

Abstract
Naproxen ((S)-6-methoxy-α-methyl-2-naphthaleneacetic acid) is a powerful non-selective non-steroidal anti-inflammatory drug that is extensively used as a prescription and over-the-counter medication. Naproxen exhibits gastrointestinal toxicity, but its cardiovascular toxicity may be reduced compared with other drugs in its class. Despite the fact that naproxen has been marketed for many years, the molecular basis of its interaction with cyclooxygenase (COX) enzymes is unknown. We performed a detailed study of naproxen-COX-2 interactions using site-directed mutagenesis, structure-activity analysis, and x-ray crystallography. The results indicate that each of the pendant groups of the naphthyl scaffold are essential for COX inhibition, and only minimal substitutions are tolerated. Mutation of Trp-387 to Phe significantly reduced inhibition by naproxen, a result that appears unique to this inhibitor. Substitution of S or CH2 for the O atom of the p-methoxy group yielded analogs that were not affected by the W387F substitution and that exhibited increased COX-2 selectivity relative to naproxen. Crystallization and x-ray analysis yielded structures of COX-2 complexed to naproxen and its methylthio analog at 1.7 and 2.3 Å resolution, respectively. The combination of mutagenesis, structure analysis, and x-ray crystallography provided comprehensive information on the unique interactions responsible for naproxen binding to COX-2.
Keywords: Cyclooxygenase (COX) Pathway, Drug Action, Enzyme Inhibitors, Site-directed Mutagenesis, X-ray Crystallography, Naproxen
Introduction
Cyclooxygenase (COX)4 enzymes are the targets for inhibition by a diverse array of non-steroidal anti-inflammatory drugs (NSAIDs), which contain functional groups, such as arylacetic acids, arylpropionic acids, β-ketoenols, and diarylheterocycles. Investigation of the molecular determinants of inhibition by different classes of compounds reveals that the protein residues in the active site maintain similar orientations and that each chemical class forms distinct sets of interactions within the active site (1). Compounds with nanomolar binding affinity (and, in many cases, COX-2 selectivity) have been successfully designed for multiple chemical series, despite their diverse binding modes.
Naproxen is one of the oldest and largest selling NSAIDs (Fig. 1). It was introduced in prescription form as Naprosyn in 1976 and as the over-the-counter drug Aleve in 1994. It exhibits analgesic, anti-pyretic, and anti-inflammatory activity and was recently reported to be effective in the prevention of bladder cancer progression even when administered several weeks after the tumor-initiating agent (2). Naproxen is a non-selective NSAID that inhibits both COX-1 and COX-2 with comparable IC50 values (3). It exhibits significant gastrointestinal side effects, but recent mounting evidence suggests that it does not exert cardiovascular side effects when administered in the higher doses that provide sustained inhibition of platelet COX-1 throughout the dosing interval (e.g. ≥500 mg twice daily) (4–6). This latter property has taken on increasing importance because evolving data suggest that the cardiovascular toxicity first exhibited by rofecoxib and celecoxib extends to other selective or non-selective inhibitors, including diclofenac, indomethacin, and ibuprofen (5, 7, 8).
FIGURE 1.

Chemical structures of NSAIDs and crystal structures of flurbiprofen and diclofenac bound in mCOX-2 active site. A, chemical structures of naproxen, flurbiprofen, diclofenac, and indomethacin. The structures of flurbiprofen (Protein Data Bank entry 3PGH; inhibitor carbon atoms colored gold) (B) and diclofenac (Protein Data Bank entry 1PXX) (C) bound at the COX-2 active site show the opposing binding modes that position their acidic groups either coordinated to the constriction residues Arg-120 and Tyr-355 at the base of the active site or to the catalytic Tyr-385 as well as Ser-530 at the top of the pocket.
Despite its long history of human use, relatively little is known of the structural determinants of naproxen interaction with the COX enzymes. No crystal structures have been reported for naproxen bound to COX-1 or COX-2, and the few naproxen analogs that have been described in the literature have only been tested in vivo (9). Relatively little information has been reported on the amino acid determinants of naproxen interaction with the COX active sites. Considering its continuing importance in the treatment of a range of inflammatory disorders and its intriguing side effect profile, we conducted an investigation of naproxen-COX interactions using site-directed mutagenesis, structure-activity analysis, and x-ray crystallography. The results reveal a novel molecular determinant of COX binding not seen with other NSAIDs and an extraordinary sensitivity to subtle chemical modification.
EXPERIMENTAL PROCEDURES
Materials
(S)-Naproxen and 6-methoxynaphthalene acetic acid were purchased from Cayman Chemical Co. (Ann Arbor, MI). 6-O-demethyl naproxen and (R)-naproxen were purchased from Sigma-Aldrich. [1-14C]Arachidonic acid was purchased from PerkinElmer Life Sciences and diluted using 0.1 n NaOH. Chemicals used for the synthesis of naproxen analogs were purchased from Sigma-Aldrich. Crystallography reagents were purchased from Hampton Research (Aliso Viejo, CA).
Enzymes
The expression and purification of recombinant murine COX-2 (mCOX-2) and human COX-2 (hCOX-2) from Sf9 cells and the purification of ovine COX-1 (oCOX-1) from ram seminal vesicles were performed as previously described (10). Site-directed mutagenesis on mCOX-2 to generate various active site mutants (V349A, V349I, V349L, R120A, R120Q, Y355F, and W387F) was performed according to published methods (11, 12). The specific activity of W387F mCOX-2 and R120Q mCOX-2 are 2.1 and 23.9 μm AA/μm enzyme/min, respectively (compared with 13.7 for WT mCOX-2); the specific activities of all other mutant enzymes have been reported previously (11, 12).
