

Abstract
In Saccharomyces cerevisiae, silent chromatin is formed at HMR upon the passage through S phase, yet neither the initiation of DNA replication at silencers nor the passage of a replication fork through HMR is required for silencing. Paradoxically, mutations in the DNA replication processivity factor, POL30, disrupt silencing despite this lack of requirement for DNA replication in the establishment of silencing. We tested whether pol30 mutants could establish silencing at either replicated or non-replicated HMR loci during S phase and found that pol30 mutants were defective in establishing silencing at HMR regardless of its replication status. Although previous studies tie the silencing defect of pol30 mutants to the chromatin assembly factors Asf1p and CAF-1, we found pol30 mutants did not exhibit a gross defect in packaging HMR into chromatin. Rather, the pol30 mutants exhibited defects in histone modifications linked to ASF1 and CAF-1-dependent pathways, including SAS-I- and Rtt109p-dependent acetylation events at H4-K16 and H3-K9 (plus H3-K56; Miller, A., Yang, B., Foster, T., and Kirchmaier, A. L. (2008) Genetics 179, 793–809). Additional experiments using FLIM-FRET revealed that Pol30p interacted with SAS-I and Rtt109p in the nuclei of living cells. However, these interactions were disrupted in pol30 mutants with defects linked to ASF1- and CAF-1-dependent pathways. Together, these results imply that Pol30p affects epigenetic processes by influencing the composition of chromosomal histone modifications.
Keywords: Chromatin, Epigenetics, Histone Acetylase, Histones, Sirtuins, ASF1, PCNA, RTT109, SAS-I, Silencing
Introduction
To regulate expression of a gene epigenetically, cells must efficiently assemble specialized chromatin at those genes during or shortly after DNA replication each cell cycle. This process ensures that the gene expression state is inherited in future generations. In the budding yeast Saccharomyces cerevisiae, an epigenetic process called silencing prevents transcription of the mating-type genes at the silent mating-type loci HMR and HML (1). When silent chromatin is first formed, the silent information regulator proteins Sir1p, Sir2p, Sir3p, and Sir4p are recruited to the HM loci through their physical interactions with proteins bound to DNA elements called silencers adjacent to the HM loci. At HMR, the four Sir proteins interact with each other and with the origin recognition complex (ORC),2 Rap1p and Abf1p bound to the E silencer (2–9). Sir2p, Sir3p, and Sir4p then spread along the chromosome and across the mating-type genes a2 and a1 at HMR as the deacetylase Sir2p removes acetyl groups from histones H3 and H4 and creates binding sites for Sir3p and Sir4p on nucleosomes (7, 9) (see also Ref. 5). Once silent chromatin is formed upon passage through S phase, the a2 and a1 genes at HMR are inactivated and their silenced state will be inherited as the genome is duplicated in subsequent generations (1, 10–14).
Although DNA replication itself is not required to establish silent chromatin in S phase (10–12), a link between DNA replication and silencing in S. cerevisiae has been reinforced by numerous studies. In yeast, many mutated or misexpressed replication-related genes affect silencing, including those encoding proteins involved in initiating DNA replication, in leading and lagging strand DNA synthesis, and in deposition of histones onto newly synthesized DNA (15–21). In this study, we explored how PCNA, an evolutionarily conserved protein central to each of these events during DNA replication, contributes to the formation of silent chromatin.
PCNA, which is encoded by the yeast POL30 gene, serves as an accessory factor for DNA polymerases δ and ϵ and acts by tethering these polymerases to their template DNA during DNA replication, thus enhancing their processivity (22–24). During DNA replication, PCNA is loaded as a trimer around DNA forming a homotrimeric, sliding, ring-shaped clamp (25, 26). RF-C is the primary clamp loader complex that mediates the association of PCNA with DNA (25, 27). In addition, PCNA tethers several other proteins to DNA, including Rad27p (FEN1 in mammals) and Cdc9p (DNA ligase I), which together process and ligate newly synthesized Okazaki fragments during lagging strand synthesis (28, 29) (for review see Refs. 30 and 31). PCNA also binds the chromatin assembly factor complex CAF-1 (20, 32–34). CAF-1 consists of three subunits (Cac1p, Cac2p, and Cac3p), acts as a chaperone for newly synthesized histones H3 and H4, contributes to replication- and repair-coupled nucleosome assembly (34–36), and can act synergistically with a second chromatin assembly factor, Asf1p, to achieve this feat (37, 38). Several of these POL30-interacting factors also affect silencing when mutated or misexpressed (17–19).
