

Abstract
Proteins fold up by coordinating the different segments of their polypeptide chain through a network of weak cooperative interactions. Such cooperativity results in unfolding curves that are typically sigmoidal. However, we still do not know what factors modulate folding cooperativity or the minimal amount that ensures folding into specific three-dimensional structures. Here, we address these issues on BBL, a small helical protein that folds in microseconds via a marginally cooperative downhill process (Li, P., Oliva, F. Y., Naganathan, A. N., and Muñoz, V. (2009) Proc. Natl. Acad. Sci. USA. 106, 103–108). Particularly, we explore the effects of salt-induced screening of the electrostatic interactions in BBL at neutral pH and in acid-denatured BBL. Our results show that electrostatic screening stabilizes the native state of the neutral and protonated forms, inducing complete refolding of acid-denatured BBL. Furthermore, without net electrostatic interactions, the unfolding process becomes much less cooperative, as judged by the broadness of the equilibrium unfolding curve and the relaxation rate. Our experiments show that the marginally cooperative unfolding of BBL can still be made twice as broad while the protein retains its ability to fold into the native three-dimensional structure in microseconds. This result demonstrates experimentally that efficient folding does not require cooperativity, confirming predictions from theory and computer simulations and challenging the conventional biochemical paradigm. Furthermore, we conclude that electrostatic interactions are an important factor in determining folding cooperativity. Thus, electrostatic modulation by pH-salt and/or mutagenesis of charged residues emerges as an attractive tool for tuning folding cooperativity.
Keywords: Biophysics, Cooperativity, Protein Conformation, Protein Folding, Protein Stability, Downhill Folding, Electrostatic Screening, Ultrafast Kinetics
Introduction
In principle, proteins fold into their biologically active three-dimensional structures by relying on two interrelated factors: stability and cooperativity. The stability of the native (folded) state comes from solvent-driven forces and specific weak interactions, such as hydrogen bonds, electrostatics, and van der Waals interactions (1). Contributions from these interactions to protein stability have been investigated thoroughly using mutational analysis (2). Cooperativity refers to the coupling of the various stabilizing interactions that make proteins (un)fold in a concerted fashion and result in sigmoidal equilibrium unfolding curves (3). Statistical mechanical arguments indicate that folding cooperativity cannot be too high because proteins are stabilized by limited numbers of weak interactions (4). The lower bound is possibly the formation of isolated α-helices, in which the energetic coupling is only local (i,i+3 hydrogen bonds). However, such small cooperativity results in broad ensembles of partly helical conformations rather than a single α-helix (5). In contrast, conventional wisdom states that folding into a specific globular three-dimensional structure must involve an all-or-none process with near-infinite cooperativity (6). This idea is supported by two major experimental observations. First, all single-domain proteins that fold in milliseconds or longer seem to adhere to a two-state (all-or-none) folding model (7). Second, seminal work during the 1980–1990s has shown that under conditions of low pH and high salt, many proteins form partly structured molten globule states (8–12). Such molten globules have a native-like secondary structure and unfold with low cooperativity, but they also lack defined tertiary structure (13).
On the other front, theory (14) and computer simulations (15–18) suggest that natural protein folding is nominally cooperative. Coarse-grained models show that proteins can fold into defined structures by a completely uncooperative process (15–18). In fact, to produce cooperative unfolding transitions, these models require ad hoc contributions from many-body terms in addition to simple pairwise interactions (19–21). However, calibration against experimental data indicates that a fairly small contribution from many-body interactions is sufficient to reproduce the broadness of the experimental unfolding curves. Interestingly, in such simulations, one can observe a direct relationship between cooperativity at the atomic level and the broadness of the macroscopic unfolding curve (22). Likewise, the simulations do not produce concerted global unfolding, as expected for an all-or-none process, but a distribution of unfolding behaviors for the various residues in the protein (23).
The critical question, then, is: what is the minimal level of cooperativity that affords folding into a well defined three-dimensional structure? Is it nearly infinite as assumed conventionally or nominal as suggested by theory and simulations? And, as a subsidiary, what are the physical factors that modulate such cooperativity?
To date, this question has not been amenable to experimental analysis. The reason is that for a two-state-like process, the unfolding curve is not necessarily proportional to the degree of energetic coupling but to the change in enthalpy upon unfolding. In other words, the actual degree of cooperativity of two-state-like folding is not measurable experimentally. However, recent work on ultra-fast folding offers an opportunity to solve this stalemate. Proteins that fold in a few microseconds cross marginal (<3 RT) free energy barriers and exhibit broader equilibrium unfolding curves (24). Under these conditions, folding occurs in the downhill regime predicted by energy landscape theory (14), where cooperativity is low and thus amenable to experimental analysis (25). Confirmation of this idea has come from the atom-by-atom NMR analysis of the thermal unfolding of BBL (26), a microsecond folding protein that has been identified as a global downhill folder in both equilibrium (27) and kinetic experiments (28). The NMR analysis of BBL has demonstrated the same connection between the broadness of the global unfolding process and the variability of atomic unfolding behaviors observed in simulations.
