

Abstract
Chronological life span is defined by how long a cell can survive in a non-dividing state. In yeast, it is measured by viability after entry into stationary phase. To date, some factors affecting chronological life span have been identified; however, the molecular details of how these factors regulate chronological life span have not yet been elucidated clearly. Because life span is a complicated phenomenon and is supposedly regulated by many factors, it is necessary to identify new factors affecting chronological life span to understand life span regulation. To this end, we have screened for long-lived mutants and identified Pma1, an essential P-type proton ATPase, as one of the determinants of chronological life span. We show that partial loss of Pma1 activity not only by mutations but also by treatment with the Pma1 inhibitory chemical vanadate resulted in the long-lived phenotype in Schizosaccharomyces pombe. These findings suggest a novel way to manipulate chronological life span by modulating Pma1 as a molecular target.
Keywords: Aging, ATPases, Enzyme Mutation, Yeast, Yeast Genetics, Yeast Metabolism, Life Span
Introduction
In the natural environment, most microorganisms exhibit only brief periods of rapid growth. Nutrient starvation is the most common natural situation, so the ability to adapt to nutrient limitation is crucial for microorganisms. Cells respond to starvation by ceasing growth and entering stationary phase and differentiate in ways that allow them to maintain viability for extended periods in the absence of nutrients (1). In yeast, the period in which the cells keep their viability after entry into stationary phase is recognized as the chronological life span (2).
In Saccharomyces cerevisiae, two of the major pathways that control chronological life span have been identified: the Ras-PKA-Msn2/4 pathway and the Sch9 pathway (3, 4). The down-regulation of either pathway promotes life span extension. Importantly, similar pathways (insulin/insulin-like growth factor I-like) regulate longevity in higher eukaryotes, suggesting a common evolutionary origin for the life span regulatory mechanisms (5, 6).
In Schizosaccharomyces pombe, Pka1 and Sck2 are regulators of chronological life span; each mutant shows a long-lived phenotype, and the pka1 sck2 double mutant displays an additive effect on chronological life span extension, suggesting that these two factors regulate related but independent pathways (7). We reported previously that a mutant of lcf1+, which encodes a long-chain fatty acyl-CoA synthetase, showed rapid loss of viability after entry into stationary phase and suggested that fatty acid utilization and/or metabolism is important to determine viability in stationary phase in fission yeast (8, 9). Recently, we identified a novel gene, ecl1+, which extends the chronological life span of S. pombe when overexpressed (10). In addition, we also identified two paralogs and one ortholog of ecl1+ in S. pombe and S. cerevisiae, respectively (11, 12). On the basis of these observations, we proposed that Ecl1 family proteins are novel regulators for chronological life span in yeast. Ecl1 family proteins seem to be specific in fungus groups, and the molecular mechanism responsible for the regulation of life span has not yet been clarified.
Because life span is supposed to be regulated by many factors and complicatedly and important factors for life span regulation have been conserved in eukaryotes, it is necessary to identify new conserved factors affecting chronological life span to understand life span regulation (13). To accomplish this, we have screened and identified a mutant that extends the chronological life span of S. pombe.
EXPERIMENTAL PROCEDURES
Strains and Media
S. pombe strain JY333 (h− leu1-32 ade6-M216) was used for mutant screening. Strains were grown in synthetic defined (SD)2 medium (0.67% yeast nitrogen base without amino acids (Difco) and 2% glucose) supplemented with the necessary growth requirements in standard amounts at 30 °C. The analysis of viability was carried out as described previously (10).
Mutation Mapping
To identify the mutation in the L18 mutant, a whole genome tiling array of S. pombe was constructed and used to map the L18 mutation. This analysis (Nandemo-Arei analysis) was carried out by GeneFrontier Corp.