The varying phenotypes of pol30 mutants (19, 20, 38–40) indicate that PCNA can influence silencing and other cellular processes by multiple mechanisms. The silencing defects of several POL30 mutants, including pol30-8, pol30-6, and pol30-79, have been linked to CAF-1- or ASF1-dependent pathways (20, 38). Each of these pol30 mutants has defects in binding the large subunit of the CAF-1 chromatin assembly factor complex, Cac1p (20). Additionally, although no direct interaction between Asf1p and Pol30p has yet been reported, Asf1p can bind to the clamp loader RF-C (41) as well as to Cac2p (32, 37, 42) and can stimulate CAF-1-dependent chromatin assembly (37, 38, 43). In addition, the mammalian Asf1 interacts with histones and the putative replicative helicase Mcm2–7 (44).
PCNA influences the construction of heritable chromatin structures in multiple organisms. Mutation of the PCNA ortholog mus209 suppresses position-effect variegation in Drosophila (45), and PCNA is linked to both DNA- and histone-modifying enzymes associated with epigenetic processes in mammals. Mammalian PCNA binds to and stimulates the activity of DNMT1, a DNA methyltransferase that maintains heritable methylation patterns on CpG islands (46–49). In vitro, PCNA and DNMT1 interact with the histone deacetylase HDAC1, which also functions in transcriptional silencing, and these proteins co-localize in vivo (46, 50, 51). PCNA also recruits human CAF-1 to DNA and serves as a mark for CAF-dependent chromatin assembly in vitro (33, 34). CAF-1, in turn, targets the methyl CpG-binding protein MBD1 and the H3-K9-specific methyltransferase SETDB1 to replication foci. When combined with DNA methylation, MBD1 and SETDB1 promote stable transcriptional silencing (30, 52). In addition, the mammalian H4-K20-specific methyltransferase SET8 binds to PCNA (53). Methylation of H4-K20, in turn, has also been associated with heterochromatin in multiple organisms (Ref. 54 and references within). Thus, PCNA plays a central role in epigenetic processes through recruiting chromatin-modifying machinery to replication foci.
Together, these observations compelled us to explore the role of POL30 in silencing and to test whether DNA replication was required for pol30-dependent silencing defects in yeast. Surprisingly, pol30 mutants were defective in establishing silencing at HMR, regardless of its replication status upon passage through S phase. Single molecule analyses (fluorescence lifetime imaging microscopy and fluorescence resonance energy transfer (FLIM-FRET)) revealed that PCNA interacted with the acetyltransferases Rtt109p and SAS-I in vivo but pol30p mutants had defects in these interactions. Rtt109p- and SAS-I-dependent histone modifications were also reduced in chromatin isolated from pol30 mutants, implying that histone acetylation defects linked to Asf1p- and CAF-dependent pathways influence silencing in these mutants.
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
Yeast strains were generated by standard genetic techniques including homologous recombination, one-step gene replacement, and plasmid shuffling (55–58). Genotypes of parental strains are described in supplemental Table 1, and plasmids used in this study are described in supplemental Table 2.
Plasmid pAK876 was derived from pAK196 (11), which contains an EcoRI-HindIII fragment of a modified HMR locus containing a synthetic silencer with four Gal4p binding sites, a Rap1p binding site, and an Abf1p binding site in place of the HMR-E silencer and the genes encoding a2 and a1 but lacking the HMR-I silencer. This modified HMR is flanked by two FRT sites oriented to permit excision of a 2.6-kb covalently closed circular double-stranded DNA molecule from chromosome III in the presence of FLP recombinase. In pAK876, a PstI-XmaI fragment within this modified HMR that contained the a2-a1 promoter region was replaced with 266 bp of heterologous DNA amplified from the 5′ region of bla from pUC19 using oALK441 5′-CATGCCTGCAGGTCGACTCTGTCGTGTAGATAACTACGATAC-3′ and oALK442 5′-TCGGTACCCGGGGATCCTCTCGACGAGCGTGACACCACGATG-3′. Excision of this modified HMR from chromosome III by FLP recombinase will also generate a 2.6-kb covalently closed circular double-stranded DNA molecule. Plasmid pAK928 (H3 K14R/H4) was derived from pWZ-414-F13 (59) by site-directed mutagenesis according to the QuikChange site-directed mutagenesis kit protocol (Stratagene, La Jolla, CA) using oligonucleotides oALK613 5′-AAATCCACTGGTGGTAGAGCCCCAAGAAAACAAT-3′ and oALK614 5′-ATTGTTTTCTTGGGGCTCTACCACCAGTGGATTT-3′; plasmid pAK929 (H3 K14R/H4 K16R) was derived from pWZ-414-F24 (59) using the same oligonucleotide pair. Plasmid pAK995 (H3 K9Q/H4) was derived from pPK189 as described above using oligonucleotides oALK753 5′-CAAACAGCTAGACAATCCACTGGTGGTAAAGCCC-3′ and oALK754 5′-GGGCTTTACCACCAGTGGATTGTCTAGCTGTTTG-3′. Plasmid pAK1008 (H3 K9Q K56R/H4) was derived from pAK995 as described above using previously described oligonucleotides oALK691 and oALK692 (40). Plasmids pAK1004 (H3 K9R,K56Q/H4) and pAK1005 (H3 K9R,K56R/H4) were derived from pAK873 (H3 K9R/H4) as described above using previously described oligonucleotides oALK691 and oALK692 or oALK642 and oALK643, respectively (40).
