

Abstract
The reactivity of flavoenzymes with dioxygen is at the heart of a number of biochemical reactions with far reaching implications for cell physiology and pathology. Flavin-containing monooxygenases are an attractive model system to study flavin-mediated oxygenation. In these enzymes, the NADP(H) cofactor is essential for stabilizing the flavin intermediate, which activates dioxygen and makes it ready to react with the substrate undergoing oxygenation. Our studies combine site-directed mutagenesis with the usage of NADP+ analogues to dissect the specific roles of the cofactors and surrounding protein matrix. The highlight of this “double-engineering” approach is that subtle alterations in the hydrogen bonding and stereochemical environment can drastically alter the efficiency and outcome of the reaction with oxygen. This is illustrated by the seemingly marginal replacement of an Asn to Ser in the oxygen-reacting site, which inactivates the enzyme by effectively converting it into an oxidase. These data rationalize the effect of mutations that cause enzyme deficiency in patients affected by the fish odor syndrome. A crucial role of NADP+ in the oxygenation reaction is to shield the reacting flavin N5 atom by H-bond interactions. A Tyr residue functions as backdoor that stabilizes this crucial binding conformation of the nicotinamide cofactor. A general concept emerging from this analysis is that the two alternative pathways of flavoprotein-oxygen reactivity (oxidation versus monooxygenation) are predicted to have very similar activation barriers. The necessity of fine tuning the hydrogen-bonding, electrostatics, and accessibility of the flavin will represent a challenge for the design and development of oxidases and monoxygenases for biotechnological applications.
Keywords: Drug Metabolism, Flavoproteins, Genetic Diseases, Oxidase, Oxygen Binding
Introduction
Flavin-containing monooxygenases (FMOs)3 form a large family of enzymes present almost ubiquitously among living organisms (1, 2, 3). They are involved in diverse biological processes such as the biosynthesis of various natural products (4) and catabolism of xenobiotics. Humans have five different isozymes that are coded by different genes and represent a key enzymatic system in drug metabolism, whose relevance almost equals that of cytochrome P450s. Among known FMO substrates, there are metabolites directly or indirectly deriving from food digestion such as trimethylamine, drugs such as tamoxifen, and toxic molecules such as nicotine. Mutations in the gene for the isozyme 3 of human FMO (FMO3), which is most abundant in the liver, are directly responsible for trimethylaminuria, also known as fish odor syndrome (TMAU). This genetic disease causes the accumulation of trimethylamine so that the body of the patients tend to have an unpleasant smell (5, 6).
Irrespectively of their biological context, FMOs always catalyze the same reaction: the oxygenation of a soft nucleophile using molecular oxygen as oxygen source and NADPH as electron donor (7) (Fig. 1A). At the heart of this very complex reaction is the stabilization of the flavin-hydroperoxide adduct resulting from the reaction of dioxygen with the two-electron reduced FAD (Fig. 1A). Equally essential is the role of NADP(H). This cofactor not only acts as the electron donor that reduces the flavin but it directly takes part also in formation/stabilization of the flavin-hydroperoxide. This notion is best illustrated by the fact that using alternative electron donors (e.g. dithionite) in place of NADPH does not support catalysis because the reaction of dioxygen with the artificially reduced flavin generates hydrogen peroxide rather than the stable flavin-hydroperoxide needed for monooxygenation (8, 9, 10). In the cellular milieu, FMOs are thought to be mainly in the flavin-hydroperoxide state, ready to attack a suitable substrate (the so-called “cocked gun”) (11).
FIGURE 1.

Properties of FMO enzymes. A, overall scheme of the reaction and chemical structure of the crucial flavin-hydroperoxide intermediate that results from the reaction of dioxygen with the C4a atom of the reduced flavin. Stabilization of this intermediate requires the presence of a bound NADP+ molecule. Therefore, NADP(H) must have two binding modes; one competent in flavin reduction by hydride transfer and one competent in intermediate stabilization (13). B, overall three-dimensional structure of mFMO bound to NADP+. The NADP-binding and FAD-binding domains are in blue and cyan, respectively. Nitrogens are in blue, oxygens in red, phosphorous atoms in brown, flavin carbons in yellow, and NADP+ carbons in cyan.