Synthesis and Characterization of Naproxen Analogs
2-(6-Methoxynapthalen-2-yl)-2-methylpropanoicacid was synthesized as described previously by Stock et al. (13). (2-(6-Ethylnapthalen-2-yl)propanoic acid and 2-(6-(methylthio)napthalen-2-yl)propanoic acid were synthesized starting from the common intermediate (S)-methyl 2-(6-(trifluoromethylsulfonyloxy)napthalen-2-yl)propanoate. This intermediate was synthesized starting from (S)-naproxen, which was converted to des-methyl naproxen under acidic conditions (14), followed by methyl ester protection of the acid group (15) and trifiate protection of the phenolic oxygen. For 2-(6-ethylnapthalen-2-yl)propanoic acid, the triflic intermediate was coupled with potassium vinyltrifluoroborate using a Suzuki reaction, and the resulting alkene was reduced to the corresponding alkane, followed by hydrolysis under basic conditions to afford the desired acid. 2-(6-(Methylthio)napthalen-2-yl)propanoic acid was synthesized starting from the triflic intermediate, which was coupled to sodium triisopropylsilanethiolate (16), which was deprotected using tetrabutylammonium fluoride followed by alkylation with iodomethane. The racemic acid was then obtained by basic hydrolysis. The enantiomers were separated by chiral HPLC using either a Chiralpak AD or Chiralpak IC column, respectively. Additional synthetic methods can be found in the supplemental material.
Standard COX Inhibition Screening Assay
Hematin-reconstituted enzyme and inhibitor were preincubated for 17 min at room temperature, followed by a 3-min incubation at 37 °C prior to the addition of 50 μm [1-14C]AA for 30 s at 37 °C. The reactions were then terminated by extraction with diethyl ether/methanol/citrate (30:4:1) and analyzed for substrate consumption by thin layer chromatography as described previously (17). All inhibitor concentrations for 50% enzyme activity (IC50) were determined by nonlinear regression analysis using GraphPad Prism software and are the average of multiple determinations of duplicate analyses. Inhibitors were prepared as stock solutions in DMSO and diluted into reaction buffer so that the final DMSO concentration was 2.5%. Reactions were run with hematin-reconstituted proteins at final enzyme concentrations adjusted to give ∼30–35% substrate consumption (mCOX-2 = 154 nm, hCOX-2 = 94 nm, oCOX-1 = 31.6 nm, V349A = 250 nm, V349I = 268 nm, V349L = 113 nm, R120A = 100 nm, R120Q = 159 nm, Y355F = 174 nm, W387F = ∼750 nm, V523I = 83 nm). AA was prepared as a stock solution in 0.1 n NaOH. For IC50 determinations using 5 μm AA, the conditions were as described for the standard assay with a lowered enzyme concentration to allow for the appropriate amount of metabolism (oCOX-1 = 7.6 nm, mCOX-2 = 40 nm).
COX Inhibition Screening Assay for a Substrate Concentration of 500 nm
The COX inhibition assay described above was modified to perform IC50 determinations in the presence of submicromolar concentrations (18). Hematin-reconstituted enzyme and inhibitor were incubated for 0 or 5 min at 37 °C before the addition of 0.5 μm [1-14C]AA. The reaction was terminated after 8 s, and substrate consumption was analyzed as described above. Enzyme concentrations were adjusted to allow ∼30–50% substrate consumption under the modified conditions (mCOX-2 = ∼20 nm, oCOX-1 = ∼15 nm).
COX-2 Crystallization
Murine recombinant COX-2 was purified from Sf21 insect cells as described previously (19). Following the initial anion exchange and gel filtration columns, anion exchange chromatography was repeated on an 8-ml Mono-Q column followed by size exclusion chromatography using the same conditions. mCOX-2 was stored at −80 °C at 1–2 mg/ml for further use. For crystallization, protein was concentrated to ∼10 mg/ml and reconstituted with 1 eq of heme from a 15 mm stock in DMSO. Hematin-reconstituted mCOX-2 was dialyzed overnight at 4 °C in exchange buffer containing 100 mm NaCl, 20 mm Na3PO4, pH 6.7, 0.6% β-OG, 0.01% NaN3. Inhibitor complexes were formed by the addition of 1 mm inhibitor from a 50 mm stock in EtOH for 15–20 min on ice immediately before setting up hanging drop crystallization trays. mCOX-2 crystals were grown as previously described with the following alterations to the procedure (20). The crystallizations were conducted in darkness, and crystals were transferred into a cryosolution of a diffusion-equilibrated sitting drop crystallization experiment containing a 1:1 ratio of COX-2 inhibitor complex and well solution (28% polyethylene glycol monomethyl ether 550, 100 mm MgCl2, 50 mm EPPS, pH 8.0) that was set up at the time of the initial crystallization.
Crystals were flash frozen, and data were collected at the Southeastern Regional Collaborative Access Team beam line 22-ID or the Life Sciences Collaborative Access Team beam line 21-ID-F at the Advanced Photon Source at Argonne National Laboratory. The mCOX-2:naproxen co-crystal belongs to space group I222 with unit cell dimensions a = 122.3 Å, b = 133.2 Å, c = 181.3 Å, and α = β = γ = 90°. The crystal diffracted X-rays to a 1.7 Å resolution. There was a single mCOX-2 dimer in the asymmetric unit. The mCOX-2:p-methylthio naproxen crystal belongs to space group P21212 with unit cell parameters a = 181.2 Å, b = 134.2 Å, c = 122.0 Å, and α = β = γ = 90°. The asymmetric unit consisted of two mCOX-2 dimers; inhibitor was bound in each monomer. The structures were determined by molecular replacement using a Pfizer high resolution structure as the search model and the program MOLREP (21). Data collection and refinement statistics can be found in supplemental Table 1. The models were refined using REFMAC5 (22) with iterated manual fitting using COOT (23). The coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 3NT1 for naproxen and 3NTB for the naproxen analog.