Plasmids pAK1105 and pAK1106 were constructed by amplifying POL30-GFP from yAK5110 using oALK1038 5′-CTGCAGCCCGGGGGATCCACTAGTTCTAGAGCGGCCGCCAATGCTACACGTGCTTGA-3′ and oALK1040 5′-GAATTCGATATCAAGCTTATCGATACCGTCGACACTAGTAGGTCTTAGTGTTGACTGTCA-3′. PCR products were digested with HindIII and BamHI and inserted into HindIII-BamHI of pRS415 or with SpeI and inserted into SpeI of pRS416. Plasmids expressing pol30-CFP mutants were generated from pAK1105 by site-directed mutagenesis according to the QuikChange site-directed mutagenesis kit protocol. pAK1107 (pol30-6-CFP) was made using oALK1026 5′-CAAGCTGCTGCTGCCTCAAGAGTTC-3′ and oALK1027 5′-GAACTCTTGAGGCAGCGACAGCTTG-3′; pAK1108 (pol30-79-CFP) using oALK1030 5′-GATGCTGATTTCGCAAAGGCTGAAGAATTACAG-3′ and oALK1031 5′-CTGTAATTCTTCAGCCTTTGCGAAATCAGCATC-3′; and pAK1126 (pol30-8-CFP) using oALK 1114 5′-TTCCAAGAATATGCATGTGCCCATCCTGTTACG-3′ and oALK 1115 5′-CGTAACAGGATGGGCACATGCATATTCTTGGAA-3′.
Strains encoding various combinations of Pol30-CFPp, Rtt109-YFPp, Sas4-YFPp, Sas5-YFPp, Trx3-CFPp, and Spc29-YFPp fusion proteins were generated by homologous recombination using PCR products generated from pKT212 or pKT211 (60) plus oligonucleotides complimentary to the 3′-ends of the specified loci (sequences available upon request) and by standard genetic crosses.
Cell Cycle Experiments
Cell cycle experiments were conducted as described previously (11, 61). In these experiments, silencing was regulated by expressing Gal4-Sir1p from the methionine-regulated promoter MET3. Cells were grown in synthetic complete medium lacking histidine and tryptophan and containing 2% glucose and methionine to repress Gal4-Sir1p expression. Cells were then collected by centrifugation, resuspended in synthetic complete medium lacking histidine and tryptophan and containing 65 μm methionine and 2% raffinose with 1 or 10 μg/ml α-factor, and incubated at 30 °C for approximately 3 h until >90% of the cells arrested in G1 phase and formed shmoos. To induce FLP recombinase to excise the HMR locus, galactose was added to the medium to a final concentration of 2% and cells were incubated for 1 to 1.5 h at 30 °C. Alternatively, to leave the HMR locus in the chromosome, cells were incubated further in medium containing 2% raffinose. Cells were concentrated onto 0.45-μm nitrocellulose filters (Whatman), washed with synthetic complete medium lacking histidine and tryptophan and containing 2% raffinose, resuspended in synthetic complete medium lacking histidine, tryptophan, and methionine and containing 2% raffinose plus 1 or 10 μg/ml α-factor, and incubated for 1 h at 30 °C to express Gal4-Sir1p in G1. Cells were then concentrated by filtration as described above, washed with synthetic complete medium lacking histidine, tryptophan, and methionine and containing 2% glucose, resuspended in synthetic complete medium lacking histidine, tryptophan, and methionine and containing 2% glucose plus 10 μg/ml Pronase, 30 μg/ml benomyl, and 10 μg/ml nocodazole, and incubated at 30 °C for 2 h. Samples were harvested at each time point for analysis of cell cycle arrests by microscopy or flow cytometry, and analysis of RNA to monitor silencing of a1 at the HMR locus and of DNA to monitor excision of HMR from chromosome III was conducted as described previously (11, 55, 61). Statistical analyses for cell cycle and topology assays were conducted using the Wilcoxon rank sum test with MSTAT v2.6.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation experiments were performed using two independent yeast strains for each genotype and analyzed by real-time PCR on an ABI Prism 7000 as described previously (61, 62) using anti-Pol30p (41, 63), anti-Sir2p, and anti-Sir3p antibodies (9) or anti-H3 antibodies (Abcam, catalog No. ab1791). Cells were grown in synthetic complete medium containing 2% glucose and lacking histidine or histidine and methionine. Oligonucleotides used for chromatin immunoprecipitation analyses at MAT and HMR have been described previously (40, 61, 62), and sequences are available upon request.
