support from the National Science Foundation (Collaborative Research in Chemistry Nos. CHE-0404699 and CHE-0908265). A.J.L. acknowledges support from the National Science Foundation CAREER Program (No. CBET-0952875) and Princeton University.
Supporting Material
References
- 1.Pavlova O.A., Severinov K.V. Posttranslationally modified microcins. Russ. J. Genet. 2006;42:1380–1389. [PubMed] [Google Scholar]
- 2.Wilson K.A., Kalkum M., Darst S.A. Structure of microcin J25, a peptide inhibitor of bacterial RNA polymerase, is a lassoed tail. J. Am. Chem. Soc. 2003;125:12475–12483. doi: 10.1021/ja036756q. [DOI] [PubMed] [Google Scholar]
- 3.Bayro M.J., Mukhopadhyay J., Ebright R.H. Structure of antibacterial peptide microcin J25: a 21-residue lariat protoknot. J. Am. Chem. Soc. 2003;125:12382–12383. doi: 10.1021/ja036677e. [DOI] [PubMed] [Google Scholar]
- 4.Rosengren K.J., Clark R.J., Craik D.J. Microcin J25 has a threaded sidechain-to-backbone ring structure and not a head-to-tail cyclized backbone. J. Am. Chem. Soc. 2003;125:12464–12474. doi: 10.1021/ja0367703. [DOI] [PubMed] [Google Scholar]
- 5.Duquesne S., Destoumieux-Garzón D., Rebuffat S. Two enzymes catalyze the maturation of a lasso peptide in Escherichia coli. Chem. Biol. 2007;14:793–803. doi: 10.1016/j.chembiol.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Bierbaum G., Jansen A. Tying the knot: making of lasso peptides. Chem. Biol. 2007;14:734–735. doi: 10.1016/j.chembiol.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Rebuffat S., Blond A., Peduzzi J. Microcin J25, from the macrocyclic to the lasso structure: implications for biosynthetic, evolutionary and biotechnological perspectives. Curr. Protein Pept. Sci. 2004;5:383–391. doi: 10.2174/1389203043379611. [DOI] [PubMed] [Google Scholar]
- 8.Pavlova O., Mukhopadhyay J., Severinov K. Systematic structure-activity analysis of microcin J25. J. Biol. Chem. 2008;283:25589–25595. doi: 10.1074/jbc.M803995200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolb H.C., Finn M.G., Sharpless K.B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Rostovtsev V., Green L.G., Sharpless K. A stepwise Huisgen cycloaddition process: copper (I)-catalyzed regioselective ligation of azides and terminal alkynes. Agnew. Chem. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 11.Tornøe C.W., Christensen C., Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 12.Wang Q., Chan T.R., Finn M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 13.Wu P., Fokin V. Catalytic azide-alkyne cycloaddition: reactivity and applications. Aldrichim Acta. 2007;40:7–17. [Google Scholar]
- 14.Cheung W.L., Pan S.J., Link A.J. Much of the microcin J25 leader peptide is dispensable. J. Am. Chem. Soc. 2010;132:2514–2515. doi: 10.1021/ja910191u. [DOI] [PubMed] [Google Scholar]
- 15.Coifman R.R., Lafon S., Zucker S.W. Geometric diffusions as a tool for harmonic analysis and structure definition of data: diffusion maps. Proc. Natl. Acad. Sci. USA. 2005;102:7426–7431. doi: 10.1073/pnas.0500334102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coifman R.R., Lafon S. Diffusion maps. Appl. Comput. Harmon. Anal. 2006;21:5–30. [Google Scholar]
- 17.Belkin M., Niyogi P. Laplacian eigenmaps for dimensionality reduction and data representation. Neural Comput. 2003;15:1373–1396. [Google Scholar]
- 18.Dyke J., Groves A., Moutinho A. Study of the thermal decomposition of 2-azidoacetic acid by photoelectron and matrix isolation infrared spectroscopy. J. Am. Chem. Soc. 1997;119:6883–6887. [Google Scholar]
- 19.Chan T.R., Hilgraf R., Fokin V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 20.Lewis W.G., Magallon F.G., Finn M.G. Discovery and characterization of catalysts for azide-alkyne cycloaddition by fluorescence quenching. J. Am. Chem. Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 21.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 22.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 23.Berman H.M., Battistuz T., Zardecki C. The Protein DataBank. Acta Crystallogr. D Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 24.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 25.Okabe T., Kawata M., Mikami M. Replica-exchange Monte Carlo method for the isobaric–isothermal ensemble. Chem. Phys. Lett. 2001;335:435–439. [Google Scholar]
- 26.Hukushima K., Nemoto K. Exchange Monte Carlo method and application to spin glass simulations. J. Phys. Soc. Jpn. 1996;65:1604–1608. [Google Scholar]
- 27.Patriksson A., van der Spoel D. A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 2008;10:2073–2077. doi: 10.1039/b716554d. [DOI] [PubMed] [Google Scholar]