BBL is the peripheral subunit binding domain from the subunit 2 of the Escherichia coli oxo-glutarate reductase complex (29). This little domain (40 residues) is involved in the sequential binding to subunits 1 and 3 (29) in a coordinated process for which its low folding cooperativity may be critical (30). Here, we take advantage of the folding properties of BBL to experimentally explore the limits of folding cooperativity. We focus on the role of electrostatic interactions. Interactions between charged residues contribute to protein stability (31), and, because they act over proportionally long distances, electrostatics also are likely to affect cooperativity (32). Previous work has shown that acidic pH destabilizes folded BBL, which reaches complete unfolding at pH 3.0 (27). The key to this process appears to be the protonation of two buried histidines (His13 and His37 with pKa values of 6.4 and 5.3 (33)).
Therefore, to investigate the electrostatic effects on the folding cooperativity of BBL (i.e. determined from the broadness of the unfolding curve and the changes in folding kinetics), we revisited the combination of acid denaturation plus salt addition. The idea is simple; by varying pH and salt concentration, we should be able to screen the electrostatic interactions of BBL while tuning up its folding stability.
EXPERIMENTAL PROCEDURES
Protein Samples and Concentration Determination
Naf-BBL was produced as indicated previously (34). Protein samples at acidic pH were prepared in citrate buffer and neutral pH samples in phosphate buffer, each at 20 mm. Buffer reagents and salts were of chemical grade, purchased from Sigma-Aldrich. The concentration of salt in the protein samples was estimated by weight and refractive index measurements. Samples for infrared (IR) spectroscopic measurements were prepared in deuterated buffers, with D2O purchased from Cambridge Isotope Laboratories (Andover, MA). Protein concentration was determined by UV absorbance spectroscopy on a Cary100 Bio spectrophotometer from Varian. Naf-BBL concentrations were estimated as the average from determinations at 266 and 280 nm using molar absorptivities of 3,595 m−1·cm−1 at 266 nm and 5,526 m−1·cm−1 at 280 nm.
Far-UV CD
CD experiments were performed on a Jasco J-815 spectropolarimeter at a protein concentration of ∼50 μm in 20 mm buffer. Equilibrium thermal unfolding was monitored by collecting CD spectra from 268 to 368 K every 5 K with 1-nm resolution in a cuvette of 1-mm path length. Reversibility was ensured by comparing spectra at 298 K collected before and after each ramp.
Phenomenological Fits to Equilibrium Unfolding Curves
CD thermal unfolding data were fitted to the sigmoidal unfolding curve,
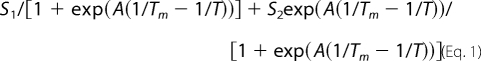 |
where A is the sharpness of the curve (smaller A indicates lower cooperativity), Tm is the temperature at which the signal decreases by 50%, and S1 and S2 are the signals of the pre- and post-transition baselines, which were assumed to be salt-independent and linearly dependent on temperature. CD chemical denaturation experiments were fitted to the sigmoidal unfolding curve,
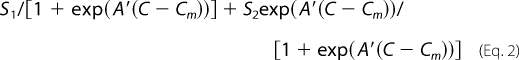 |
where A′ is the sharpness of the curve, C is the concentration of denaturant, Cm is the concentration of denaturant at which the signal decreases by 50%, and S1 and S2 are the baselines, which are assumed to be linear functions of chemical denaturant.
NMR
NMR one-dimensional, two-dimensional 1H-1H nuclear Overhauser spectroscopy (NOESY),2 two-dimensional 1H-1H total correlation spectroscopy, and 1H/15N correlation via heteronuclear zero and double quantum coherence (SOFAST-HMQC) spectra were collected for Naf-BBL at 283 K. NOESY spectra were acquired with a 150-ms mixing time. Protein samples were prepared at 2 mm in the presence of 10% D2O and 10 mm 4,4-dimethyl-4-silapentane-1-sulfonic acid. 1H/15N SOFAST-HMQC experiments were performed in isotopic natural abundance using ∼12,000 scans.
Laser-induced Nanosecond IR Temperature-jump Spectroscopy
IR temperature-jump experiments were performed on an improved home-built version of the instrument described by Gai and co-workers (35) (see supplemental “Experimental Procedures” for details).
RESULTS
Salt-induced Stabilization and Refolding of Acid-denatured BBL
At pH 3, BBL is fully unfolded. However, the addition of a neutral monovalent salt such as LiCl under such acidic conditions leads to a progressive increase in the α-helical content of protonated BBL as probed by CD (Fig. 1A). Above 1 m salt, the helical CD signal of BBL exhibited typical sigmoidal unfolding curves with temperature, which suggested that protonated BBL refolds at low temperatures. Moreover, the apparent temperature denaturation midpoint (Tm) increases as more salt is added. The Tm increase is manifested in Fig. 1A as a right shift of the unfolding curve. Quantitative estimates, obtained from either the maximum in the derivative of the unfolding curve using the method of Naganathan and Muñoz (36) or by fitting the unfolding experiments to phenomenological sigmoidal curves (Equation 1), showed that the increase in Tm tapers off at LiCl concentrations above 2 m (Fig. 1B). These experiments suggest that the addition of LiCl stabilizes a folded form of protonated BBL, leading to complete refolding at concentrations above 1 m. Similar refolding was observed upon addition of other salts with larger cationic radii, such as NaCl and CsCl (supplemental Fig. S2).
FIGURE 1.