RNA Analyses
Total RNA was isolated from logarithmically growing cells, a1 mRNA levels relative to control SCR1 levels were analyzed by RNA blot assays or quantitative real-time PCR as described previously. Oligonucleotide sequences used to generate a1 and SCR1 probes and to conduct quantitative real-time PCR are available upon request (11, 61, 62, 64).
Confocal Fluorescence Lifetime Imaging Analysis
Yeast were grown logarithmically in synthetic complete medium with 2% glucose to an A600 of 0.8 prior to FLIM-FRET analysis. FLIM measurements (65) were performed on single living cells (n = 100–200) with a confocal setup using an inverted Olympus IX71 microscope (Center Valley, PA) equipped with a picosecond 40-MHz pulsed 467-nm diode laser (Microtime 200, PicoQuant GmbH, Berlin) for excitation at 3–10 microwatts of power (66). The laser beam was focused on the sample using an apochromatic 60× water immersion objective (1.2 N.A.); the emitted fluorescence was collected by the same objective and separated from the excitation beam by a dichroic mirror using appropriate band-pass filters (Omega Optical, Brattleboro, VT). Single-photon avalanche photodiodes (SPAD; SPCM-AQR, PerkinElmer Life Sciences) were used to record the emitted photons, and fluorescence was measured using the time-correlated single photon counting (TCSPC) module in the time-tagged time-resolved (TTTR) mode (Time Harp200, PicoQuant). To obtain fluorescence lifetimes, TCSPC decay curves were fitted by a double exponential function using SymphoTime software (version, 5.13, PicoQuant). The donor lifetimes were calculated in the absence and presence of the acceptor and FRET efficiency (E) and distance (R) between two energy-transferring molecules were then obtained from
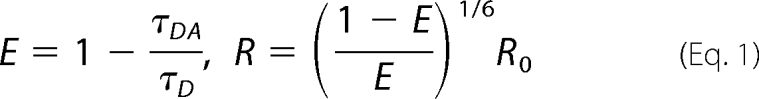 |
where τDA and τD are the donor excited state lifetime in the absence and presence of acceptor, respectively. R0 is the Förster distance (the distance at which 50% energy transfer between donor and acceptor exists). For CFP and YFP, this distance is 52 Å (67).
Extraction of Chromatin-associated Histones
Histones were isolated from the chromatin fraction of nuclei derived from logarithmic yeast cultures as described previously (40).
Protein Blot Analyses
Chromatin fractions or whole cell extracts from each strain were separated on 15% SDS-polyacrylamide gels and transferred to PVDF membranes (Bio-Rad). Membranes were blocked with a 1:1 dilution of Odyssey blocking buffer (Li-Cor Biosciences, catalog No. 927-40000) and 1× phosphate-buffered saline (137 mm NaCl, 2.7 mm KCl, 5.4 mm NaH2PO4, and 1.47 mm KH2PO4). For analysis of chromatin-associated histones, protein blots were probed with anti-acetylhistone H3 (Lys-9) antibodies (1:1000; Cell Signaling Technology, catalog No. 9671) or anti-Acetyl-Histone H4 (Lys-16) antibodies (1:5000; Upstate, catalog No. 07-329) using Alexa Fluor anti-rabbit IgG as the secondary antibody (1:2000 for analysis of H3-K9ac or 1:10,000 for analysis of H4-K16ac; Molecular Probes, catalog No. A21109). Membranes were stripped with 0.2 m NaOH at room temperature and reprobed with anti-histone H3 antibodies (1:30,000; Abcam, catalog No. ab1791) using Alexa Fluor anti-rabbit IgG as the secondary antibody (1:40,000). For analysis of whole cell extracts, protein plots were probed as above except with dilutions of 1:20,000 for anti-PCNA antibodies (41, 63) and 1:20,000 for anti-histone H3 antibodies. Antibodies were diluted in 50% Odyssey blocking buffer, 0.5× PBS, and 0.1% Tween 20 prior to use. Blots were analyzed using an Odyssey infrared imager and Odyssey software v1.2 according to manufacturer's instructions (Li-Cor Biosciences, Odyssey User Guide, version 1.2). Statistical analyses for quantitative protein blots were conducted using the Wilcoxon rank sum test.