The peculiar catalytic role of NADP(H) in oxygenation makes FMOs a very attractive system to study the more general problem of oxygen reactivity in flavoenzymes. Because it has been established that the nicotinamide cofactor remains bound to enzyme during oxygenation, NADP(H) can be exploited as a probe to gain insight into the reaction of dioxygen with the reduced flavin and to interrogate the elements that promote the stabilization of the crucial flavin-hydroperoxide. This concept represents the starting point of our study that builds on the previous analysis of the crystal structure and basic biochemical properties of the FMO from Methylophaga sp. strain SK1 (mFMO, Fig. 1B) (12). This bacterial enzyme shares many features with the human FMO3 including a considerable degree of sequence identity (33%), a similar substrate specificity, and the ability to form a stable flavin-hydroperoxide intermediate (13). At the same time, mFMO is better suited for detailed structural and enzymological investigations because it is a soluble protein (i.e. not membrane-associated). The aim of our study is 2-fold: (i) to gain insight into the astonishing properties of FMOs that combine in a single active center two cofactors and a complex sequence of catalytic steps that lead to the formation of an “activated-form” of dioxygen (flavin-hydroperoxide, Fig. 1A), and (ii) to translate this knowledge into a molecular rational for the effects of mutations found in patients affected by TMAU. For these purposes, we have probed the mFMO catalytic properties by altering both the active site amino acids by site directed mutagenesis and the NADP(H) substrate by using analogues of this cofactor. Such a “double-engineering” strategy has revealed that very fine details in the stereochemical and hydrogen-bonding environment around the flavin determine the efficiency of the reaction with dioxygen and the nature of the resulting products.
EXPERIMENTAL PROCEDURES
Protein Purification and Reagents
All reagents were purchased from Sigma-Aldrich. Mutagenesis, protein expression, and purification were carried out using standard protocols as previously described (13). All the experiments were carried out using the E158A/E159A mutant. This surface mutation does not affect the enzymatic properties but is essential for the growth of suitable crystals (13). Throughout the text, we shall refer to this protein as the wild type.
Steady-state Kinetics
The steady-state activity of the mFMO variants was measured spectrophotometrically by monitoring the decrease of NADPH in time at 340 nm (ϵ340 nm = 6.22 mm−1 cm−1). The reaction mixture (1.0 ml) typically contained 50 mm Tris/HCl pH 8.0, 100 mm NaCl, 5% (v/v) glycerol, 3 mm β-mercaptoethanol, 0–200 μm NADPH, 0–2 mm trimethylamine, and 0.2–0.6 μm mFMO. Trimethylamine was chosen because it is the best substrate for mFMO (13) and is commonly used for FMO activity evaluation (7). The oxidase activity was probed in the same conditions except for the absence of trimethylamine. The corresponding kuncoupling (Table 1) was measured by monitoring the consumption of NADPH (spectrophotometrically) and oxygen (using an oxygen electrode, Hansatech Oxygraph). Furthermore, the production of hydrogen peroxide was verified using the well-known amplex red/peroxidase assay as described (14). Reduced NADP analogues (3-acetylpyridine adenine dinucleotide and thioNADP) were generated enzymatically using glucose-6-phosphate dehydrogenase or phosphite dehydrogenase. In a typical experiment, solutions (150 μm) of reduced dinucleotides were produced by overnight incubation in 50 mm Tris-HCl pH 8.0, 150 μm glucose-6-phosphate or phosphite, and 1 unit of dehydrogenase. Dinucleotide reduction was monitored spectrophotometrically. Enzymatic activities were then carried out as with NADPH. For 3-acetylpyridine adenine dinucleotide (APADP), the commercially available reduced form was used as additional check (OYC Europe). Lack of a monooxygenase activity was inferred from the absence of an increased rate of oxygen or NADPH consumption upon addition of trimethylamine. When no monooxygenase activity was detected with trimethylamine, a very sensitive colorimetric assay using indole (whose monooxygenation product spontaneously dimerises to indigo) was used as an additional check for oxygenation activity. Kinetic parameters were determined by measuring at ≥5 different substrate concentrations and the observed rates were measured in duplicate or triplicate. Michaelis-Menten kinetic parameters were obtained by fitting the data using Origin software (version 7.0 for Windows) and were within 15% standard deviation.
TABLE 1.
Steady-state and presteady-state kinetic parameters
| Steady state |
Titration, Kd,NADP+ | Presteady state |
||||||
|---|---|---|---|---|---|---|---|---|
| Km,TMA | Km,NADPH | kcat | kuncoupling | Kd,NADPH | kred | kox | ||
| μm | μm | s−1 | s−1 | μm | μm | s−1 | m−1s−1 | |
| WT | 7 | 18.6 | 3.3 | 0.5 | 21 | <10 | 18.4 | 65,000 |
| WTa | NDb | NDb | 0.4 | 0.2 | – | – | – | – |
| WTc | – | – | – | 0.13 | – | – | – | – |
| N78K | – | >160 | – | >0.07 | >250 | >250 | >0.1 | 670 |
| N78S | – | 3.6 | – | 0.7 | 22 | <10 | 6.7 | 5,000 |
| N78D | – | 85 | – | 0.19 | >250 | 270 | 1.9 | 2,200 |
a Performed using 150 μm thioNADPH and monitoring oxygen consumption.
b ND, not determined.
c Performed using 150 μm APADPH and monitoring oxygen consumption.