Insertion of a Kanamycin (Km) Resistance Marker Downstream of the pma1+ Gene
To carry out linkage analysis, a Km resistance marker was inserted at 1824–1835 bp downstream of the termination codon of the pma1+ gene as described previously (14). Both the upstream and downstream regions of the desired insertion region were PCR-amplified using primers F1 (AGAAGTTATCGTGAGCTACG) and F2 (TTAATTAACCCGGGGATCCGGAAATCATTGATTTATCTATATAC) and primers R1 (GTCTTGGTCTGGTATCAACG) and R2 (GTTTAAACGAGCTCGAATTCCATGGATAAGCTGCTAATCCATAAT), respectively, and both fragments were purified. After mixing both DNA fragments with pFA6a-kanMX6, PCR was performed with primers F1 and R1. JY336 (h+ leu1-32 ade6-M210) was transformed with the amplified DNA fragment, and stable G418-resistant transformants were selected. The construct on the chromosome was then confirmed by PCR using the appropriate primers.
Preparation of an Anti-Pma1 Antibody
An antibody for Pma1 was prepared by immunizing a rabbit with peptide MMNGKPKESRNQRSIEDL, which corresponds to amino acids 886–903 of the Pma1 protein and was purchased from Sigma.
Pma1 ATPase Assay
An ATPase assay was carried out essentially as described (15). The assay was carried out with or without 0.1 mm sodium vanadate, and the released inorganic phosphate was determined using the Phospho C-Test Wako kit (Wako Co.). The vanadate-sensitive ATPase activity was determined and expressed as Pma1 activity.
Assay of Glucose Concentration
Cells were grown in SD medium; 20 μl of each culture was sampled; and the concentration of glucose in the medium was assayed using a glucose CII-Test Wako kit (Wako Co.).
RESULTS AND DISCUSSION
Isolation of a Mutant That Stays Viable after Entry into Stationary Phase
To isolate long-lived mutants, wild-type cells (strain JY333) were grown in SD liquid medium for 7 days, and an aliquot was transferred into the same fresh medium. It is known that the viability of wild-type cells decrease to 1/10,000 within 5 days in SD medium, so we repeated the 7-day incubation five times (8, 10). From this old culture, we isolated one spontaneous mutant, named L18, which showed an increase in viability after entry into stationary phase. As shown in Fig. 1A, wild-type cells decreased viability to 1/1000 over 3 days of incubation. On the other hand, the L18 mutant kept its viability for a long period compared with wild-type cells. It should be noted that the calculated doubling time at logarithmic growth phase for L18 and JY333 was identical, i.e. 209 and 205 min, respectively, under this growth condition. Next, to confirm that the L18 mutant increases chronological survival under other growth conditions, we analyzed the viability of the cells in water. Cells grown in SD medium to logarithmic growth phase (A600 = 1) were collected and then suspended in water. As shown in Fig. 1B, the viability of the L18 mutant was higher than that of wild-type cells. On the basis of these results, we concluded that L18 is a chronologically long-lived mutant of S. pombe.
FIGURE 1.

Phenotypes of the mutant that increases cell viability after entry into stationary phase. A and B, cell growth (upper panels) and cell viability (lower panels) of wild-type (○) and L18 mutant (●) cells in SD medium (A) and in H2O (B) were monitored. The data shown represent the mean ± S.D. of three independent experiments. C, cell morphology at various growth phases. Wild-type (○) and L18 mutant (●) cells were sampled at each time point indicated (arrows a–c) in the growth curve and subjected to microscopic observation.
Phenotypic Characterization of the L18 Mutant
To understand why the L18 mutant showed a long-lived phenotype, cell morphology was monitored with cell growth in SD medium. As shown in Fig. 1C, there was no difference in cell morphology between wild-type and L18 mutant cells at logarithmic growth phase. However, after entry into stationary phase, wild-type cells showed an abnormal morphology, such as broken or shrunken shapes. On the other hand, the L18 mutant showed a normal morphology, although the cells became a little bit longer. This phenotypic difference in morphology might be reflected in the different viability after entry into stationary phase.