RESULTS
NSAIDs appear to follow a multistep kinetic mechanism for inhibition of COX enzymes (see Reaction 1). Initial bimolecular association of the inhibitor with the enzyme is followed by a slower intramolecular step that results in a more tightly bound complex. In the case of aspirin, the intramolecular step results from acetylation of the active site residue, Ser-530, but in all other cases the inhibitor-enzyme association is non-covalent (24, 25). The magnitude of the individual rate constants determines the apparent type of inhibition (Reaction 1).
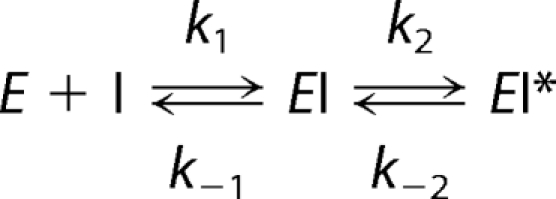 |
Inhibitors with a low k2/k−2 ratio appear to be rapid, reversible inhibitors, whereas inhibitors with a high k2/k−2 ratio appear to be slow, tightly binding inhibitors. The extent of reversibility is revealed by the existence of plateaus for maximal inhibition; rapidly reversible inhibitors are unable to completely inhibit COX activity, so their maximal inhibition plateaus at non-zero values. In contrast, slowly reversible inhibitors completely inhibit COX activity.
Previous kinetic studies suggest that naproxen exhibits time-independent inhibition of COX-1 and “mixed” inhibition of COX-2; “mixed” inhibition is defined as an initial time-dependent loss of enzyme activity followed by a non-zero plateau (26). This type of inhibition is characteristic of a weakly binding, readily reversible inhibitor. We explored the time dependence of COX inhibition by naproxen using a very low concentration of AA (500 nm; ∼0.1 Km). In the absence of a preincubation, the IC50 value for naproxen inhibition of oCOX-1 was ∼5.6 μm, and nearly 100% inhibition was achieved at 25 μm inhibitor (Fig. 2A). For mCOX-2, the extent of inhibition was very low so that an IC50 value could not be determined at concentrations of up to 25 μm naproxen (Fig. 2B). Following a 3-min incubation of COX and naproxen prior to the addition of AA, we observed substantial concentration-dependent inhibition of AA turnover for both COX isoforms. Naproxen inhibited oCOX-1 with an IC50 value of 340 nm and mCOX-2 with an IC50 value of 180 nm and demonstrated greater than 80% inhibition in the presence of 500 nm AA (Fig. 2). These data suggest the existence of a significant time-dependent component of naproxen inhibition of both COX-1 and COX-2. This time dependence was observed with higher concentrations of AA as well (1, 10, and 50 μm; supplemental Fig. 1). The time course for inhibition was too rapid to enable us to calculate rate constants for inhibition of either enzyme.
FIGURE 2.

Effect of preincubation of enzyme and inhibitor on COX inhibition by naproxen. Closed circles (●) represent incubations in which naproxen (0.05–25 μm) and AA (500 nm) were added simultaneously to COX. For the incubations represented by open circles (○), COX was preincubated with naproxen (0.05–4 μm) for 5 min before the addition of 500 nm AA. A is representative of incubations with oCOX-1, and B represents reactions with mCOX-2. The reaction with substrate was allowed to proceed for 8 s before quenching. Substrate consumption was analyzed by TLC as described. Each data point is the mean of at least two experiments in duplicate. Error bars, S.E.
In our subsequent studies, we utilized an IC50 assay designed for time-dependent inhibitors to elucidate tightly binding interactions critical for the formation of the naproxen-mCOX-2 complex. Under our standard assay conditions, enzyme and inhibitor were preincubated for 20 min prior to the addition of a saturating concentration of substrate (50 μm). In this assay, the inhibition of oCOX-1 activity reached a plateau at ∼50% inhibition, and an IC50 value could not be determined at concentrations up to 25 μm. Naproxen appeared to be a slightly more potent inhibitor of mCOX-2 in that an IC50 value of 0.90 μm (∼70% inhibition) could be measured (Table 1). An inhibition assay was performed using hCOX-2 (data not shown), and the IC50 was determined to be 0.75 μm with an inhibition curve that plateaued at ∼55%.
TABLE 1.
Determination of IC50 values of naproxen analogs with WT COX
Each naproxen analog was screened against purified oCOX-1 and mCOX-2 as described under “Experimental Procedures” for COX inhibition assays. no inhib., less than 10% inhibition up to inhibitor concentrations of 25 μm. The number in parentheses represents the extent of inhibition, indicating where the plateau for inhibition is reached for each inhibitor.

Probing Naproxen-COX-2 Interactions
Arylcarboxylic acid inhibitors bind in one of two orientations in the COX active site (Fig. 1). Flurbiprofen binds in the canonical fashion with its carboxylate moiety ion-paired and hydrogen-bonded to the constriction site residues Arg-120 and Tyr-355 (Fig. 1B) (20, 27). In contrast, diclofenac binds in an inverted orientation in which its carboxylate is hydrogen-bonded to the side chains of Tyr-385 and Ser-530 (Fig. 1B) (11). These two orientations can be discriminated by mutations of constriction site residues, which abolish inhibition by flurbiprofen but have no effect on inhibition by diclofenac. Mutation of Tyr-355 to Phe in mCOX-2 abolished inhibition by naproxen (Fig. 3A), whereas mutation of Arg-120 to Gln slightly increased the potency of inhibition as exhibited by an improved IC50 and a greater extent of inhibition (∼90%). Mutation of Arg-120 to Ala resulted in a complete loss of enzyme inhibition by naproxen (Fig. 3A). Together, these results imply that the carboxylate group of naproxen binds at the constriction site in the canonical orientation, coordinated to Tyr-355 and Arg-120.
FIGURE 3.