Salt-induced refolding of protonated BBL measured by far-UV CD. A, circles indicate mean residue ellipticity at 222 nm (α-helical signal) at pH 3 with varying concentrations of LiCl. Red lines are phenomenological fits to sigmoidal curves (i.e. Equation 1) to guide the eye. B, plot of the midpoint temperature as a function of LiCl concentration: (light blue circles and left scale) pH 3, (red circles and right scale) pH 7. C, BBL α-helix signal at 222 nm, pH 3, and 268 K as a function of salt concentration: circles, LiCl; triangles, NaCl; and inverted triangles, CsCl. The black line is a linear fit to the composite data from 1 to 6 m to guide the eye. The red line indicates the CD signal of native BBL at pH 7 and 268 K. Inset, far-UV CD spectra at 268 K for pH 7 (red), pH 3 plus 1 m LiCl (blue), and pH 3 plus 5.3 m LiCl (green).
The response of protonated BBL to salt was quite strong, with 0.25 m being sufficient to boost the helical signal to >50% of the native signal at the lowest temperature (Fig. 1C). At ∼1 m salt, the low temperature CD spectrum of protonated BBL was nearly identical to that of native BBL at pH 7 and at the same temperature (Fig. 1C, red line and inset). From this point onward, the α-helix signal at 222 nm increased linearly upon further addition of salt. This increase reports a global increase in α-helix content, as can be assessed by comparison of the CD spectrum at 1 and 5.3 m salt (Fig. 1C, inset). It is thus reasonable to assume that the linear α-helix increase we observed above 1 m salt corresponds to the native baseline of BBL, which has been seen to change proportionally to the degree of destabilization induced by a variety of factors such as temperature (34), urea (37), and pH (33).
Protonated BBL Folds into the Native Three-dimensional Structure in the Presence of Salt
CD is a very sensitive folding probe for α-helical proteins such as BBL. However, CD monitors only secondary structure, and thus, it is possible that in the presence of high salt, protonated BBL becomes as α-helical as native BBL without folding into a well defined tertiary structure. That is, the addition of salt at low pH could be inducing a molten globule intermediate rather than the folded state, as it has been seen for many other proteins (11, 38).
NMR is the most powerful spectroscopic technique to distinguish between a folded protein and a molten globule. The NMR spectrum of a molten globule lacks signal dispersion, resembles that of an unfolded protein, and does not show long range NOEs (39). Fig. 2 summarizes the results from the NMR analysis of BBL. Fig. 2A shows the two-dimensional SOFAST-HMQC spectra recorded in N15 natural abundance of BBL at pH 7 (red), pH 3 with 2 m LiCl (light blue), and pH 3 without salt (dark blue). In the absence of salt, the HMQC of protonated BBL showed the typical fingerprint of an unfolded protein with most amide protons clustered in the 8–8.5 ppm range (dark blue). In turn, upon addition of salt, the spectrum changed quite drastically (light blue), showing much larger dispersion (7.5–9.5 ppm) and a clearly identifiable pattern of highly dispersed proton signals (i.e. the labeled cross-peaks). The HMQC fingerprint of protonated BBL in 2 m LiCl was, in fact, quite similar to that observed for native BBL at pH 7 (red). The small differences of 0.1–0.25 ppm between the two are most likely due to the strong intrinsic effects of pH on NH and 15N chemical shifts. The deviations of the Hα chemical shifts from typical random coil values (conformational shifts), which report on the backbone structure and are less sensitive to pH, were almost identical for pH 7 and 3 plus 2 m salt (Fig. 2B). Furthermore, the 1.0–0.0 ppm spectral region that is characteristic of aliphatic protons involved in tertiary contacts had many peaks at pH 3 plus 2 m LiCl, whereas there were no peaks in this region at pH 3 alone (Fig. 2C). Finally, the most definitive evidence of formation of the BBL native tertiary structure was provided by the NOESY spectrum, which showed the same long range NOEs that are expected from the native structure at neutral pH. The top and bottom right panels of Fig. 2 highlight examples of these native tertiary contacts between residues conforming the hydrophobic core of BBL: Leu10, Leu15, Ala17, Ile20, Leu29, Val34, and Leu38 and the two buried histidines (His13 and His37). The results from the NMR analysis demonstrated unambiguously that protonated BBL stabilized by salt and BBL at neutral pH fold into essentially the same three-dimensional structure.
FIGURE 2.

Structural analysis of the salt-refolded state of BBL by NMR. A, H1-N15 SOFAST-HMQC spectra measured at 283 K with 15N natural abundance for native BBL at pH 7 (red), native BBL at pH 3 and 2 m LiCl (light blue), and acid-denatured BBL at pH 3 (dark blue). The labels indicate some cross-peaks that are characteristic of the BBL native state. B, Hα conformational shifts calculated as the difference between the chemical shift of the protein and the typical random coil values for the same residues. Light blue, data at pH 3 plus 2 m LiCl; red, data at pH 7. C, the aliphatic region in the one-dimensional proton NMR spectrum of BBL at pH 3 with 2 m LiCl (light blue) and without (dark blue). The upper and lower panels on the right show fragments of the NOESY spectrum of BBL at 283 K, pH 3, and 2 m LiCl. NOEs between side chain protons of hydrophobic residues indicating critical tertiary contacts in the native structure of BBL are marked in boxes. The residues of BBL involved in these contacts are as follows: Leu10, His13, Leu15, Ala17, Ile20, Leu29, Val34, His37, and Leu38.