In S. cerevisiae, it has been reported that chronological long-lived mutants exhibit oxidative stress resistance phenotypes (3). Previously, we also presented data that cells overexpressing Ecl1 family proteins, such as Ecl1, Ecl2, and Ecl3, are resistant to oxidative stress (10–12). Hence, we analyzed oxidative stress sensitivity by spotting cells on a plate containing H2O2. As shown in Fig. 2A, L18 mutant cells were more resistant to oxidative stress (5 mm H2O2) compared with wild-type cells. Because the catalase encoded by the ctt1+ gene is known to protect cells from the toxic effects of H2O2, we monitored the expression of ctt1+ mRNA. As shown in Fig. 2B, ctt1+ mRNA was highly expressed in L18 mutant cells compared with wild-type cells, which is in good agreement with the phenotype. It is known that ctt1+ is controlled by transcription factors Prr1 and Pap1 (16). Prr1 is a response regulator that is involved in the His-Asp phosphorelay system. Pap1 is controlled by the stress-activated MAPK Sty1, which responds to a range of environmental stresses. Thus, the L18 mutant may cause stress signals that activate the Sty1 MAPK and/or the His-Asp phosphorelay system to induce ctt1+ expression through Pap1 and/or Prr1, respectively. We analyzed other phenotypes, such as sensitivity to high osmolarity (2 m sorbitol) and heat stress (37 °C); however, no difference was found between wild-type and L18 mutant cells (data not shown).
FIGURE 2.

The L18 mutant is resistant to oxidative stress. A, wild-type and L18 mutant cells were spotted on an SD plate with or without 5 mm H2O2 with serial dilution. The plates were incubated for 4 days at 30 °C and photographed. B, total RNAs were isolated from wild-type and L18 mutant cells grown in SD medium until mid-log phase and subjected to Northern blot analysis with a radiolabeled ctt1+ probe.
Identification of the Mutation Site in the L18 Mutant
To identify the mutation in L18, the whole genome mutation mapping method was adopted using tiling arrays of S. pombe. The results suggested that one mutation arose at nucleotides 3,873,842–3,873,898 on chromosome 1 of the L18 mutant. This region corresponds to the coding sequence of the pma1+ gene, which encodes the P-type proton ATPase. We then sequenced the pma1+ gene of L18 and identified the C-to-A missense mutation that causes the A270D substitution (Fig. 3, A and B). We named this mutation allele pma1-L18. We then determined whether the pma1-L18 mutation is responsible for the chronologically long-lived phenotype of L18 as follows. The Km resistance cassette was inserted in the chromosome at 1824–1835 bp downstream of the termination codon of the wild-type pma1+ gene, and then the Km-resistant strain was mated with the L18 mutant to analyze the linkage between the Km resistance phenotype and the chronologically long-lived phenotype. The phenotype of the progeny after crossing revealed that 100% of the chronologically long-lived cells (n = 20) were Km-sensitive, indicating that the pma1-L18 mutation and the locus in which the Km resistance cassette was inserted are closely located on the chromosome. Next, we cloned the wild-type pma1+ gene on a plasmid, introduced it into the L18 mutant, and confirmed that the long-lived phenotype of L18 was partially complemented (data not shown). Moreover, after crossing the L18 mutant with the wild-type strain, we randomly isolated both long-lived cells (n = 4) and non-long-lived cells (n = 4) and sequenced their chromosomal regions corresponding to the pma1-L18 mutation. We confirmed that all of long-lived cells had the pma1-L18 mutation and that all of non-long-lived cells had the wild-type pma1+ allele. On the basis of these results, we concluded that the pma1-L18 mutation is the causative mutation that confers the long-lived phenotype.
FIGURE 3.

L18 has a mutational lesion in the pma1+ gene that causes a decrease in Pma1 ATPase activity. A, the pma1 gene of the L18 mutant was sequenced, and an identified mutation (C-to-A substitution) is shown (arrowhead). B, schematic representation of the topology of the Pma1 protein in the plasma membrane. The mutation site (A270D) is indicated (star). aa, amino acids. C, Pma1 activities in wild-type (open bars) and L18 mutant (closed bars) cells were assayed using a cell lysate prepared from cells grown in SD medium to A600 = 1 or 2. The data shown represent the mean ± S.D. of three independent experiments. D, Pma1 activities in wild-type (open bars) and L18 mutant (closed bars) cells were assayed using a cell lysate prepared from cells grown in SD medium containing the indicated concentrations of glucose. E, expression profile of Pma1 with growth. Wild-type and L18 mutant cells were grown in SD medium at 30 °C. Total RNAs were isolated from cells at each absorbance (OD) indicated and subjected to Northern blot analysis with a radiolabeled pma1+ probe (upper panel). An ethidium bromide-stained gel showing total RNA is presented as a loading control (middle panel). Wild-type and L18 mutant cells were grown as described above, and a cell lysate was prepared. The amount of Pma1 protein was analyzed by Western blotting using Pma1 antiserum (lower panel).