Inhibition of mCOX-2 active site mutants by naproxen and nonselective NSAIDs. A, naproxen (0.25–25 μm) was preincubated with WT mCOX-2 (○) or R120A (▴), R120Q (▵), and Y355F (♦) mCOX-2 mutant enzymes for 20 min prior to the addition of substrate (50 μm) for 30 s at 37 °C. B, naproxen (●), indomethacin (■), diclofenac (▴), or flurbiprofen (♦) was preincubated with mCOX-2 W387F for 20 min prior to the addition of substrate (50 μm). Inhibitor concentrations ranged from 0.25 to 25 μm. Each reaction was terminated and substrate consumption was analyzed by TLC as described. Data points represent the mean of duplicate determinations. Data points for naproxen against W387F represent the mean of five independent experiments in duplicate. Error bars, S.E.
In order to rigorously examine the binding mode predicted from these studies, we determined the co-crystal structure of naproxen bound to mCOX-2 at 1.7 Å resolution, the highest resolution COX structure to date and among the highest resolution membrane protein structures described. The topology of the COX dimer and active site resemble those of previous studies, although we have resolved additional solvent, ion, and detergent molecules not observed in lower resolution structures. Somewhat surprisingly, the residues lining the active site were observed in single conformations, despite the fact that many of them were bordered only by solvent. The resolution of this structure enabled the identification of a β-OG molecule lying on the external side of the constriction, at the base of the funnel-shaped entrance to the active site (lobby region), similar to high resolution structures of COX-1 (e.g. Protein Data Bank entry 2AYL) (28). A solvent molecule is often observed hydrogen-bonded to Tyr-385 and Ser-530 in the active site; however, when this density was fitted with a water molecule in the current structure, a 5σ residual electron density peak remained at that position. When fitted with a chlorine atom, the residual peak disappeared, and the atom refined to a temperature factor similar to neighboring protein and inhibitor atoms. A peak in the anomalous difference map at the same location provided further evidence that the peak was a chlorine atom. Although the physiological significance of chloride binding at this position is unknown, chloride ions have been used previously to identify the binding site of molecular oxygen in various proteins, including dioxygenases (29, 30). This raises the possibility that the chloride ion may be indicative of the position of molecular oxygen prior to incorporation into COX substrates.
Strong electron density was observed for a single orientation of naproxen binding within the COX-2 active site, making no contacts in the COX-2 side pocket or lobby region (Fig. 4). As predicted by the mutagenesis data, the binding mode of naproxen is similar to that of other members of the 2-arylpropionic acid family of NSAIDs with the carboxylate group of naproxen participating in hydrogen-bonding interactions with Arg-120 (2.8 and 2.9 Å) and Tyr-355 (2.5 Å) at the base of the active site. The remainder of the interactions between the compound and protein were van der Waals contacts. The (S)-α-methyl group of naproxen inserts into the hydrophobic cleft adjacent to Val-349, whereas the naphthyl backbone of naproxen makes hydrophobic contacts with Ala-527, Gly-526, and Leu-352. Interestingly, the side chain of Leu-352 adopts an alternate conformation from that observed in the co-crystal structures of flurbiprofen, indomethacin, and diclofenac bound to mCOX-2. The p-methoxy group of naproxen is oriented toward the apex of the COX active site and forms van der Waals interactions with Trp-387 and Tyr-385.
FIGURE 4.

Crystal structure of naproxen bound to mCOX-2. A, difference electron density map (FO − FC) contoured at 3.5σ of the COX-2 active site prior to the addition of naproxen to the model or modification of side chain positions in the binding pocket. This and other molecular graphics images were composed with PyMOL (Delano Scientific). B, stereoview of the crystal structure of naproxen (blue carbon atoms) bound at the COX-2 active site reveals that it forms extensive van der Waals contacts within the binding pocket and hydrogen-bonds, similar to flurbiprofen, to the side chains of Tyr355 and Arg120. The inhibitor does not enter the side pocket into which the phenyl sulfonamide or phenyl sulfone moieties of diaryl heterocyclic compounds protrude.
As described above, the mCOX-2:naproxen crystal structure indicates that the (S)-α-methyl group of naproxen is oriented in a conformation similar to that of the α-methyl group of flurbiprofen and makes hydrophobic contacts with Val-349 as well as Leu-359 (20). To further probe these interactions, we quantified the ability of naproxen to inhibit V349A, V349L, and V349I mutants, and we synthesized a series of α-substituted naproxen analogs. Naproxen inhibited V349A mCOX-2 with a potency and extent of inhibition similar to those of WT mCOX-2 (IC50 = 3.5 μm, 75% inhibition), but the V349I and V349L mutants were both more sensitive to inhibition (IC50 = 0.28 and 0.35 μm, greater than 95% inhibition) (supplemental Fig. 2). The increase in inhibition observed when naproxen was tested against V349I or V349L could arise from increased hydrophobic interactions between the α-methyl group and residue 349. To test this hypothesis, 2-des-methyl naproxen was assayed against the WT and V349I/L mutant enzymes. Elimination of the methyl group resulted in a significant decrease in inhibition of WT COX as well as the V349I/L mCOX-2 mutants (maximum inhibition of ∼20% at concentrations up to 25 μm) (Table 1 and data not shown). The addition of bulk at the α-position also resulted in a loss of potency because the α-ethyl analog of naproxen displayed no inhibition of either wild-type COX-1 or COX-2 enzymes in our standard IC50 assay (Table 1). Similarly, bulkier substitutions at the α-position of flurbiprofen result in a complete loss of inhibition of COX-1 (31). Thus, the (S)-α-methyl group is a critical determinant of naproxen efficacy, and it cannot be replaced with smaller (hydrogen) or larger (ethyl) substituents.