Salt Plus Protonation Results in Less Cooperative Unfolding for BBL
At 2 m LiCl, protonated BBL unfolded with an apparent Tm of ∼320 K, which was almost identical to that exhibited by BBL at pH 7 with no salt. The agreement in Tm was manifested by a coincidence in the position of the maximum of the derivative of the equilibrium unfolding curves (Fig. 3A) and confirmed by fitting to Equation 1 (Fig. 1B).
FIGURE 3.

Comparing the thermal unfolding of salt-stabilized BBL at pH 3 and native BBL at pH 7. A, first derivative of the CD thermal unfolding curves of BBL at pH 7 (red circles) compared with those at pH 3 and 2 m of LiCl (blue circles), NaCl (blue triangles), and CsCl (blue inverted triangles). Black curves connect the data points to guide the eye. B, CD signal at 222 nm of the BBL high temperature (368 K) denatured state at pH 3 and 2 m LiCl, NaCl, and CsCl. Symbols are as described in A. The red line corresponds to the CD signal at pH 7 and the same temperature. The black line is a fit to an exponential function to guide the eye. Inset, far-UV CD spectra of BBL at 363 K and pH 7 (red), pH 3 plus 1 m LiCl (blue), or pH 3 plus 5 m LiCl (green).
However, despite the similarity in apparent Tm, the thermal unfolding process of protonated BBL at 2 m LiCl was much broader than that at pH 7 with no salt. This was again readily apparent in the derivative of the unfolding curve, which showed that for the former, the maximum reached lower values, whereas the tails were higher (Fig. 3A). Quantitative determination of the sharpness of the unfolding curve (i.e. from parameter A obtained by fitting the curves to Equation 1) indicated that the thermal unfolding of protonated BBL at 2 m LiCl was almost 2-fold broader (relative sharpness of ∼0.6; Table 1). Therefore, in conditions of salt that match the apparent Tm to that exhibited by BBL at neutral pH, protonated BBL unfolded by a significantly broader process. It also is important to note that, to a first order approximation, Tm and curve broadness were both independent of the type of salt employed (see Fig. 3). This indicated that the effects on the unfolding of BBL induced by addition of salt were generic and thus mostly independent of salt type.
TABLE 1.
Relative sharpness of BBL unfolding
| Salt |
|||
|---|---|---|---|
| 0 m | 2.0 m | 4.0 m | |
| pH 7 | 1.0 | 0.67 | 0.71 |
| pH 3 + LiCl | n.f. | 0.62 | 0.75 |
| pH 3 + CsCl | n.f. | 0.52 | 0.82 |
| pH 3 + NaCl | n.f. | 0.51 | |
Values correspond to the parameter A (equation 1) relative to pH 7 and no salt. N.f. indicates that BBL is not folded at the lowest experimental temperature.
A possible explanation for an apparently broader unfolding curve is that the end states (folded and/or unfolded) have different properties at pH 7 and 3 plus 2 m salt. However, this does not seem to be the case. NMR showed that BBL folds to the same native structure under both conditions (Fig. 2). The high temperature unfolded state also exhibited a similar CD spectrum and thus a similar degree of residual α-helical structure, whether BBL was at neutral pH or protonated plus LiCl, NaCl, or CsCl (Fig. 3B). Therefore, salt-stabilized protonated BBL unfolds by temperature through a process that is intrinsically broader, or less cooperative.
BBL Unfolding Cooperativity Is Proportional to the Degree of Electrostatic Screening and Independent of Protonation Status
Having ruled out a structural change, the decreased cooperativity observed in Fig. 3A could be caused by a combination of three factors: 1) protonation of the buried side-chains of His13 and His37; 2) screening of electrostatic interactions; and 3) solvent-driven forces caused by organization of water molecules around the highly concentrated ions (kosmotropic effect). To elucidate the roles of these three factors, we compared the effect of very high salt concentrations at acidic and neutral pH. We found that in the presence of 4 m LiCl, the thermal unfolding of BBL is virtually identical at both pH values (Fig. 4A). Moreover, these curves differed from that at pH 7 with no salt in that they shifted to the right and had increased α-helical content in the pretransition baseline (Fig. 4A). From the derivative of the unfolding curves, we found that the presence of 4 m LiCl increases the apparent Tm up to ∼332 K and also makes the unfolding process equally broader for neutral and acidic pH (Fig. 4B and Table 1).
FIGURE 4.

Salt-induced refolding of BBL is independent of its ionization status. A, CD thermal unfolding curves of BBL in the presence of 4 m LiCl and at pH 3 (blue) and pH 7 (orange). Red circles correspond to the thermal unfolding curve at pH 7 without salt. B, first derivates of the thermal unfolding curves shown in A (same color scheme). Black curves connect the data points to guide the eye in both panels.
These results have a straightforward explanation if we assume that the main factor determining the stabilization and decreased cooperativity of BBL by salt is the screening of electrostatic interactions. The agreement between the thermal unfolding curves at the two pH values (Fig. 4A) directly rules out the protonation of buried ionizable groups. This is in stark contrast to the critical role that protonation of buried histidines plays in the formation of a molten-globule intermediate in apomyoglobin (40).