Analysis of the ATPase Activity of the L18 Mutant
Pma1 is an essential P-type ATPase in the plasma membrane and functions as a hydrogen ion pump (17, 18). To characterize the nature of the Pma1-L18 protein, the H+-ATPase activity of the L18 mutant was analyzed. A whole cell extract was prepared, and H+-ATPase activity was assayed with or without vanadate. Because plasma membrane H+-ATPase is sensitive to vanadate, we estimated the vanadate-sensitive H+-ATPase activity as plasma membrane H+-ATPase activity. Cells were grown to mid-logarithmic phase (A600 = 1) and to stationary phase (A600 = 2), and H+-ATPase activities were analyzed. As shown in Fig. 3C, the H+-ATPase activity of the L18 mutant was about one-half of that of the wild-type cells at both growth phases. In S. cerevisiae, it is known that the activity of Pma1 H+-ATPase is regulated by glucose, i.e. when glucose is added to carbon-starved cells, ATPase activity increases (19, 20). We analyzed the S. pombe Pma1 activity in response to glucose concentration. Wild-type and L18 mutant cells were grown in SD medium until logarithmic growth phase and then transferred into SD medium containing various concentrations of glucose. Cells were grown in each medium for 90 min, and H+-ATPase activity was analyzed. As shown in Fig. 3D, the H+-ATPase activity of wild-type cells seemed to be regulated by glucose, i.e. the ATPase activity that was low in medium containing no glucose increased in response to glucose concentration. In the L18 mutant, the activity also increased in response to glucose. However, the activity was lower than that of the wild-type cells in medium containing 0.5–2% glucose. These results suggested that the Pma1-L18 protein has a defect in activity but not in regulation by glucose. We concluded that the L18 mutant has low H+-ATPase activity.
Expression Profile of pma1+ mRNA and the Pma1 Protein
To determine the expression profile of the pma1+ gene, wild-type and L18 mutant cells were grown in SD medium, and the pma1+ mRNA level was analyzed at several points during cell growth by Northern hybridization. As shown in Fig. 3E, the expression of pma1+ mRNA was constant during the periods analyzed, and there was no obvious difference between L18 mutant and wild-type cells. Next, we analyzed the amount of Pma1 protein by Western blotting under the same growth conditions. As shown in Fig. 3E, very similar amounts of the Pma1 protein were expressed in both wild-type and L18 mutant cells. On the basis of these analyses, we concluded that there is no difference in the amount and stability of the Pma1 protein expressed in L18 mutant and wild-type cells. This means that the specific activity for H+-ATPase of the Pma1-L18 protein is lower than that of the wild-type Pma1 protein.
The pma1-L18 Mutant Consumes Less Glucose
We next addressed why the pma1-L18 mutation extends chronological life span. Previously, it was reported that, in the S. pombe pma1 mutant, which has a defect in ATPase activity, the rate and extent of extracellular acid secretion are lower compared with wild-type cells (13). Thus, we hypothesized that the difference in acidification of the growth medium might explain the phenotype of L18, but this was not the case. We analyzed the pH change in the culture medium at several points during cell growth. As shown in Fig. 4A, pH decreased with cell growth, but there was no obvious difference between wild-type and L18 mutant cells.
FIGURE 4.