The majority of NSAIDs of the 2-arylpropionic acid family are marketed as racemic mixtures but naproxen is sold exclusively as the (S)-enantiomer. The (S)-enantiomer is significantly more potent than the (R)-enantiomer in inflammatory models in vivo, which is typical of members of the 2-arylpropionic acid class of inhibitors (9). In our assay, the (R)-enantiomer did not inhibit oCOX-1 or mCOX-2 to any appreciable extent at concentrations up to 25 μm (Table 1). Previous studies suggest that the strict stereoselectivity of the 2-arylpropionic acid class of inhibitors is due to unfavorable steric interactions with Tyr-355 when the methyl group of the inhibitor is in the (R)-stereochemistry (27, 32, 33). However, the lack of inhibition observed with the des-methyl naproxen analog raised the possibility that the inability of the (R)-enantiomer to inhibit is due to the absence of the (S)-methyl group. To address this possibility, the α,α-dimethyl analog was synthesized and tested for its ability to inhibit mCOX-2 (13). Assuming that this compound occupies the active site in a manner analogous to naproxen, the (S)-methyl group should be in position to interact with the pocket below Val-349 in the COX active site. However, the α,α-dimethyl analog was completely inactive against oCOX-1 or mCOX-2 (Table 1). This is consistent with previous reports demonstrating that a dimethyl substitution for the α-methyl group of flurbiprofen eliminates COX-1 inhibition (31). Although the presence of an (R)-methyl substituent clearly eliminates inhibition, the (S)-methyl group makes key interactions within the COX active site that are essential for binding and inhibition.
The crystal structure shows the p-methoxy group of naproxen is oriented toward the top of the hydrophobic channel within the COX active site. However, the importance of the hydrophobic interactions between the p-methoxy substituent and surrounding residues remained unclear. To probe the importance of the p-methoxy group, we synthesized a series of analogs with different substituents in the para position. The p-hydroxy analog (O-demethyl naproxen), which is the major in vivo metabolite of naproxen, was a very weak inhibitor, exhibiting roughly 30% inhibition up to 25 μm (Table 1) (34). This analog would place a polar, hydrogen bond-donating group within an entirely hydrophobic pocket, consistent with the reduction in inhibition. The o-ethoxy analog was completely inactive against both COX-1 and COX-2 (Table 1). In this case, the distance from the new terminal methyl group in any of its preferred rotamers to adjacent protein atoms is probably insufficient to accommodate the additional substituent. The results with these analogs suggest important and specific interactions for the methoxy group at the top of the active site channel.
The naproxen-COX-2 crystal structure shows that the p-methoxy group interacts with Trp-387 by van der Waals contacts with two carbon atoms of the side chain, Cζ2 (3.4 Å) and Cη2 (3.6 Å), the latter being the position of the side chain unique spatially to tryptophans. Trp-387 is located at the top of the COX active site near the catalytic residue, Tyr-385, and has been shown to be a critical residue for the proper positioning of AA within the active site to yield the cyclooxygenase product, PGG2. The W387F mCOX-2 mutant enzyme forms relatively low amounts of PGG2 but increased amounts of the uncyclized product, 11-hydroxyeicosatetraenoic acid (35). We tested the W387F mutant for sensitivity to naproxen inhibition. A higher protein concentration was used because of the mutant's reduced catalytic activity. Naproxen had a minimal inhibitory effect on W387F, exhibiting only 25% inhibition at 25 μm (Fig. 3B). The W387F mutation has not been studied with other inhibitors, so we tested it against several other carboxylate-containing NSAIDs. Surprisingly, the IC50 values for diclofenac, flurbiprofen, and indomethacin against the W387F mutant enzyme were similar to previously reported values against wild-type enzyme (∼87, ∼120, and ∼250 nm, respectively) (Fig. 3B). Diclofenac and indomethacin only form a single contact point to the Trp side chain, to the Cζ2 position, one closely mimicked by phenylalanine (11, 20). Although flurbiprofen also forms interactions with both carbon atoms of the Trp residue, they originate from a phenyl ring of the inhibitor that is already buttressed by other interactions, making the one with the Trp perhaps less important for binding. In the case of naproxen, the oxygen of the p-methoxy group lies within van der Waals contact range only of the Trp side chain. Therefore, the interaction between Trp-387 and naproxen appears to be unique among carboxylate-containing NSAIDs.
To further probe the interaction between the methoxy group and Trp-387, we synthesized two naproxen analogs, in which an ethyl or methylthio group was substituted for p-methoxy to introduce variations in size and polarity as well as to eliminate the possibility of hydrogen-bonding interactions. The methylthio analog has been reported to exhibit anti-inflammatory activity in vivo but has not been tested in vitro. The ethyl analog has not been reported. Both the p-ethyl and p-methylthio analogs were able to inhibit wild-type mCOX-2 to the same extent as naproxen (IC50 = 0.67 and 0.77 μm in our standard IC50 assay) (Table 1). Interestingly, both analogs exhibited a loss of potency compared with naproxen when tested against oCOX-1 so that no IC50 value could be determined at inhibitor concentrations up to 25 μm (Table 1). We also observed an increase in COX-2 selectivity at reduced substrate concentrations (500 nm and 5 μm AA) (supplemental Table 2). More remarkable however, both analogs inhibited W387F as well as they inhibited wild-type enzyme (Fig. 5).
FIGURE 5.

Inhibition of WT and W387F mCOX-2 by naproxen and naproxen analogs. Following a 20-min preincubation of naproxen (A), p-ethyl naproxen (B), or a p-methylthio naproxen (C) with mCOX-2 (●) or W387F mCOX-2 (○), [1-14C]AA (50 μm) was added and allowed to react for 30 s prior to termination with organic solvent. Concentrations of inhibitors ranged from 0.25 to 25 μm. Product formation was measured by TLC as described. Each data point is the mean of at least two independent experiments. Error bars, S.E.