A kosmotropic effect should be proportional to salt concentration (41), whereas electrostatic screening depends exponentially on the ionic strength according to the Debye-Hückel theory. The thermal stabilization that we observed at both pH values produced an abrupt increase in apparent Tm at low ionic strengths, which leveled off above 2 m (Fig. 1B). The trend was exactly as expected from Debye-Huckel. At an ionic strength of 4 m electrostatic screening should be complete, thus explaining why under these conditions BBL exhibits the same properties regardless of its protonation status (Fig. 4). Moreover, the increase in Tm induced by salt at pH 7 indicated that the electrostatic contributions at neutral pH are also mildly destabilizing.
The same arguments apply to the changes in the unfolding cooperativity of BBL. It is clear that the protonation of the two buried histidines had no direct effect on the broadness of the unfolding curve (compare orange and blue in Fig. 4), contrary to previous arguments (33). Furthermore, the unfolding of protonated BBL was similarly broad at 2 m and 4 m LiCl. This was consistent with the expectation of ∼90% electrostatic screening at 2 m ionic strength. In turn, kosmotropic effects should produce significantly broader unfolding at 4 m because the salt concentration is doubled. If anything, the thermal unfolding of protonated BBL at 2 m was slightly broader (see Table 1), which could be due to heat capacity effects on the overall unfolding enthalpy given the ∼12 K lower Tm compared with the experiments at 4 m salt.
Further evidence for the role of electrostatic screening arose from chemical denaturation experiments using the ionic chaotrope guanidinium chloride as denaturant. As with temperature, the guanidinium chloride denaturation of BBL at pH 7 and 3 plus 2 m salt produced sigmoidal unfolding curves (supplemental Fig. S3) that had identical denaturation midpoints (i.e. Cm = 2.1 m as determined by fitting to Equation 2). However, the denaturation curve of protonated BBL in the presence of 2 m LiCl was only ∼10% broader because guanidinium chloride should screen electrostatic interactions as it denatures the protein. Thus, near the denaturation midpoint, electrostatic screening was already at ∼90% for pH 7 (∼2.1 ionic strength (I)) and complete for pH 3 plus 2 m salt (I ∼ 4.1 m). In other words, the guanidinium chloride denaturation experiment is equivalent to the thermal denaturation experiment in the presence of 4 m salt (Fig. 4) because in both cases, electrostatic interactions were almost fully screened. On the other hand, if kosmotropic effects were responsible, the unfolding curve at pH 3 plus 2 m LiCl should be much broader than at pH 7 without salt.
Effects of Protonation and Salt on the Ultra-fast Folding Kinetics of BBL
All previous results focus on stability and cooperativity from a thermodynamic viewpoint. The analysis of the effects of salt and protonation on the folding relaxation kinetics of BBL offers two additional advantages: 1) the comparison between the folding relaxation rates of BBL at pH 7 and 3 under conditions where thermal stability and solution viscosity are matched (e.g. 4 m LiCl) provides an independent test of whether BBL folds through the same mechanism when is protonated and at neutral pH; and 2) with these experiments, we can investigate the kinetic manifestations of the less cooperative equilibrium unfolding induced by salt screening of electrostatic interactions.
To measure the folding relaxation kinetics of BBL, we performed nanosecond resolution laser-induced IR temperature-jump experiments measuring the degree of α-helical structure from the amide I band absorbance at 1632 cm−1 (42). The kinetic relaxation after ∼10 K jumps resulted in decays that could be well fitted to single exponential curves at all conditions. In the presence of 4 m LiCl, the relaxation time at 325 K (near the Tm) was ∼13 μs for both pH 7 and 3 (Fig. 5A; orange and blue, respectively). However, at pH 7, in the absence of salt, the relaxation time was only ∼3 μs (red in Fig. 5A). This pattern was maintained at all other temperatures, with the two sets of experiments at 4 m LiCl producing nearly identical rates and the experiments at pH 7 without salt rendering rates ∼4 times faster (Fig. 5B).
FIGURE 5.

Ultrafast relaxation kinetics of BBL measured by the nanosecond IR temperature-jump method. A, normalized time-dependent changes in the amide I band absorbance at 1632 cm−1 (ΔABS) after temperature-jumps of ∼10 K to a final temperature of 325 K. Red, decay at pH 7 without salt; orange, decay at pH 7 plus 4 m LiCl; blue, decay at pH 3 plus 4 m LiCl. The black lines show the fits to a single exponential function. B, relaxation rates as a function of temperature for the three experimental conditions of A (color scheme as described in A). The data points shown at pH 7 (red) are the average of three measurements, whereas both data sets at 4 m LiCl correspond to single measurements.
The agreement between the relaxation rates of the two sets of experiments at 4 m LiCl provided kinetic confirmation that, once electrostatic interactions are screened out, BBL folds to the native three-dimensional structure via the same mechanism whether protonated or not. It follows that acidic pH unfolds BBL by generic destabilization due to solvent accessible electrostatic repulsions and not because it introduces charges in solvent protected residues within the protein core. This is again different from the apomyoglobin case, in which protonation of a buried histidine greatly speeds up unfolding (43). The practical implication is that the acid unfolding of BBL can be completely reverted by simply increasing the ionic strength, in contrast to previous examples of acid unfolding (11).