Colony Color and Mating Assays
Colony color assays, conducted using two independent yeast strains for each genotype, were performed as outlined in Fig. 4 and Refs. 40 and 68. Briefly, logarithmically growing yeast containing ADE2 integrated between the E and I silencers at HMR were plated at a density of ∼300 cells/plate on rich medium (YPD) plates, incubated at 30 °C for 2 days, and stored at 4 °C for approximately 3 days prior to collecting images with a Leica MZ125 microscope and SPOT 4.1.1 imaging software. In this assay, red colonies indicated that HMR::ADE2 was silenced, white colonies indicated HMR::ADE2 was expressed, pink colonies indicated a defect in maintaining or inheriting silencing, and sectored colonies indicated a defect in establishing silencing at HMR::ADE2. Patch mating assays were conducted with two independent clones for each genotype as described in Fig. 5. Quantitative mating assays were performed in triplicate as described in Table 7 and supplemental Table 6 (40, 62, 68). Mating indicates that the HMR locus was silenced.
FIGURE 4.

POL30-dependent histone modifications influence silencing. A, map of HMR::ADE2. B and C, overlapping silencing defects in histone H3 K9R and pol30-8 mutants. D, overlapping silencing defects in histone H3 K9R and cac1Δ double mutants. Colony color assays were used to monitor silencing of the ADE2 gene integrated at HMR. Cells were grown logarithmically, plated onto rich medium (YPD), incubated at 30 °C for 2 days, and then stored at 4 °C for 3 days prior to acquiring images (see “Experimental Procedures”).
FIGURE 5.

Loss of H3-K9 acetylation restores silencing at HMRae**. MATα HMRae** strains expressing the indicated histone H3 mutants were grown on minimal medium with supplements for 24 h at 30 °C and then replicated to rich medium (YPD) or to MATa lawns (JRY2726) on minimal medium and incubated for 2 days at 30 °C. Restoration of silencing prevents expression of a1 from HMRae**, thereby enabling the MATα cells to mate and grow as diploids on minimal medium.
TABLE 7.
Hypoacetylated histone mutants rescue silencing at HMRae**
| Histones | HMR | Relative efficiency of matinga |
|---|---|---|
| H3/H4 | HMR | 1 |
| H3 K9R,K14R/H4 K16R | HMR | 0.73 ± 0.11 |
| H3/H4 | HMRae** | 0.011 ± 0.0051 |
| H3 K9R/H4 | HMRae** | 0.067 ± 0.032 |
| H3 K14R/H4 | HMRae** | 0.074 ± 0.055 |
| H3/H4 K16R | HMRae** | 0.070 ± 0.034 |
| H3 K14R/H4 K16R | HMRae** | 0.074 ± 0.060 |
| H3 K9R,K14R/H4 | HMRae** | 0.20 ± 0.12 |
| H3 K9R,K14R/H4 K16R | HMRae** | 0.16 ± 0.15 |
a The efficiency of mating of yeast containing MATα HMR plus wild-type histones H3 and H4 to tester strain JRY2726 (MATa) was determined relative to their plating efficiency on minimal (YM) medium containing supplements (45 ± 9.4%, n = 3) and was set to 1. The mating efficiency of each strain relative to MATα HMR is shown. Mean ± S.D., n = 3. Combining single mutations with each other correlated with enhanced silencing at HMRae** (p = 0.067; Jonckheere-Terpstra trend test).