The difference in sensitivity of W387F mCOX-2 to naproxen and the p-ethyl and p-methylthio analogs prompted us to crystallize the complex of mCOX-2 with the p-methylthio naproxen derivative. A structure of this complex was refined at 2.3 Å resolution (Fig. 6). Like naproxen, the inhibitor is bound entirely within the main channel of the COX active site. The closest equivalent atoms (distance = 0.2 Å) between the two compounds are the sulfur and oxygen atoms of the p-methylthio and p-methoxy groups, respectively. The carboxylate tails of the compounds differ greatest in position, with the p-methylthio-substituted compound extending ∼0.5 Å less deeply into the binding site. The p-methylthio naproxen analog adopts a binding conformation similar to that of naproxen, maintaining many of the same interactions with surrounding residues. For example, the carboxylate makes hydrogen-bonding interactions with Arg-120 (2.9 and 3.0 Å) and Tyr-355 (2.5 Å), and the (S)-α-methyl group makes hydrophobic contacts with Val-349 and Leu-359. The naphthyl backbone participates in van der Waals interactions with Ala-527 and Gly-526, whereas the methylthio substituent at the 6-position contacts Tyr-385 and Trp-387. In contrast to naproxen, the p-methylthio naproxen analog is not within van der Waals distances of Leu-352. This difference arises from the Leu side chain exhibiting different side chain conformation in the two structures, with that observed in the methylthio analog being consistent with that seen in the previously published NSAID:mCOX-2 co-crystal structures (three of four monomers). No explanation for the rotation of the residue in the naproxen structure or its failure to rotate in the methylthio derivative structure is readily apparent. Comparison of the two crystal structures also indicates that Val-523 makes hydrophobic contacts with the naphthyl backbone of the p-methylthio naproxen analog but does not contact naproxen. This observation results from the relative shift of the compounds within the active site.
FIGURE 6.

Crystal structure of p-methylthio naproxen bound to mCOX-2. A, stereoview of the (FO − FC) difference electron density map contoured at 3.0σ prior to the addition of the inhibitor to the model. B, stereoview of the p-methylthio naproxen analog bound within the mCOX-2 active site. The carboxylate participates in hydrogen-bonding interactions with Arg-120 and Tyr-355 at the base of the active site; this interaction is represented by the dashed yellow lines.
DISCUSSION
Naproxen has been a Food and Drug Administration-approved drug since the mid-1970s, but the precise mechanism by which naproxen interacts with COX is unknown. We utilized kinetic studies as well as an extensive mutagenesis study, exploration of the structure-activity relationship, and x-ray crystallography to identify the molecular determinants of COX inhibition by naproxen. By using a combined approach to probe the importance of naproxen-COX-2 interactions, we were able to elucidate key interactions that would not have been identified by one technique alone. We found critical interactions between the inhibitor and constriction site residues as well as a novel interaction with Trp-387 (Fig. 3). Substitution of an ethyl or methylthio group for the p-methoxy substituent generated COX-2-preferring naproxen analogs that were unaffected by mutation of Trp-387 to Phe (Fig. 5). We determined the x-ray crystal structures of both naproxen and the p-methylthio analog bound to mCOX-2. The combination of mutagenesis, chemical elaboration of naproxen analogs, and structural studies clearly defined the contribution of protein and inhibitor atoms to affinity (Figs. 4 and 6).
Naproxen is a relatively simple molecule with only three functional groups distributed on opposite ends of the naphthyl scaffold. Our data indicate that each of these substituents is required for potent inhibition of both COX isoforms and that very little structural variation is tolerated. Mutagenesis data suggest that one possibility regarding the nature of the interaction between the carboxylate moiety of naproxen and mCOX-2 is that the carboxylate interacts with Arg-120 via hydrogen bonding rather than ion-pairing interactions (Fig. 3A). In contrast, the crystal structure of flurbiprofen in complex with mCOX-2 indicates that the carboxylate forms a salt bridge with the guanidinium group of Arg-120 (20) Furthermore, previous studies have shown a 1000-fold increase in the IC50 value for flurbiprofen against R120Q oCOX-1 compared with wild-type enzyme, suggesting that ion-pairing interactions are more important for inhibition by flurbiprofen than naproxen (36).
The α-methyl group of naproxen appears to be involved in critical interactions with the COX enzymes. Introduction of a range of substituents of varying size and stereochemistry at the α-position suggests that the steric requirements for this interaction are stringent, whereas removal of the α-methyl group also results in a dramatic loss of potency (Table 1). Our data indicate that the α-methyl group inserts into a small hydrophobic cleft below Val-349, which may serve to anchor naproxen within the mCOX-2 active site and thereby reinforce the canonical binding orientation. The x-ray crystal structures of other 2-arylpropionic acids and the diaryl heterocyclic compound, SC-558, bound to the COX enzymes indicate that the α-methyl group (or 4-trifluoromethyl, in the case of SC-558) is bound in a similar fashion to the naproxen structure (20, 37). Carboxylate-containing COX inhibitors without a methyl group in the α-position utilize alternate interactions to reinforce binding within the COX active site. For example, the indolyl-2′-methyl group of indomethacin inserts into a hydrophobic pocket above Val-349 lined by Ala-527, Ser-530, and Leu-531 to form a tightly bound complex (12, 20). Similarly, whereas diclofenac binds in an inverted orientation with the carboxylate coordinated to Ser-530 and Tyr-385, a chlorine atom on the lower aniline ring also inserts into the hydrophobic pocket above Val-349 (11).