The difference between the relaxation rates at 4 m LiCl and the rates measured at pH 7 without salt offers additional clues about the source of the marginal unfolding cooperativity of BBL and its connection with electrostatic interactions. We discovered that upon addition of 4 m salt, BBL exhibited a broader equilibrium unfolding (Fig. 4B) together with a 4-fold slower relaxation rate (Fig. 5). The increase in solvent viscosity by addition of 4 m LiCl is 1.8 (44, 45), and thus, it only accounts for a fraction of the slowdown; even if we assumed a maximal exponent of 1 for the viscosity dependence of the folding rate (46). From the viewpoint of a classical activated folding scenario, these results are seemingly incompatible with one another. Under such a scenario, the free energy barrier is proportional to the enthalpy of unfolding, so that the same conditions that make the equilibrium unfolding curve broader should speed up the rate (47).
In contrast, our results are easily explained in the context of global downhill folding. For this scenario, the folding free energy surface has only one minimum that shifts along the reaction coordinate as denaturational stress increases (e.g. higher temperature) (34). At the denaturation midpoint, global downhill folding kinetics is described as diffusion on a single-welled surface with the minimum located halfway between the fully folded and unfolded states. The folding relaxation time on such a surface is approximately proportional to the inverse of the well curvature squared (28). The longer relaxation times observed in the presence of salt would then signify a broader well, consistent with the decreased equilibrium unfolding cooperativity. If we assume that the broadness of the thermal unfolding curve is linearly related to the curvature of the well, the relaxation time expected for BBL at 4 m salt would be ∼1.8 times longer (i.e. 1/(0.752)). This factor, added to the viscosity effect discussed above, places the rates expected for 4 m salt in good agreement with the experimental observation. Thus, the effect of salt on the ultrafast folding kinetics of BBL supported the global downhill scenario for this protein and confirmed that the marginal folding cooperativity characteristic of this scenario is amenable to further reduction by electrostatic screening.
DISCUSSION
The acid-denatured form of the ultrafast folding protein BBL can be fully refolded to its native structure by addition of salt. To a first order approximation, this effect is generic in that it is observed for different salts, such as LiCl, NaCl, and CsCl with minimal differences that seem related to salt solubility (Fig. 3). In general, salt addition has two effects. First, salt stabilizes BBL native structure by Debye-Hückel screening of the mostly repulsive electrostatic interactions present at pH 3. Second, high salt concentrations produce significantly broader unfolding transitions and thus decrease the folding cooperativity of BBL.
Control experiments reveal that the addition of salt also stabilizes the native state at neutral pH, following again the Debye-Hückel dependence on ionic strength. This result indicates that the net electrostatic interactions at neutral pH are also repulsive, although to a lesser degree than at pH 3. Remarkably, upon complete electrostatic screening by salt, BBL exhibits the same stability (as judged by the Tm) and equally broad unfolding at both neutral and acidic pH. Moreover, laser temperature-jump experiments show that the broader equilibrium unfolding process induced by salt goes together with slower folding-unfolding kinetics. These results have profound implications for our understanding of the downhill ultrafast folding regime; probe the physical limits of folding cooperativity; and shed light onto the role of electrostatics in determining such cooperativity.
A first critical result is that BBL folds up into its native structure even when it is protonated, provided that the stabilization deficit resulting from electrostatic repulsions is compensated by salt. The fact that salt screening completely reverts the acid denaturation of BBL indicates that all ionizable groups titrating between pH 7 and 3 are somewhat accessible to the solvent in the native ensemble, including the partly buried His13 and His37, which are involved in many tertiary contacts in the native structure (see bottom right panels in Fig. 2).
Furthermore, we find that the salt-induced stabilization of BBL results in proportional increases in the α-helical content of its native state (the pretransition baseline (34)) at both acidic and neutral pH (Figs. 1C and 4A). This proportionality is identical to that observed with temperature (Figs. 1A and 4A) or chemical denaturant (37). Inspection of recent mutational data on BBL (48) hints at a similar relationship between the α-helix fraying of the native baseline and the decrease in stability induced by mutation. Here, it is important to note that the variability in the pretransition CD signal indicates true structural changes and thus is not a phenomenological baseline (49). This intriguing observation is easily explained in the context of global downhill folding. Along these lines, the changes in helical content of the BBL native ensemble reflect the shift of the single well toward more disorder as environmental conditions become less stabilizing (50). Recently, it has been proposed that histidine protonation in BBL induces a partially structured intermediate that folds via a different mechanism from that of the native protein (33, 51). Our results demonstrate that acid pH unfolds BBL by generic destabilization of the native state, not by inserting charges in solvent-inaccessible positions within the protein core. Therefore, the structural changes upon histidine protonation observed in molecular simulations of BBL (51) merely reflect the conformational changes of the native ensemble in response to mild destabilization that are characteristic of global downhill folding.
A related observation is that the decrease in BBL unfolding cooperativity induced by salt results in a slightly slower (∼4-fold) microsecond folding relaxation (Fig. 5). It is impossible to explain this result with a classical view of protein folding as an activated (barrier crossing) process, because in this scenario the lower cooperativity implies faster folding kinetics. However, what seems paradoxical for activated folding does in fact agree exactly with the behavior expected for global downhill folding. A downhill relaxation rate is, to a first order approximation, proportional to the curvature of the diffusing surface squared. The slower relaxation at 4 m salt would thus indicate diffusion on a broader surface, consistent with the less cooperative unfolding observed in equilibrium. This argument is identical to the theoretical explanation of why downhill folding relaxation rates reach a shallow minimum near the chemical denaturation midpoint (52).