RESULTS
pol30 Mutants Have Defects in Establishing Silencing on Non-replicated Templates
Although DNA replication is not required to establish silencing (10–12) (see also Refs. 14, 69, and 70), mutations in POL30 paradoxically lead to silencing defects (20, 38). It was possible that pol30 mutants prevented the establishment of silencing in a replication-dependent manner (i.e. through disruption of Sir association at HMR during passage of a replication fork). Alternatively, pol30 mutants may have had a defect in a replication-coupled process that resulted in a cellular state incompatible with silencing-replicated as well as non-replicated genes. Therefore, we asked whether silencing at a non-replicated HMR could be established in pol30 cells as opposed to a replicated HMR. To conduct these experiments, we introduced POL30 or pol30-6, pol30-8, or pol30-79 mutations (71, 72) into yeast containing a regulatable HMR locus (10, 11) (Fig. 1A). This HMR contains a synthetic E silencer with four Gal4p binding sites, a Rap1p and an Abf1p binding site (HMR-GalSS), and the a1 and a2 genes, but it lacks the I silencer. Sir protein association and silencing at this HMR can be regulated by expressing Gal4-Sir1p via the methionine-repressible promoter MET3 (9–11, 61). This regulatable HMR is also flanked by FRT binding sites for the Flp1p recombinase and can be excised from the chromosome upon inducing FLP1 via the GAL10 promoter (11, 61). When excised from the chromosome, this HMR, which lacks its own origin of DNA replication, is not replicated upon passage through S phase, yet it can still be silenced in a Sir protein-dependent manner by the time cells reach G2/M (11).
FIGURE 1.

pol30 mutants are defective in silencing a regulatable HMR locus. A, a regulatable HMR locus. The HMR contained a modified E silencer, HMR-GalSS, which comprised four Gal4 binding sites in place of the ORC binding site plus a Rap1p and an Abf1p binding site and the a1 and a2 genes, but lacked the I silencer. Expression of the chimeric Gal4-Sir1p via the MET3-repressible promoter in these cells enabled Sir protein association at HMR. Upon induction of Flp1p from the GAL10 promoter, Flp1p binds to the FRT sites that flank HMR and excises HMR from chromosome III as a 2.6-kb double-stranded circular DNA molecule. B, experimental strategy. Cells grown in methionine lacked Gal4-Sir1p and expressed a1 mRNA. Cells were arrested in G1 with α-factor. Flp1p expression was then induced with galactose to excise HMR from the chromosome in G1. Next, expression of Flp1p was repressed, and Gal4-Sir1p was induced in G1 in medium containing raffinose and lacking methionine. Finally, cells expressing Gal4-Sir1p were released from G1 and allowed to progress through the cell cycle until G2/M where they were rearrested with benomyl plus nocodazole.
Initially, to confirm POL30 mutants-affected silencing at this modified HMR, we monitored silencing in pol30 versus POL30 cells by measuring a1 mRNA expression in the presence or absence of Gal4-Sir1p (Table 1). This analysis indicated that pol30 mutants were partially derepressed at HMR relative to POL30 cells, which is in support of previous observations (20, 38). We then evaluated the role of PCNA in silent chromatin formation by monitoring the establishment of silencing in POL30, pol30-8, and pol30-79 cells in the presence and absence of a replication fork at the modified HMR shown in Fig. 1A (Table 2). pol30-6 mutants were not used for these cell cycle analyses because of their more severe growth defects (20, 38, 71). As outlined in Fig. 1B, POL30, pol30-8, or pol30-79 cells lacking Gal4-Sir1p, and thus expressing a1 from HMR, were arrested in G1 with α-factor. Each initial culture was divided in two, and Flp1p was induced in one of the cultures to excise HMR; then Gal4-Sir1p was induced in both G1-arrested cultures. Cells were then released from G1 into S phase and rearrested at G2/M with benomyl and nocodozole. Steady state levels of a1 mRNA were used to measure the transcriptional status of HMR because a1 mRNA has a short half-life (10–14, 61, 73) (Table 2), excision of HMR was confirmed by DNA blots, and cell cycle arrests were confirmed by microscopy or flow cytometry (data not shown). To monitor the establishment of silencing in each strain, the level of a1 mRNA during the G2/M arrest was compared with that observed during the G1 arrest by RNA blot analyses (Table 2). POL30 cells established silencing at the replicated chromosomal HMR upon passage through S phase, as observed previously (11, 61). In contrast, pol30-8 and pol30-79 cells were defective in establishing silencing at the chromosomal HMR relative to POL30 cells (p = 0.025 for each comparison, n = 3). Similarly, POL30 cells established silencing at the non-replicated extrachromosomal HMR upon passage through S phase (see also Ref. 11, 61), whereas both pol30-8 and pol30-79 cells were also defective in establishing silencing at the extrachromosomal HMR relative to POL30 cells (p = 0.025 for each comparison, n = 3). Analogous negative control experiments using medium containing methionine to repress Gal4-Sir1p expression throughout the time course indicated that similar levels of a1 mRNA were expressed in G2/M relative to G1 in the absence of Gal4-Sir1p (data not shown). These results indicated that pol30-8 and pol30-79 mutants were defective in establishing silencing in both the presence and absence of DNA replication through HMR. Moreover, as this modified HMR lacked an ORC binding site, PCNA was required under conditions in which ORC and replication initiation were not. This defect in establishing silencing could have been caused by a defect that occurred either in cis at HMR or in trans throughout the yeast nucleus or genome.