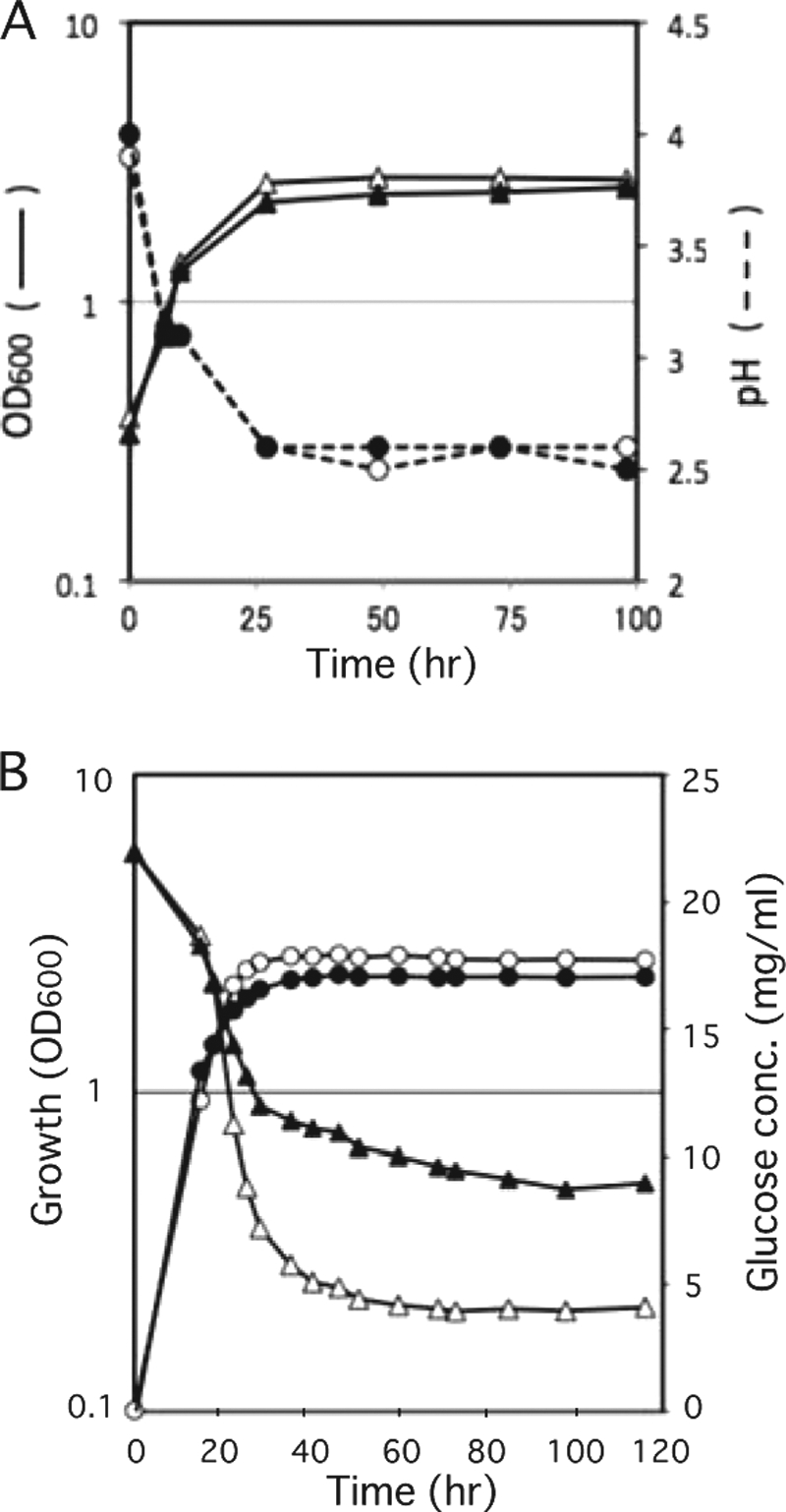
pH change and consumption of glucose in growth medium. A, cells were grown in SD medium at 30 °C, and changes in pH in the growth medium of wild-type (○) and L18 mutant (●) cells were analyzed. The growth of wild-type (△) and L18 mutant (▴) cells was monitored at A600. B, cells were grown in SD medium at 30 °C, and concentrations of glucose in the medium of wild-type (△) and L18 mutant (▴) cells were analyzed. The growth of wild-type (○) and L18 mutant (●) cells was monitored at A600.