- 28.Ferguson A.L., Panagiotopoulos A.Z., Kevrekidis I.G. Systematic determination of order parameters for chain dynamics using diffusion maps. Proc. Natl. Acad. Sci. USA. 2010;107:13597–13602. doi: 10.1073/pnas.1003293107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.García A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992;68:2696–2699. doi: 10.1103/PhysRevLett.68.2696. [DOI] [PubMed] [Google Scholar]
- 30.Amadei A., Linssen A.B.M., Berendsen H.J.C. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 31.Hegger R., Altis A., Stock G. How complex is the dynamics of peptide folding? Phys. Rev. Lett. 2007;98:028102–028104. doi: 10.1103/PhysRevLett.98.028102. [DOI] [PubMed] [Google Scholar]
- 32.Hummer G., Kevrekidis I.G. Coarse molecular dynamics of a peptide fragment: free energy, kinetics, and long-time dynamics computations. J. Chem. Phys. 2003;118:10762–10773. [Google Scholar]
- 33.Das P., Moll M., Clementi C. Low-dimensional, free-energy landscapes of protein-folding reactions by nonlinear dimensionality reduction. Proc. Natl. Acad. Sci. USA. 2006;103:9885–9890. doi: 10.1073/pnas.0603553103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhuravlev P.I., Materese C.K., Papoian G.A. Deconstructing the native state: energy landscapes, function, and dynamics of globular proteins. J. Phys. Chem. B. 2009;113:8800–8812. doi: 10.1021/jp810659u. [DOI] [PubMed] [Google Scholar]
- 35.Sanbonmatsu K.Y., García A.E. Structure of Met-enkephalin in explicit aqueous solution using replica exchange molecular dynamics. Proteins. 2002;46:225–234. doi: 10.1002/prot.1167. [DOI] [PubMed] [Google Scholar]
- 36.Tournier A.L., Smith J.C. Principal components of the protein dynamical transition. Phys. Rev. Lett. 2003;91:208106. doi: 10.1103/PhysRevLett.91.208106. [DOI] [PubMed] [Google Scholar]
- 37.Roweis S.T., Saul L.K. Nonlinear dimensionality reduction by locally linear embedding. Science. 2000;290:2323–2326. doi: 10.1126/science.290.5500.2323. [DOI] [PubMed] [Google Scholar]
- 38.Tenenbaum J.B., de Silva V., Langford J.C. A global geometric framework for nonlinear dimensionality reduction. Science. 2000;290:2319–2323. doi: 10.1126/science.290.5500.2319. [DOI] [PubMed] [Google Scholar]
- 39.Coifman R.R., Shkolnisky Y., Singer A. Graph Laplacian tomography from unknown random projections. IEEE Trans. Image Process. 2008;17:1891–1899. doi: 10.1109/TIP.2008.2002305. [DOI] [PubMed] [Google Scholar]
- 40.Nadler B., Lafon S., Kevrekidis I.G. Diffusion maps, spectral clustering and eigenfunctions of Fokker-Planck operators. In: Weiss Y., Schölkopf B., Platt J., editors. Advances in Neural Information Processing Systems. Vol. 18. MIT Press; Cambridge, MA: 2006. pp. 955–962. [Google Scholar]
- 41.Sonday B.E., Haataja M., Kevrekidis I.G. Coarse-graining the dynamics of a driven interface in the presence of mobile impurities: effective description via diffusion maps. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2009;80:031102–031111. doi: 10.1103/PhysRevE.80.031102. [DOI] [PubMed] [Google Scholar]
- 42.Maiorov V.N., Crippen G.M. Size-independent comparison of protein three-dimensional structures. Proteins. 1995;22:273–283. doi: 10.1002/prot.340220308. [DOI] [PubMed] [Google Scholar]
- 43.Pan S.J., Cheung W.L., Link A.J. Engineered gene clusters for the production of the antimicrobial peptide microcin J25. Protein Expr. Purif. 2010;71:200–206. doi: 10.1016/j.pep.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 44.Knappe T.A., Linne U., Marahiel M.A. Isolation and structural characterization of capistruin, a lasso peptide predicted from the genome sequence of Burkholderia thailandensis E264. J. Am. Chem. Soc. 2008;130:11446–11454. doi: 10.1021/ja802966g. [DOI] [PubMed] [Google Scholar]
- 45.Yang S., Onuchic J.N., Levine H. Folding time predictions from all-atom replica exchange simulations. J. Mol. Biol. 2007;372:756–763. doi: 10.1016/j.jmb.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 46.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 47.Seborg D.E., Edgar T.F., Mellichamp D.A. John Wiley & Sons; New York: 1989. Process Dynamics and Control. [Google Scholar]
- 48.Ma A., Dinner A.R. Automatic method for identifying reaction coordinates in complex systems. J. Phys. Chem. B. 2005;109:6769–6779. doi: 10.1021/jp045546c. [DOI] [PubMed] [Google Scholar]
- 49.Peters B., Trout B.L. Obtaining reaction coordinates by likelihood maximization. J. Chem. Phys. 2006;125:054108. doi: 10.1063/1.2234477. [DOI] [PubMed] [Google Scholar]
- 50.Peters B., Beckham G.T., Trout B.L. Extensions to the likelihood maximization approach for finding reaction coordinates. J. Chem. Phys. 2007;127:034109. doi: 10.1063/1.2748396. [DOI] [PubMed] [Google Scholar]
- 51.Knappe T.A., Linne U., Marahiel M.A. Insights into the biosynthesis and stability of the lasso peptide capistruin. Chem. Biol. 2009;16:1290–1298. doi: 10.1016/j.chembiol.2009.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.