TABLE 1.
pol30 mutants are defective in silencing
| Gal4-Sir1p | Relative efficiency of a1 mRNA expressiona |
|||
|---|---|---|---|---|
| POL30 | pol30-8 | pol30-6 | pol30-79 | |
| − | 1 | 1 | 1 | 1 |
| + | 0.053 ± 0.036 | 0.096 ± 0.0022 | 0.36 ± 0.066 | 0.17 ± 0.015 |
a Silencing at HMR was monitored in logarithmic POL30 and pol30 cells. For each strain, the relative level of a1 mRNA was calculated as the ratio of PhosphorImager units as follows: [(a1/SCR1 in the absence or presence of Gal4-Sir1p)/(a1/SCR1 in the absence of Gal4-Sir1p)]. The data have been normalized to the ratios observed in the absence of Gal4-Sir1p to combine data from independent experiments: mean ± S.D., n = 3.
TABLE 2.
pol30 mutants are defective in establishing silencing in the absence of DNA replication
| Arrest | HMRa | Relative efficiency of a1 mRNA expressionb |
||
|---|---|---|---|---|
| POL30 | pol30-8 | pol30-79 | ||
| G1 | Not replicated | 1 | 1 | 1 |
| G2/M | Not replicated | 0.16 ± 0.092 | 0.32 ± 0.075 | 0.49 ± 0.17 |
| G1 | Replicated | 1 | 1 | 1 |
| G2/M | Replicated | 0.15 ± 0.031 | 0.41 ± 0.20 | 0.36 ± 0.13 |
a HMR was either excised in G1 (Not replicated) or left within the chromosome (Replicated).
b Establishment of silencing was monitored in POL30 and pol30 cells. For each strain, the relative level of a1 mRNA expressed in G1 and in G2/M was normalized to an internal control transcript, SCR1. The data have been normalized to the ratios observed during the G1 arrest, and the levels in G2/M are expressed relative to those in G1 to combine data from independent experiments: mean ± S.D., n = 3 (11).
Lack of Evidence for Marking HMR with POL30
One possible explanation for the defects in establishing silencing in the pol30 mutants is that Pol30p influences silencing through a replication-independent function of PCNA at HMR during the cell cycle in which silencing was established. Consistent with this idea, PCNA that is left on DNA after replication in a SV40 DNA replication system in vitro can recruit CAF-1 to that DNA for chromatin assembly (33). Further, PCNA co-purifies with the elongator complex in vitro and in vivo (74), suggesting that a variety of chromosomal processes other than DNA synthesis can be affected by the presence of PCNA.
We reasoned if Pol30p had been preferentially left at the HMR locus during the S phase prior to the one in which we monitored the establishment of silencing and then remained at HMR, or if Pol30p simply preferentially associated with HMR in general, Pol30p would be enriched at HMR relative to other loci before its excision from chromosome III. To test this possibility, we performed ChIP analyses with anti-Pol30p antibodies (63) to test whether Pol30p was preferentially enriched at HMR in G1-arrested cells. In these ChIP assays, Pol30p was not preferentially bound to HMR relative to MAT in G1 either in the absence of Gal4-Sir1p or upon expression of Gal4-Sir1p in G1 (supplemental Fig. 1). We had shown previously that this anti-Pol30p antibody can detect the association of Pol30p at stalled replication forks by ChIP (41, 75), and control experiments indicated PCNA was expressed efficiently in both POL30 and pol30 cells (supplemental Fig. 2). Thus, Pol30p was not preferentially localized to HMR in a Sir-independent or -dependent manner prior to the establishment of silencing. We therefore tested whether other aspects of chromatin composition were altered in the mutants.