Finally, the thermal unfolding of BBL in the presence of 4 m salt extends over a broad temperature range (Fig. 4B), indicating an extremely low cooperativity. Here, it is important to emphasize that under these conditions, the unfolding process does start from the native ensemble of BBL and not from a molten globule or partially folded intermediate. Therefore, the effect of salt addition in the folding of BBL is drastically different from the classical stabilization of molten globule states by combination of salt and acidic pH (8–12). The fact that in the presence of salt BBL folds/unfolds with decreased cooperativity is remarkable, given that the unfolding of BBL in physiological conditions is already extremely broad. These results are an experimental demonstration that proteins can fold to their native structure with nominal cooperativity, exactly as predicted by theory (53) and observed in computer simulations (17, 18, 54).
From salt titrations, both at acidic and neutral pH, we can conclude that the source of the decreased cooperativity is the cancellation of electrostatic interactions by Debye-Hückel ionic screening. The net electrostatic interactions are repulsive (even at pH 7), suggesting that they enhance the folding cooperativity of BBL by preferentially destabilizing partially folded conformations. Similar electrostatic effects on folding cooperativity have been described recently for lysozyme and α-lactoalbumin (32). Thus, electrostatic interactions, which act over longer distances than all other noncovalent interactions, emerge as a general mechanism to tune folding cooperativity. This mechanism, which appears to be used by evolution, puts at our fingertips a simple tool for engineering folding barriers and cooperativity in single-domain proteins.
Acknowledgment
We thank Dr. Eva de Alba for assistance with the NMR measurements.
This work was supported by Marie Curie Excellence Grant MEXT-CT-2006-042334 and Grants BFU2008-03237 and CONSOLIDER CSD2009-00088 from the Spanish Ministry of Science and Innovation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures,” Table 1, and Figs. S1–S3.
- NOESY
- nuclear Overhauser spectroscopy
- TOCSY
- total correlation spectroscopy
- SOFAST-HMQC
- heteronuclear zero and double quantum coherence.
REFERENCES
- 1.Dill K. A. (1990) Biochemistry 29, 7133–7155 [DOI] [PubMed] [Google Scholar]
- 2.Fersht A. R. (1998) in Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Freeman W. H. ed) 1st Ed., W. H. Freeman, New York [Google Scholar]
- 3.Poland D. C., Scheraga H. A. (1965) Biopolymers 3, 401–419 [Google Scholar]
- 4.Kouza M., Li M. S., O'brien E. P., Jr., Hu C. K., Thirumalai D. (2006) J. Phys. Chem. A. 110, 671–676 [DOI] [PubMed] [Google Scholar]
- 5.Poland D. C., Scheraga H. A. (1965) J. Chem. Phys. 43, 2071–2074 [DOI] [PubMed] [Google Scholar]
- 6.Aune K. C., Tanford C. (1969) Biochemistry 8, 4586–4590 [DOI] [PubMed] [Google Scholar]
- 7.Jackson S. E. (1998) Fold. Des. 3, R81–91 [DOI] [PubMed] [Google Scholar]
- 8.Goto Y., Fink A. L. (1989) Biochemistry 28, 945–952 [DOI] [PubMed] [Google Scholar]
- 9.Goto Y., Nishikiori S. (1991) J. Mol. Biol. 222, 679–686 [DOI] [PubMed] [Google Scholar]
- 10.Fink A. L., Calciano L. J., Goto Y., Nishimura M., Swedberg S. A. (1993) Protein Science. 2, 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fink A. L., Calciano L. J., Goto Y., Kurotsu T., Palleros D. R. (1994) Biochemistry 33, 12504–12511 [DOI] [PubMed] [Google Scholar]
- 12.Hamada D., Kidokoro S., Fukada H., Takahashi K., Goto Y. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 10325–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ptitsyn O. B. (1995) Adv. Protein Chem. 47, 83–229 [DOI] [PubMed] [Google Scholar]
- 14.Bryngelson J. D., Onuchic J. N., Socci N. D., Wolynes P. G. (1995) Prot. Struct. Func. and Gen. 21, 167–195 [DOI] [PubMed] [Google Scholar]
- 15.Camacho C. J., Thirumalai D. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 6369–6372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan H. S., Bromberg S., Dill K. A. (1995) Philos. Trans. R. Soc. Lond. B Biol. Sci. 348, 61–70 [DOI] [PubMed] [Google Scholar]
- 17.Abkevich V. I., Gutin A. M., Shakhnovich E. I. (1995) J. Mol. Biol. 252, 460–471 [DOI] [PubMed] [Google Scholar]