Presteady-state Kinetics
Reductive and oxidative half-reactions of mFMO proteins were analyzed using a stopped-flow apparatus from Applied Photophysics Ltd. (model SX17MV) equipped with a diode array detector. Spectral data (300–720 nm) were collected at time intervals of 2.5 ms. The obtained spectra were analyzed by means of numerical integration methods using Pro-K software (Applied Photophysics Ltd.), yielding individual observed rate constants and deconvoluted spectra (Table 1 and Fig. 2). All experiments described below were performed at 25 °C in the same buffer used for steady-state experiments using 8–12 μm enzyme (after mixing). Each reaction condition was measured in triplicate. Rate constants obtained by fitting were within 20% standard deviation. Anoxic conditions were achieved by flushing the system and solutions with N2 and removing traces of oxygen upon addition of 10 mm glucose and a catalytic amount of glucose oxidase. Reduction of mFMO by NADPH was measured anoxically at various concentrations of the reduced nicotinamide coenzyme. The observed rates increased hyperbolically with increasing NADPH concentration and could be fitted with Equation 1 using non-linear regression analysis (Origin version 7.0 for Windows).
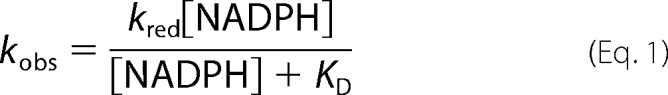 |
For measuring the oxidation of reduced mFMO, the enzyme was first anoxically reduced outside the stopped-flow apparatus by adding a slight 1.2-fold excess of NADPH. Only in the case of the N78K mutant a 10-fold excess of NADPH was needed to obtain the reduced enzyme form, due to the relatively poor NADPH affinity of this mutant. The reduced enzyme was subsequently mixed with oxygenated buffer containing equimolar amounts of NADP+. By varying the oxygen concentration (125, 360, and 595 μm), the oxygen dependence of the rates for the reoxidation events was probed. The affinity for NADP+ (KD, NADP+) was determined by measuring the change in the flavin absorbance spectrum upon titration of 10 μm of each mFMO variant in 50 mm Tris-HCl, pH 7.5 with 0–500 μm NADP+ in the same buffer.
FIGURE 2.

Presteady-state investigation of mFMO. A, spectra of deconvoluted enzyme species observed during the oxidative half-reaction in the (A) wild-type and (B) N78S proteins. Anerobic enzymes (12–15 μm protein in 50 mm Tris/HCl, pH 8.0, 100 mm NaCl, 5% glycerol, 3 mm β-mercaptoethanol) solutions were anaerobically reduced with NADPH and subsequently mixed with oxygenated buffer (125 μm oxygen). Numerical integrations (Pro-K software, Applied Photophysics Ltd) yielded a three-step model for wild-type protein in which the first step reflects the formation of the flavin-hydroperoxide (A → B = 8 s−1). The rates for the B → C and C → D are 4 s−1 and 0.5 s−1, respectively. Their values are independent of oxygen concentrations. The analogous kinetic data of N78S mutant were best fitted using a single-step model (Table 1).
Crystallization and Structure Determination
All crystals were obtained using microbatch technique under 100% paraffin oil at 4 °C by mixing equal volumes of 8 mg protein/ml in 25 mm Tris/HCl, pH 8.0, 250 mm NaCl, 1 mm NADP+ (or analog), and 20% (w/v) PEG4000 in 100 mm Na/HEPES pH 7.5 as described (13). Data were collected at the European Synchrotron Radiation Facility (Grenoble, France) and processed with the CCP4 package (15). The structures were solved by difference Fourier methods. Phases were further improved by density 4-fold averaging with DM (16). The programs COOT (17) and Refmac5 (18) were used for model rebuilding and refinement, which was carried out using strict non-crystallographic symmetry restraints (Table 2). Figures were generated using PyMOL (DeLano, 2002 on World Wide Web).
TABLE 2.
Crystallographic data collection and refinement statistics
| N78S | N78D | N78K | APADP+ | ThioNADP+ | |
|---|---|---|---|---|---|
| PDB code | 2xlp | 2xlr | 2xls | 2xlt | 2xlu |
| Unit cell (Å; space group P61) | a = b = 219.7 c = 131.4 | a = b = 214.1 c = 152.8 | a = b = 219.8 c = 131.2 | a = b = 219.7 c = 130.6 | a = b = 220.7 c = 130.5 |
| Resolution (Å) | 2.8 | 2.5 | 3.0 | 2.2 | 2.6 |
| Rsyma,b (%) | 11.5 (48.1) | 10.2 (49.9) | 14.2 (49.9) | 10.0 (33.5) | 12.5 (40.9) |
| Completenessb (%) | 99.9 (100) | 100 (100) | 100 (100) | 99.9 (100) | 99.9 (100) |
| Unique reflections | 88,448 | 129,218 | 72,308 | 180,729 | 110,842 |
| Redundancyb | 5.0 (5.0) | 3.7 (3.8) | 3.7 (3.7) | 4.3 (4.3) | 5.0 (4.9) |
| I/σb | 10.5 (3.2) | 8.9 (2.9) | 9.0 (3.6) | 11.3 (4.0) | 9.7 (3.7) |
| N° of atoms | 15,209 | 15,348 | 15,065 | 16,288 | 15,494 |
| Average B value (Å2) | 40.4 | 41.8 | 31.9 | 24.6 | 28.5 |
| Rcrystc (%) | 19.3 | 21.4 | 18.4 | 17.4 | 21.0 |
| Rfreec (%) | 21.4 | 24.4 | 21.1 | 20.2 | 25.2 |
| Rms bond length (Å) | 0.016 | 0.024 | 0.022 | 0.014 | 0.020 |
| Rms bond angles (°) | 1.7 | 1.9 | 1.9 | 1.6 | 2.0 |
a Rsym = Σ|Ii − <I>|/ΣIi, where Ii is the intensity of ith observation and <I> is the mean intensity of the reflection.