A key interaction between naproxen and Trp-387 was uncovered during our mutagenesis screen by the finding that the W387F mutant was largely insensitive to naproxen inhibition. This interaction appears to be unique to naproxen because the same mutation had no appreciable effect on inhibition of mCOX-2 by diclofenac, flurbiprofen, or indomethacin (Fig. 3B). The interaction with Trp-387 may result from a combination of hydrophobic packing of the methyl group and electrostatic interactions with the polarized methoxy group. This region of the enzyme appears to be strict in its ability to bind functional groups; the p-hydroxy and p-ethoxy analogs were very weak inhibitors, whereas a methylene or sulfur substitution for the oxygen atom of the p-methoxy group of naproxen generated potent inhibitors of WT mCOX-2. Unlike naproxen, the p-ethyl and p-methylthio analogs are effective inhibitors of W387F mCOX-2. This suggests that either the interaction with Trp-387 is not required for inhibition by the naproxen analogs or that they are able to interact more effectively with W387F COX-2 than is naproxen. Crystal structures of naproxen and the p-methylthio naproxen analog show the substituents at the 6-position oriented in a very similar fashion at the top of the COX active site, providing no definitive basis for differential inhibition of W387F mCOX-2. Moreover, with the exception of Leu-352, there are no dramatically different interactions throughout the rest of the active site. The substitution of Phe for Trp at position 387 creates a larger active site for the mutant enzyme compared with WT mCOX-2. The ability of the p-ethyl and p-methylthio naproxen analogs to inhibit W387F mCOX-2 as well as WT suggests that the analogs may be able to adopt an alternate conformation in the larger active site of W387F mCOX-2, compensating for the loss of the interaction with Trp-387 in the wild-type enzyme.
It is clear that there is great diversity and subtlety in the types of molecular interactions that result in COX inhibition and selectivity for isoforms. Even viewed from this perspective, our discoveries of the effect of methylene and sulfur substitution for the oxygen atom of the p-methoxy group of naproxen seem extraordinary. A single atom substitution changes the sensitivity of the inhibitor to single active site mutations and increases their COX-2 selectivity. Mutation of Val-523 to Ile in COX-2 greatly reduces the selectivity of potent COX-2-selective inhibitors like celecoxib and rofecoxib. It has previously been reported that a V523I mutation in the hCOX-2 background completely abrogates inhibition of hCOX-2 by naproxen as measured by a prostaglandin E2 ELISA (38). Consistent with these results, naproxen was not an effective inhibitor of V523I mCOX-2 in our standard IC50 assay; the maximal extent of inhibition was ∼20% (data not shown). The crystal structure of the p-methylthio naproxen analog bound to mCOX-2 is suggestive of hydrophobic interactions between Val-523 and the proximal ring of the napthyl backbone. Thus, steric interactions at the top of the channel may put additional pressure on Val-523 near the base of the active site so that inhibition of COX-1, where a bulkier isoleucine is located at residue 523, is significantly more difficult. Consistent with this hypothesis, the naproxen analogs were unable to significantly inhibit V523I mCOX-2.
The elucidation of the critical interactions between naproxen and mCOX-2 described herein may be useful for the specific design of more potent or selective naproxen analogs. In fact, substitution of sulfur or a methylene group for the methoxy oxygen of naproxen moderately increases its COX-2 selectivity. Such minimally altered analogs may be useful in dissecting the importance of isoform selectivity in cardiovascular toxicity or in the generation of gastrointestinal-sparing chemopreventive agents. Finally, the COX-2-naproxen crystal structure represents the highest resolution structure reported for either COX-1 or COX-2, in complex with an inhibitor or a substrate. The higher resolution provided by this structure may provide additional insights into the structural basis of COX catalysis and inhibition.
Supplementary Material
Acknowledgments
We are grateful to Pfizer Inc. and Karen Seibert for the release of internal protocols and reagents for the COX-2 crystallography. Data were collected at the Southeastern Regional Collaborative Access Team beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Life Sciences Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38.
This work was supported, in whole or in part, by National Institutes of Health Research Grant CA89450 and Center Grant GM15431.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2 and Figs. 1 and 2.
The atomic coordinates and structure factors (codes 3NT1 and 3NTB) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- COX
- cyclooxygenase
- mCOX-2 and hCOX-2
- mouse and human COX-2, respectively
- oCOX-1
- ovine COX-1
- AA
- arachidonic acid
- NSAID
- non-steroidal anti-inflammatory drug
- EPPS
- N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid
- PGG2
- prostaglandin G2.
REFERENCES
- 1.Blobaum A. L., Marnett L. J. (2007) J. Med. Chem. 50, 1425–1441 [DOI] [PubMed] [Google Scholar]
- 2.Lubet R. A., Steele V. E., Juliana M. M., Grubbs C. J. (2010) J. Urol. 183, 1598–1603 [DOI] [PubMed] [Google Scholar]