Another possibility was the defect in nutrient (glucose) uptake in the L18 mutant. In S. cerevisiae, Pma1 H+-ATPase is responsible for H+-dependent nutrient uptake (20). Pma1 ATPase acts physiologically to pump protons out of the cell, creating the electrochemical gradient that drives solute uptake by an array of H+-coupled co-transporters. In S. pombe, glucose uptake was described to be energy-dependent, driven by the plasma membrane ATPase-generated electrochemical gradient (21). We compared the consumption of glucose with cell growth between wild-type and L18 mutant cells (Fig. 4B). The L18 mutant consumed less glucose compared with wild-type cells. The difference in glucose consumption is likely to be caused by the difference in the Pma1 activity that provides energy for glucose uptake. On the basis of these observations, we propose that the L18 mutant suffers somewhat during glucose-limiting conditions, so the physiology of the L18 mutant has been changed to adapt to these conditions.
In S. pombe, chronological aging is accelerated by glucose signaling; cells bearing mutations in genes controlling this pathway, such as pka1 and sck2, live longer (7). In S. cerevisiae, deletion of the genes SCH9, CYR1, and RAS2, which mediate glucose signaling, extends the chronological life span (4, 22). To investigate the relationship between the pma1-L18 mutation and glucose signaling in more detail, we analyzed cell viability in a glucose-limiting medium. The analysis of viability in a glucose-limiting medium is known as a calorie restriction experiment, and calorie restriction is a condition that extends life span in a variety of species (23, 24). As shown in Fig. 5 (A and B), calorie restriction extended the viability of both wild-type and L18 mutant cells. Under the calorie restriction condition, there was little difference in viability between wild-type and L18 mutant cells.
FIGURE 5.

Effect of calorie restriction and vanadate, a Pma1 inhibitor, on cell viability after entry into stationary phase. A and B, effect of calorie restriction. Wild-type and L18 mutant cells were grown in SD medium (2% glucose) or SD low-glucose medium (0.5% glucose). Cell growth (A) and viability (B) were monitored. ○, wild-type cells in SD medium; △, wild-type cells in SD low-glucose medium; ●, L18 mutant in SD medium; ▴, L18 mutant in SD low-glucose medium. The data shown represent the mean ± S.D. of three independent experiments. C and D, effect of vanadate. Wild-type (C) and L18 mutant (D) cells were grown in SD medium (○) or SD medium containing 100 μm (●) and 300 μm (△) vanadate, and cell viability was monitored. The data shown represent the mean of ± S.D. three independent experiments.
On the basis of these results, we propose the following scenario explaining the long-lived phenotype of the L18 mutant. In the L18 mutant, the reduced Pma1 activity causes some defect in glucose uptake. This might cause physiological changes that are equivalent to the changes caused by calorie restriction. In this study, we have presented evidence indicating that Pma1 is a factor that affects chronological life span. This highlights the new possibility for the regulation of chronological life span by modulating Pma1 activity. We attempted to reduce Pma1 activity by adding vanadate, an inhibitor of the P-type ATPase. If Pma1 activity was reduced by vanadate, we hypothesized that chronological life span was extended. This was the case, as shown in Fig. 5C. When wild-type cells were grown in medium containing various concentrations of vanadate, cell viability increased in a concentration-dependent manner (Fig. 5C). On the other hand, the viability of the L18 mutant was not affected by vanadate (Fig. 5D).
In summary, we have presented evidence showing a link between Pma1 and life span for the first time. This suggests that the Pma1 ATPase activity is crucial for the determination of chronological life span in S. pombe and implies that chronological life span could be manipulated by modulating Pma1 activity in other organisms. The possibility to regulate life span in this novel way needs to be verified in future experiments.
Acknowledgments
We thank M. Maeshima (Nagoya University) for helpful discussions and K. Ichinose (our laboratory) for technical assistance.
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to H. A.) and by Research for Promoting Technological Seeds from the Japan Science and Technology Agency (JST) and the Nagase Science and Technology Foundation (to H. A.).