b Values in parentheses are for reflections in the highest resolution shell.
c Rcryst = Σ|Fobs − Fcalc|/ΣFobs where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. Rcryst and Rfree were calculated using the working and test set, respectively.
RESULTS
Functional Properties of Wild-type mFMO
To provide a starting point for our studies, we have first investigated the enzymatic properties of the wild-type enzyme. Steady-state kinetic analysis revealed that, similar to mammalian FMOs, trimethylamine, and NADPH are efficient substrates (Table 1). Furthermore, it was found that the enzyme has considerable NADPH oxidase activity as gathered from the rate of NADPH depletion in the absence of an organic substrate (kuncoupling = 0.5 s−1, Table 1). The ability of mFMO to act as NADPH oxidase was further confirmed by measuring the formation of hydrogen peroxide, which approximately equaled the amount of consumed NADPH.
The presteady-state analysis using the stopped-flow technique showed that mFMO is rapidly and almost fully reduced by NADPH (kred in Table 1). The reductive-half reaction data also indicated that the enzyme has a high affinity for the reduced nicotinamide coenzyme. Titration of the oxidized mFMO revealed a somewhat lower affinity for NADP+ (Table 1). The NADPH-reduced protein was employed to monitor the oxidative half-reaction of mFMO, which can be best described by a three-step process (rate constants are listed in Table 1 and Fig. 2A). At first, the relatively stable flavin-hydroperoxide (or flavin peroxide) intermediate is rapidly formed (spectrum B in Fig. 2A) with a typical absorbance maximum at around 360 nm. Next, the intermediate is converted to a second species (spectrum C), which might represent a conformational change occurring during re-oxidation and/or protonation of the intermediate (i.e. conversion of flavin peroxide into flavin hydroperoxide). A partly similar spectral intermediate has been observed also in the oxidative half-reaction of the pig-liver FMO (10). Finally, the enzyme is completely re-oxidized (spectrum D). It is of note that the actual reaction of the flavin with oxygen (i.e. formation of the flavin-hydroperoxide; A → B in Fig. 2A) linearly depends on the oxygen concentration as typically found in flavin-dependent oxidases and monooxygenases (rate constant kox of 6.5 × 104 m−1 s−1, Table 1). By repeating the experiments with 1 mm trimethylamine, it was found that kox does not depend on the binding of substrate. This is in contrast with flavoprotein hydroxylases and P450 monooxygenases but is fully in line with previous kinetic studies on eukaryotic FMOs (10). Consistent with this mode of function, the three-dimensional structure of mFMO exhibits a two-domain organization that defines a large cleft at the domain interface (Fig. 1B). At the bottom of this cleft, a small pocket is defined by the nicotinamide moiety of NADP+, the side chain of Asn-78, and the N5-C4a flavin atoms. This is the site where dioxygen reacts with the reduced flavin forming the flavin-hydroperoxide, which remains exposed to the incoming substrate (Fig. 3A).
FIGURE 3.

Structural data on mFMO mutants. The coloring is as in Fig. 1B with the carbons of highlighted residues in green. A, structure of N78S mutation does not perturb the protein conformation. The picture shows the superposition of the N78S structure onto that of the wild-type enzyme in the same orientation as in Fig. 1B. The superposition yields a root-mean-square deviation of 0.16 Å for 443 Cα atoms (with reference to monomer A of the tetrameric enzyme). For the sake of clarity, only the side chain of Ser-78 of the mutant enzyme is shown (carbons in green). The wild-type structure is colored as in Fig. 1B. The carboxamide group of NADP+ is oriented with its -NH2 group pointing toward the flavin ring as to make H-bonds (dashed lines) with the flavin N5 and O4 atoms. We cannot fully rule out that upon flavin reduction, the carboxamide flips so that its carbonyl oxygen (rather than the amide) would point toward the flavin. However, this would cause an unfavorable short contact between the NADP+ oxygen and flavin O4 and between the NADP+ nitrogen and Arg-413. B, structure of the N78D mutant exhibits an altered NADP+ binding mode with the nicotinamide ring flipped out toward the surface. This alteration does not involve any large conformational change in the protein (root-mean-square deviation of 0.37 Å). The coloring is as in Fig. 1B with the carbons of Asp-78 in green. C, N78K mutant displays a disordered nicotinamide ring (which is therefore not shown) and Tyr-212 oriented toward the flavin to partly occupy the nicotinamide site. To illustrate the underlying conformational change, the Tyr-212 side chain and NADP+ in the positions as observed in the wild-type enzyme are shown as black thin sticks. The terminal oxygen of Lys-78 is not fully ordered in the crystal structure. It might form a H-bond with the NADP+ ribose.