- 3.Laneuville O., Breuer D. K., Dewitt D. L., Hla T., Funk C. D., Smith W. L. (1994) J. Pharm. Exp. Ther. 271, 927–934 [PubMed] [Google Scholar]
- 4.Ray W. A., Varas-Lorenzo C., Chung C. P., Castellsague J., Murray K. T., Stein C. M., Daugherty J. R., Arbogast P. G., García-Rodríguez L. A. (2009) Circ. Cardiovasc. Qual. Outcomes 2, 155–163 [DOI] [PubMed] [Google Scholar]
- 5.Kearney P. M., Baigent C., Godwin J., Halls H., Emberson J. R., Patrono C. (2006) BMJ 332, 1302–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capone M. L., Tacconelli S., Sciulli M. G., Anzellotti P., Di Francesco L., Merciaro G., Di Gregorio P., Patrignani P. (2007) J. Pharmacol. Exp. Ther. 322, 453–460 [DOI] [PubMed] [Google Scholar]
- 7.Graham D. J., Campen D., Hui R., Spence M., Cheetham C., Levy G., Shoor S., Ray W. A. (2005) Lancet 365, 475–481 [DOI] [PubMed] [Google Scholar]
- 8.McGettigan P., Henry D. (2006) JAMA 296, 1633–1644 [DOI] [PubMed] [Google Scholar]
- 9.Harrison I. T., Lewis B., Nelson P., Rooks W., Roszkowski A., Tomolonis A., Fried J. H. (1970) J. Med. Chem. 13, 203–205 [DOI] [PubMed] [Google Scholar]
- 10.Rowlinson S. W., Crews B. C., Lanzo C. A., Marnett L. J. (1999) J. Biol. Chem. 274, 23305–23310 [DOI] [PubMed] [Google Scholar]
- 11.Rowlinson S. W., Kiefer J. R., Prusakiewicz J. J., Pawlitz J. L., Kozak K. R., Kalgutkar A. S., Stallings W. C., Kurumbail R. G., Marnett L. J. (2003) J. Biol. Chem. 278, 45763–45769 [DOI] [PubMed] [Google Scholar]
- 12.Prusakiewicz J. J., Felts A. S., Mackenzie B. S., Marnett L. J. (2004) Biochemistry 43, 15439–15445 [DOI] [PubMed] [Google Scholar]
- 13.Stock N., Munoz B., Wrigley J. D., Shearman M. S., Beher D., Peachey J., Williamson T. L., Bain G., Chen W., Jiang X., St-Jacques R., Prasit P. (2006) Bioorg. Med. Chem. Lett. 16, 2219–2223 [DOI] [PubMed] [Google Scholar]
- 14.Gant T. G., Sarshar S., Woo S. H. (June12, 2007) International Patent WO/2007/140189
- 15.Arewång C. J., Lahmann M., Oscarson S., Tidén A. K. (2007) Carbohydr. Res. 342, 970–974 [DOI] [PubMed] [Google Scholar]
- 16.Kovács I., Bélanger-Gariépy F., Shaver A. (2003) Inorg. Chem. 42, 2988–2991 [DOI] [PubMed] [Google Scholar]
- 17.Kalgutkar A. S., Crews B. C., Rowlinson S. W., Garner C., Seibert K., Marnett L. J. (1998) Science 280, 1268–1270 [DOI] [PubMed] [Google Scholar]
- 18.Boutaud O., Aronoff D. M., Richardson J. H., Marnett L. J., Oates J. A. (2002) Proc. Natl. Acad. Sci. 99, 7130–7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens A. M., Pawlitz J. L., Kurumbail R. G., Gierse J. K., Moreland K. T., Stegeman R. A., Loduca J. Y., Stallings W. C. (1999) J. Cryst. Growth 196, 350–355 [Google Scholar]
- 20.Kurumbail R. G., Stevens A. M., Gierse J. K., McDonald J. J., Stegeman R. A., Pak J. Y., Gildehaus D., Miyashiro J. M., Penning T. D., Seibert K., Isakson P. C., Stallings W. C. (1996) Nature 384, 644–648 [DOI] [PubMed] [Google Scholar]
- 21.Vagin A., Teplyakov A. (1997) J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 22.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 23.Emsley P., Cowtan K. (2004) Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24.Rome L. H., Lands W. E. (1975) Proc. Natl. Acad. Sci. 72, 4863–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blobaum A. L., Marnett L. J. (2007) J. Biol. Chem. 282, 16379–16390 [DOI] [PubMed] [Google Scholar]
- 26.Gierse J. K., Koboldt C. M., Walker M. C., Seibert K., Isakson P. C. (1999) Biochem. J. 339, 607–614 [PMC free article] [PubMed] [Google Scholar]
- 27.Picot D., Loll P. J., Garavito R. M. (1994) Nature 367, 243–249 [DOI] [PubMed] [Google Scholar]
- 28.Gupta K., Selinsky B. S., Loll P. J. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 151–156 [DOI] [PubMed] [Google Scholar]
- 29.Colloc'h N., Gabison L., Monard G., Altarsha M., Chiadmi M., Marassio G., Sopkova-de Oliveira Santos J., El Hajji M., Castro B., Abraini J. H., Prangé T. (2008) Biophys. J. 95, 2415–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steiner R. A., Janssen H. J., Roversi P., Oakley A. J., Fetzner S. (2010) Proc. Natl. Acad. Sci. 107, 657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peretto I., Radaelli S., Parini C., Zandi M., Raveglia L. F., Dondio G., Fontanella L., Misiano P., Bigogno C., Rizzi A., Riccardi B., Biscaioli M., Marchetti S., Puccini P., Catinella S., Rondelli I., Cenacchi V., Bolzoni P. T., Caruso P., Villetti G., Facchinetti F., Del Giudice E., Moretto N., Imbimbo B. P. (2005) J. Med. Chem. 48, 5705–5720 [DOI] [PubMed] [Google Scholar]
- 32.Loll P. J., Picot D., Ekabo O., Garavito R. M. (1996) Biochemistry 35, 7330–7340 [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharyya D. K., Lecomte M., Rieke C. J., Garavito M., Smith W. L. (1996) J. Biol. Chem. 271, 2179–2184 [DOI] [PubMed] [Google Scholar]
- 34.Miners J. O., Coulter S., Tukey R. H., Veronese M. E., Birkett D. J. (1996) Biochem. Pharmacol. 51, 1003–1008 [DOI] [PubMed] [Google Scholar]
- 35.Thuresson E. D., Lakkides K. M., Smith W. L. (2000) J. Biol. Chem. 275, 8501–8507 [DOI] [PubMed] [Google Scholar]
- 36.Rieke C. J., Mulichak A. M., Garavito R. M., Smith W. L. (1999) J. Biol. Chem. 274, 17109–17114 [DOI] [PubMed] [Google Scholar]
- 37.Selinsky B. S., Gupta K., Sharkey C. T., Loll P. J. (2001) Biochemistry 40, 5172–5180 [DOI] [PubMed] [Google Scholar]
- 38.Gierse J. K., McDonald J. J., Hauser S. D., Rangwala S. H., Koboldt C. M., Seibert K. (1996) J. Biol. Chem. 271, 15810–15814 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.