- SD
- synthetic defined
- Km
- kanamycin.
REFERENCES
- 1.Werner-Washburne M., Braun E., Johnston G. C., Singer R. A. (1993) Microbiol. Rev. 57, 383–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roux A. E., Chartrand P., Ferbeyre G., Rokeach L. A. (2010) J. Gerontol. A Biol. Sci. Med. Sci. 65, 1–8 [DOI] [PubMed] [Google Scholar]
- 3.Fabrizio P., Longo V. D. (2003) Aging Cell 2, 73–81 [DOI] [PubMed] [Google Scholar]
- 4.Fabrizio P., Pozza F., Pletcher S. D., Gendron C. M., Longo V. D. (2001) Science 292, 288–290 [DOI] [PubMed] [Google Scholar]
- 5.Longo V. D., Fabrizio P. (2002) Cell. Mol. Life Sci. 59, 903–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo V. D., Kennedy B. K. (2006) Cell 126, 257–268 [DOI] [PubMed] [Google Scholar]
- 7.Roux A. E., Quissac A., Chartrand P., Ferbeyre G., Rokeach L. A. (2006) Aging Cell 5, 345–357 [DOI] [PubMed] [Google Scholar]
- 8.Oshiro T., Aiba H., Mizuno T. (2003) Mol. Genet. Genomics 269, 437–442 [DOI] [PubMed] [Google Scholar]
- 9.Fujita Y., Mita S., Ohtsuka H., Aiba H. (2007) Biosci. Biotechnol. Biochem. 71, 3041–3047 [DOI] [PubMed] [Google Scholar]
- 10.Ohtsuka H., Mita S., Ogawa Y., Azuma K., Ito H., Aiba H. (2008) FEMS Yeast Res. 8, 520–530 [DOI] [PubMed] [Google Scholar]
- 11.Ohtsuka H., Ogawa Y., Mizuno H., Mita S., Aiba H. (2009) Biosci. Biotechnol. Biochem. 73, 885–889 [DOI] [PubMed] [Google Scholar]
- 12.Azuma K., Ohtsuka H., Mita S., Murakami H., Aiba H. (2009) Biosci. Biotechnol. Biochem. 73, 2787–2789 [DOI] [PubMed] [Google Scholar]
- 13.Smith E. D., Kennedy B. K., Kaeberlein M. (2007) Mech. Ageing Dev. 128, 106–111 [DOI] [PubMed] [Google Scholar]
- 14.Krawchuk M. D., Wahls W. P. (1999) Yeast 15, 1419–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Briskin D., Leonard R., Hodges T. (1987) Methods Enzymol. 148, 542–558 [Google Scholar]
- 16.Ohmiya R., Kato C., Yamada H., Aiba H., Mizuno T. (1999) J. Biochem. 125, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 17.Ulaszewski S., Van Herck J. C., Dufour J. P., Kulpa J., Nieuwenhuis B., Goffeau A. (1987) J. Biol. Chem. 262, 223–228 [PubMed] [Google Scholar]
- 18.Ghislain M., De Sadeleer M., Goffeau A. (1992) Eur. J. Biochem. 209, 275–279 [DOI] [PubMed] [Google Scholar]
- 19.Serrano R. (1983) FEBS Lett. 156, 11–14 [DOI] [PubMed] [Google Scholar]
- 20.Lecchi S., Nelson C. J., Allen K. E., Swaney D. L., Thompson K. L., Coon J. J., Sussman M. R., Slayman C. W. (2007) J. Biol. Chem. 282, 35471–35481 [DOI] [PubMed] [Google Scholar]
- 21.Höfer M., Nassar F. R. (1987) J. Gen. Microbiol. 133, 2163–2172 [Google Scholar]
- 22.Fabrizio P., Liou L. L., Moy V. N., Diaspro A., Valentine J. S., Gralla E. B., Longo V. D. (2003) Genetics 163, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guarente L., Picard F. (2005) Cell 120, 473–482 [DOI] [PubMed] [Google Scholar]
- 24.North B. J., Sinclair D. A. (2007) Trends Biochem. Sci. 32, 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]