The N78S Mutant
The position of Asn-78 in direct contact with the C4a atom of the flavin and close to the NADP+ nicotinamide ring. This indicated that this side chain could play a key role in the reaction with dioxygen (Figs. 1 and 3A). Furthermore, two mutations affecting the corresponding residue in human FMO3 have been identified in TMAU patients (19, 20, 21). Thus, Asn-78 represented an obvious target for site-directed mutagenesis.
At first, we generated the N78S mutation, which is one of the mutations known to be the cause of TMAU. It is a relatively conservative amino acid replacement that does not greatly perturb the bulkiness of the side chain and maintains a group capable of H-bonding interactions. Nevertheless, we found that the mutant is inactive against various potential FMO substrates (trimethylamine, nicotine, methimazole, and indole). The mutant was further investigated for its reactivity toward NADPH and oxygen. Steady-state kinetics showed that N78S has oxidase activity with a Km for NADPH of about 4 μm and a rate constant (kuncoupling) of 0.7 s−1, which is very similar to that of the wild-type (Table 1). Stopped-flow kinetics confirmed that the mutant enzyme readily reacts with NADPH. It also demonstrated that the enzyme reacts with oxygen albeit less efficiently than the wild-type (13-fold lower kox, Table 1) and, most importantly, without any detectable flavin-hydroperoxide intermediate (Fig. 2B). The kinetic data for the re-oxidation reaction could be best fitted by a one-step process implying that, if the flavin-hydroperoxide forms, it decays rapidly being unable to sustain substrate oxygenation.
The crystallographic analysis of the protein in complex with NADP+ indicated no conformational changes both in the overall structure and locally in the active site (Fig. 3A). In essence, placing a Ser in place of an Asn in position 78 slightly widens the oxygen site at the flavin C4a-N5 locus. The only detectable alteration outlined by the crystallographic data concerns the NADP+ nicotinamide ring, which is poorly defined by the electron density whereas the ADP-ribose moiety is clearly visible in the density map. This feature indicates that the nicotinamide is partly disordered. The main lesson from the N78S analysis is that an apparently small alteration in the flavin C4a environment is sufficient to convert FMO into a relatively efficient oxidase, explaining why the corresponding N61S in human FMO3 leads to enzyme deficiency in patients affected by TMAU (20, 21).
Adding Charges: N78D and N78K
Having analyzed the effect of a small hydrophilic side chains at position 78, we further probed this site by introducing charged side chains. We generated and studied the mutations N78D and N78K, the latter also being found in TMAU patients (19). Both mutants turned out to be unable to catalyze trimethylamine monooxygenation but exhibited drastically different enzymatic properties.
N78D functions as moderately efficient oxidase (kuncoupling of 0.19 s−1) although it binds NADPH with a >20-fold worse affinity than the wild-type (Table 1). As for the N78S protein, the presteady-state kinetics showed that the N78D mutant is unable to effectively stabilize the flavin-hydroperoxide with the re-oxidation process occurring in one step without detectable intermediates (kox of 2200 m−1 s−1). These functional properties can be evaluated in light of the crystallographic analysis of the mutant enzyme bound to NADP+. The structure displays a peculiar feature: the nicotinamide is flipped out and hosted at the rim of the active site cleft (Fig. 3B). This binding mode is brought about by a rotation of the nicotinamide ribose, which does not affect the conformation of the ADP fragment of NADP+. In this position, the cofactor carboxamide is engaged in a H-bond with the Asp-78 side chain, which compensates for the nicotinamide positive charge. As a result, the flavin N5 atom remains exposed to solvent without the shielding exerted by NADP+ as in the wild-type protein (Fig. 3A). Although this binding mode does not require significant conformational changes in the active site residues, it is evident that such a modified NADP+-binding drastically alters the structure, shape, and electrostatics of the active site which becomes unable to promote accumulation of the flavin-hydroperoxide intermediate. This fully supports the idea that proper NADP+ binding is an essential element for the oxygenation reaction.