- 18.Klimov D. K., Thirumalai D. (1998) Fold. Des. 3, 127–139 [DOI] [PubMed] [Google Scholar]
- 19.Kaya H., Chan H. S. (2003) J. Mol. Biol. 326, 911–931 [DOI] [PubMed] [Google Scholar]
- 20.Ejtehadi M. R., Avall S. P., Plotkin S. S. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 15088–15093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki Y., Onuchic J. N. (2005) J. Phys. Chem. B. 109, 16503–16510 [DOI] [PubMed] [Google Scholar]
- 22.Li M. S., Klimov D. K., Thirumalai D. (2004) Phys. Rev. Lett. 93, 268107. [DOI] [PubMed] [Google Scholar]
- 23.Klimov D. K., Thirumalai D. (2002) J. Comput. Chem. 23, 161–165 [DOI] [PubMed] [Google Scholar]
- 24.Muñoz V. (2007) Annu. Rev. Biophys. Biomol. Struct. 36, 395–412 [DOI] [PubMed] [Google Scholar]
- 25.Muñoz V., Sadqi M., Naganathan A. N., de Sancho D. (2008) HFSP J. 2, 342–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sadqi M., Fushman D., Muñoz V. (2006) Nature 442, 317–321 [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Mira M. M., Sadqi M., Fischer N., Sanchez-Ruiz J. M., Muñoz V. (2002) Science 298, 2191–2195 [DOI] [PubMed] [Google Scholar]
- 28.Li P., Oliva F. Y., Naganathan A. N., Muñoz V. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perham R. N. (2000) Annu. Rev. Biochem. 69, 961–1004 [DOI] [PubMed] [Google Scholar]
- 30.Naganathan A. N., Doshi U., Fung A., Sadqi M., Muñoz V. (2006) Biochemistry 45, 8466–8475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez-Ruiz J. M., Makhatadze G. I. (2001) Trends Biotechnol. 19, 132–135 [DOI] [PubMed] [Google Scholar]
- 32.Halskau O., Jr., Perez-Jimenez R., Ibarra-Molero B., Underhaug J., Muñoz V., Martinez A., Sanchez-Ruiz J. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8625–8630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arbely E., Rutherford T. J., Sharpe T. D., Ferguson N., Fersht A. R. (2009) J. Mol. Biol. 387, 986–992 [DOI] [PubMed] [Google Scholar]
- 34.Naganathan A. N., Perez-Jimenez R., Sanchez-Ruiz J. M., Muñoz V. (2005) Biochemistry 44, 7435–7449 [DOI] [PubMed] [Google Scholar]
- 35.Huang C. Y., Getahun Z., Zhu Y. J., Klemke J. W., DeGrado W. F., Gai F. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 2788–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naganathan A. N., Muñoz V. (2008) Biochemistry 47, 6752–6761 [DOI] [PubMed] [Google Scholar]
- 37.Oliva F. Y., Muñoz V. (2004) J. Am. Chem Soc. 126, 8596–8597 [DOI] [PubMed] [Google Scholar]
- 38.Fink A. L., Oberg K. A., Seshadri S. (1998) Fold. Des. 3, 19–25 [DOI] [PubMed] [Google Scholar]
- 39.Alexandrescu A. T., Evans P. A., Pitkeathly M., Baum J., Dobson C. M. (1993) Biochemistry 32, 1707–1718 [DOI] [PubMed] [Google Scholar]
- 40.Barrick D., Hughson F. M., Baldwin R. L. (1994) J. Mol. Biol. 237, 588–601 [DOI] [PubMed] [Google Scholar]
- 41.Collins K. D., Washabaugh M. W. (1985) Q Rev. Biophys. 18, 323–422 [DOI] [PubMed] [Google Scholar]
- 42.Callender R., Dyer R. B. (2002) Curr. Opin. Struct. Biol. 12, 628–633 [DOI] [PubMed] [Google Scholar]
- 43.Jamin M., Geierstanger B., Baldwin R. L. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6127–6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wimby J. M., Berntsson T. S. (1994) J. Chem. Eng. Data 39, 68–72 [Google Scholar]
- 45.Mao S. D., Duan Z. H. (2009) Int. J. Thermophys. 30, 1510–1523 [Google Scholar]
- 46.Ansari A., Jones C. M., Henry E. R., Hofrichter J., Eaton W. A. (1992) Science 256, 1796–1798 [DOI] [PubMed] [Google Scholar]
- 47.Naganathan A. N., Muñoz V. (2005) J. Am. Chem. Soc. 127, 480–481 [DOI] [PubMed] [Google Scholar]
- 48.Neuweiler H., Sharpe T. D., Rutherford T. J., Johnson C. M., Allen M. D., Ferguson N., Fersht A. R. (2009) J. Mol. Biol. 390, 1060–1073 [DOI] [PubMed] [Google Scholar]
- 49.Sadqi M., Fushman D., Muñoz V. (2007) Nature 445, E17–E18 [DOI] [PubMed] [Google Scholar]
- 50.Muñoz V. (2002) Int. J. Quantum Chem. 90, 1522–1528 [Google Scholar]
- 51.Settanni G., Fersht A. R. (2009) J. Mol. Biol. 387, 993–1001 [DOI] [PubMed] [Google Scholar]
- 52.Naganathan A. N., Doshi U., Muñoz V. (2007) J. Am. Chem. Soc. 129, 5673–5682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onuchic J. N., Luthey-Schulten Z., Wolynes P. G. (1997) Annu. Rev. Phys. Chem. 48, 545–600 [DOI] [PubMed] [Google Scholar]
- 54.Knott M., Chan H. S. (2004) Chem. Phys. 307, 187–199 [Google Scholar]