The N78K mutant exhibited strongly altered catalytic properties and remarkable structural features. In addition of being inactive as monooxygenase, the protein is very inefficient in using NADPH for flavin reduction (Table 1). Equally impaired is the reaction of the NADPH-reduced enzyme with oxygen, which occurs without any observable intermediate at a rate (kox of 670 m−1 s−1), which is not dissimilar to that of the free flavin in water. The structural data further emphasize the large perturbation caused by the mutation (Fig. 3C). The Lys-78 side chain points toward the nicotinamide ribose possibly forming a H-bond with the ribose hydroxyl groups (the NΕ atom of Lys-78 has only weak electron density). The nicotinamide of NADP+ is disordered, lacking any significant electron density. This feature is coupled to a conformational change in the position of Tyr-212, which swings into active site pointing toward to interior of the protein (Fig. 3C). This conformation of Tyr-212 combined with the bulkiness of Lys-78 makes the flavin C4a very crowded and inaccessible, which accounts for the poor reactivity of this mutant with dioxygen.
The structure of the N78K mutant indicates that Tyr-212 may function as an active-site backdoor. In this scenario, the conformation found in the N78K protein may well correspond to that present in the NADP+-free enzyme. Thus, the flavin of the unliganded enzyme is predicted to be partly shielded by Tyr-212, which, upon nicotinamide cofactor binding, swings out to position itself above the nicotinamide ring, establishing favorable stacking interactions with the nicotinamide ring (Fig. 3, A and C). Tyr-212 could also play a role in product release, displacing NADP+. A fascinating observation concerning this aspect is that the function of Tyr-212 of mFMO is reminiscent of that played by an active-site tyrosine in human glutathione reductase, a flavoenzyme that uses NADPH as flavin reductant and shares the same dinucleotide-binding topology as mFMO (22).
Modifying the Substrate: NADPH Analogues
Having established how critical is the environment around the flavin C4a atom for the oxygen reaction and for attaining proper NADP+ binding, we investigated the effects on catalysis exerted by alterations in the NADP(H) substrate. To this aim, we employed two NADP(H) analogues, thioNADP(H) and APADP(H) (Fig. 4A). Both compounds, in the oxidized form, bind tightly to the protein as inferred by the significant increase (5–7 °C) in the protein melting temperature measured by ThermoFAD method as reported (23).
FIGURE 4.

Binding of NADP+ analogues to mFMO. A, chemical formulas of the two compounds using for this study (APADP+ on the left and thioNADP+ on the right). B, structure of the complex with APADP+ in approximately the same orientation and coloring scheme as in Fig. 1B. The carbons of APADP+ are in green. H-bonds are shown as dashed lines. C, comparison between the binding of NADP+ and APADP+ using a composite picture that shows the protein surface of the wild-type protein bound to NADP+ (carbons in cyan) and FAD (carbons in yellow). The pyridine ring of APADP+ is superimposed to highlight its different position at the rim of the active-site cleft surface. The NADP+ and APADP+ complexes are very similar (root-mean-square deviation of 0.21 Å for the Cα atoms).
The crystal structure of the complex with thioNADP+ revealed that the dinucleotide binds exactly as NADP+. The only noticeable difference concerns the pyridine ring, which is disordered as gathered from the lack of well-resolved electron density in all four crystallographically independent subunits present in the crystal asymmetric unit (Table 2). We found that thioNADPH is an efficient substrate both in the oxidase and in the monooxygenase assays (Table 1). Thus, replacement of the oxygen atom of the nicotinamide with a bulkier sulfur does not seem to significantly affect catalysis (Fig. 4A).
APADP(H) turned out to be a more insightful compound to probe the enzymatic properties. First of all, the reduced form of this cofactor analog (both commercially obtained or generated enzymatically) is unable to sustain any monooxygenase activity. Instead, we found that it can function as substrate in the oxidase assay with a rate constant close to that measured with NADPH (kuncoupling in Table 1) implying that APADPH can reduce the flavin. The complex of mFMO with APADP+ produced crystals of very good quality resulting in excellent electron density maps that were even better than those obtained with the NADP+-bound proteins. The cofactor analog binds with the ADP-ribose group in the standard conformation (Fig. 4B). Conversely, the pyridine ring is positioned at the entrance of the active site cleft in a binding mode, which is almost identical to that observed for NADP+ in the structure of the N78D mutant (Figs. 3B and 4, B and C). This conformation enables Asn-78 to form a H-bond interaction with the carbonyl group of APADP+, leaving the flavin ring unprotected. Combining these biochemical and structural data leads to conclusion that the reduced APADPH must be able to adopt the conformation required for reducing the flavin (Fig. 1A). However, after flavin reduction, the pyridine moiety moves away from the flavin. A reason for the differing binding modes between NADP+ and APADP+ might be that the acetyl group of APADP+ is unable to donate a H-bond to the flavin N5 (Figs. 1A, 3A, and 4B). This can be expected to favor the “flipped out” conformation which allows the APADP+ acetyl group to H-bond to Asn-78 (Fig. 4B). As for the N78D mutant, such a “out” conformation is unable to support oxygenation because it does not generate the proper environment for flavin-hydroperoxide stabilization and, possibly, because it may hamper the access of the organic substrate to the flavin (Fig. 4C). On the other hand, the flavin remains accessible to dioxygen and thereby competent in the oxidase reaction.
DISCUSSION
The specific objective of our study was to investigate the reaction of FMO with dioxygen using site-directed mutagenesis and NADP(H) analogues as dissecting tools. The general aim was to improve our knowledge of the molecular basis of TMAU and to advance our current understanding of the oxygen reactivity in flavoenzymes. Along these lines, we can draw several conclusions.
The analysis of the Asn-78 mutants indicates that at least some of the TMAU-causing mutations target the Achilles heel of the enzyme: the intrinsic “fragility” of the crucial flavin-hydroperoxide intermediate, which, if not appropriately stabilized, causes the conversion of the enzyme into an oxidase.
In this scenario, the fact that the devil is in the detail is best illustrated by the N78S variant (Figs. 2A and 3A). This mutation does not affect the steric hindrance and hydrophilic nature of the side chain and does not cause any detectable change in the active site conformation. Nevertheless, it fully inactivates the enzyme. Evidently, the fine details in the environment of the C4a atom (Fig. 1A) can make a tremendous difference in the flavin reactivity. In this case, it appears that a Ser side chain in position 78 is unable to provide the precisely positioned H-bond partner required to stabilize the flavin-hydroperoxide, whose terminal oxygen can accept and donate H-bonds (Figs. 1A and 3A). Therefore, the intermediate does not form at all or, more likely, it decays too rapidly to sustain monooxygenation (Fig. 2B). As a result, the mutant enzyme retains (albeit less efficient) reactivity with oxygen but only to function as oxidase and not as monooxygenase (Table 1).
The N78D mutant illustrates the role of electrostatics. This isosteric mutation positions a negative charge close to the N5 with to a 30-fold reduction in the kox (Table 1). As observed for other flavoenzymes, it is likely that this mutation disfavors the first step of the oxygen reaction; the one-electron transfer leading to the formation of flavin semiquinone-superoxide caged radical pair that subsequently generates the flavin-hydroperoxide and/or hydrogen peroxide (24, 25).
Monooxygenation requires that NADP+ binds in the proper conformation as outlined by the study of the N78D mutant and the experiments with the APADP+ analog (Figs. 3B and 4B). The role of NADP+ is manifold: (i) together with Asn-78, it creates the niche for oxygen binding in front of the flavin C4a atom, (ii) its ribose 2′-hydroxyl group provides a potential H-bonding partner for the flavin-hydroperoxide atoms (13), and (iii) it forms key H-bond interactions with the flavin N5 and O4 atoms (Fig. 1B). This latter feature should be emphasized because it can have a crucial role. It is known that protection of the flavin N5 atom can enormously enhance the stability of the flavin-hydroperoxide to the point that the reaction of oxygen with protein-free reduced N5-alkylriboflavin forms a very stable and biocatalytically useful hydroperoxide derivative (26, 27). Although the N5 atom of the two-electron reduced flavin has a very high pKa value (around 20) (28), adequate protection of this atom is probably needed to inhibit rapid proton exchange with the solvent which may concomitantly trigger decay of the flavin-hydroperoxide. Likewise, protection of the N5 may also prevent that this atom directly takes part in the oxygen reaction for example by acting as hydrogen or proton donor.
The comparison of the NADP+ and APADP+ complexes (Fig. 4C) provides a glimpse of the likely mode of binding of the organic substrate. Indeed, as visualized by Fig. 4C, the “out” position of the APADP+ pyridine moiety corresponds to the location expected for an organic substrate (for example indole or nicotine) to be monooxygenated by the enzyme. The substrate promiscuity of FMOs, which act on a variety of diverse molecules, seems to inevitably expose these enzymes to the risk of binding the pyridine ring of the NADP(H) ligand in the catalytically incorrect positions as indicated by the mutagenesis and NADP-analog studies. In this context, it is remarkable that the mFMO appears to use a Tyr residue as a backdoor that sandwiches the nicotinamide in the H-bonding position with N5 (Fig. 3C).
The more general conclusion from this analysis is that the two alternative pathways (oxidation versus monooxygenation) along the oxygen reaction can be predicted to have very similar energy barriers so that subtle alterations in the balance of interactions around the C4a-N5 atoms of the flavin can drastically alter the outcome of the reaction of the reduced enzyme with dioxygen. This will be a crucial aspect to be considered in the design and development of FMOs and related monooxygenases for biocatalytic application.
Acknowledgment
We thank Dr. Andrea Alfieri for contributions in the earlier stages of the project.
This work was supported by the Italian Ministry of Science (PRIN08 program), Fondazione Cariplo, EU-FP7 (Oxygreen), and the American Chemical Society Petroleum Research Fund (46271-C4).
- FMO
- flavin-containing monooxygenase
- FMO3
- isozyme 3 of human FMO
- APADP
- 3-acetylpyridine adenine dinucleotide
- mFMO
- FMO from Methylophaga sp. strain SK1
- TMAU
- trimethylaminuria.
REFERENCES
- 1.Cashman J. R., Zhang J. (2006) Annu. Rev. Pharmacol. Toxicol. 46, 65–100 [DOI] [PubMed] [Google Scholar]
- 2.Krueger S. K., Williams D. E. (2005) Pharmacol. Ther. 106, 3573–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips I. R., Shephard E. A. (2008) Trends Pharmacol. Sci. 29, 294–301 [DOI] [PubMed] [Google Scholar]
- 4.Schlaich N. L. (2007) Trends Plant Sci. 12, 412–448 [DOI] [PubMed] [Google Scholar]
- 5.Al-Waiz M., Ayesh R., Mitchell S. C., Idle J. R., Smith R. L. (1987) Lancet. 1, 634–635 [DOI] [PubMed] [Google Scholar]
- 6.Dolphin C. T., Janmohamed A., Smith R. L., Shephard E. A., Phillips I. R. (1997) Nat. Genet. 17, 491–494 [DOI] [PubMed] [Google Scholar]
- 7.Ziegler D. M. (2002) Drug Metab. Rev. 34, 503–511 [DOI] [PubMed] [Google Scholar]
- 8.Beaty N. B., Ballou D. P. (1980) J. Biol. Chem. 255, 3817–3819 [PubMed] [Google Scholar]
- 9.Beaty N. B., Ballou D. P. (1981) J. Biol. Chem. 256, 4611–4618 [PubMed] [Google Scholar]
- 10.Beaty N. B., Ballou D. P. (1981) J. Biol. Chem. 256, 4619–4625 [PubMed] [Google Scholar]
- 11.Poulsen L. L., Ziegler D. M. (1979) J. Biol. Chem. 254, 6449–6455 [PubMed] [Google Scholar]
- 12.Choi H. S., Kim J. K., Cho E. H., Kim Y. C., Kim J. I., Kim S. W. (2003) Biochem. Biophys. Res. Commun. 306, 930–936 [DOI] [PubMed] [Google Scholar]
- 13.Alfieri A., Malito E., Orru R., Fraaije M. W., Mattevi A. (2008) Proc. Natl. Acad. Sci. U.S.A. 195, 6572–6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forneris F., Binda C., Vanoni M. A., Battaglioli E., Mattevi A. (2005) J. Biol. Chem. 280, 41360–41365 [DOI] [PubMed] [Google Scholar]
- 15.CCP4 (Collaborative Computational Project, Number 4) (1994) Acta. Crystallogr. D. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 16.Cowtan K. (2010) Acta. Crystallogr D. 66, 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Acta. Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vagin A. A., Steiner R. S., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov N. (2004) Acta. Crystallogr D. 60, 2284–2295 [DOI] [PubMed] [Google Scholar]
- 19.Koukouritaki S. B., Poch M. T., Henderson M. C., Siddens L. K., Krueger S. K., VanDyke J. E., Williams D. E., Pajewski N. M., Wang T., Hines R. N. (2007) J. Pharmacol Exp. Ther. 320, 266–273 [DOI] [PubMed] [Google Scholar]
- 20.Yeung C. K., Adman E. T., Rettie A. E. (2007) Arch. Biochem. Biophys. 464, 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolphin C. T., Janmohamed A., Smith R. L., Shephard E. A., Phillips I. R. (2000) Pharmacogenetics 10, 799–807 [DOI] [PubMed] [Google Scholar]
- 22.Pai E. F., Schulz G. E. (1983) J. Biol. Chem. 258, 1752–1757 [PubMed] [Google Scholar]
- 23.Forneris F., Orru R., Bonivento D., Chiarelli L. R., Mattevi A. (2009) FEBS J. 276, 2833–2840 [DOI] [PubMed] [Google Scholar]
- 24.Mattevi A. (2006) Trends Biochem. Sci. 31, 276–283 [DOI] [PubMed] [Google Scholar]
- 25.Massey V. (1994) J. Biol. Chem. 269, 22459–22462 [PubMed] [Google Scholar]
- 26.Kemal C., Chan T. W., Bruice R. C. (1977) J. Am. Chem. Soc. 99, 7272–7286 [DOI] [PubMed] [Google Scholar]
- 27.Imada Y., Iida H., Murahashi S., Naota T. (2005) Angew. Chem. Int. Ed. Engl. 44, 1704–1706 [DOI] [PubMed] [Google Scholar]
- 28.Macheroux P., Ghisla S., Sanner C., Rüterjans H., Müller F. (2005) BMC Biochem. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]