Effects of pol30 Mutants on the Topology of HMR and SIR Association at HMR
The above observations raised the possibility that both the replicated and the unreplicated HMR loci contained a preexisting pol30-dependent defect that prevented efficient silent chromatin formation during S phase. In this scenario, this pol30-dependent defect at HMR could have occurred during chromosomal replication and packaging of newly replicated DNA in the previous cell cycle, been maintained throughout G2, M, and the next G1 phase, and then interfered with silent chromatin formation in the following S phase in which the establishment of silent chromatin had been monitored.
We reasoned that the cause of the silencing defect in the pol30 mutants could be related to the nature of the nucleosomes in these cells that were deposited during replication-coupled chromatin assembly. Consistent with this notion, the silencing-defective pol30 mutants also have defects in interacting with nucleosome assembly factors (20, 38, 39), and cells lacking CAC1 are reported to have reduced levels of histone H3 at HMR and other genomic loci (76). Thus, reduced histone deposition at HMR or an altered characteristic of the chromosomal histones in the pol30 mutants may have contributed to their silencing defects. To assess the influence of pol30 mutations on the chromatin structure of HMR, we examined the topological distributions of an excised circular HMR locus that lacked the a1-a2 promoter, HMRaΔp266, in order to avoid monitoring transcription-dependent effects on topology simultaneously. However, this analysis indicated that HMR was efficiently packaged in both POL30 and pol30 cells (See supplemental “Results” and supplemental Fig. 3).
To determine whether pol30 mutants had defects in Sir protein association with HMR, despite their efficient packaging of HMR into chromatin, we conducted chromatin immunoprecipitation (ChIP) experiments to monitor Sir2p and Sir3p binding at HMR-GalSS in logarithmic POL30 and pol30 cells. In this analysis, Sir3p association at HMR-GalSS was slightly reduced in pol30-8 and pol30-6 mutants relative to POL30 cells, and Sir2p levels were also lower in pol30-8 relative to POL30 cells (Fig. 2, A and B, respectively, and supplemental Tables 3 and 4). Although some Sir levels at HMR are altered by some pol30 alleles, changes in Sir2p and Sir3p levels cannot explain the silencing defects of pol30-79 mutants. Therefore, aspects other than Sir association must contribute to silencing defects, at least in pol30-79 cells. Together, these results implied that, despite having defects in CAF-1 and ASF1-dependent pathways, pol30 mutants could efficiently package DNA into nucleosomes, but stable Sir association with HMR was compromised in these mutants. Therefore, we explored whether other aspects of nucleosomes led to the defects in silencing in pol30 mutants (see below).
FIGURE 2.

Sir protein association at HMR in POL30 and pol30 cells. Sir3 (A) and Sir2 (B) protein association at MAT (negative control locus), the synthetic silencer GalSS, and a1 at HMR was monitored in the indicated strains by chromatin immunoprecipitation using anti-Sir3p or anti-Sir2p antibodies. Co-precipitating DNA was examined by quantitative real-time PCR (see “Experimental Procedures”). The efficiency of co-precipitation of each locus in each strain was normalized and expressed relative to MAT, with MAT = 1; 2[(Sir CT − IgG CT)MAT − (Sir CT − IgG CT)locus]. An average of n = 2 is shown. Data for individual replicates are provided in supplemental Tables 3 and 4.
pol30 Mutants Have Defects in Histone Acetylation
The above observations implied that, rather than gross defects in nucleosome density or marking HMR with Pol30p, other differences such as global defects in histone modifications coupled to DNA replication may account for the silencing defects of the pol30 mutants. Global loss of histone modifications can lead to Sir relocalization throughout the genome and silencing defects (e.g. Refs. 77–79).
To determine whether pol30 mutants had defects in histone modifications, we examined acetylation of Lys-9 and Lys-56 on histone H3 and Lys-16 on histone H4 in whole cell extracts and in chromatin isolated from POL30 and pol30 cells. Like silencing in pol30 mutants (20, 38), each of these histone modifications has been linked previously to CAC1- and ASF1-dependent pathways. Acetylation of Lys-9 on histone H3 occurs on newly synthesized histone H3, is cell cycle-regulated, and peaks in S phase in an ASF1-dependent manner (80–83). H3-K9ac is mediated by the acetyltransferases Rtt109p and Gcn5p (82, 84, 85). Acetylation of Lys-56 on histone H3 by Rtt109p also peaks during S phase and requires ASF1 (75, 86–93). Also, acetylation of Lys-16 on histone H4 is mediated in part by the acetyltransferase complex SAS-I (94, 95), which interacts physically with both CAF-1 and Asf1p (96–98). Cells lacking the SAS-I subunits encoded by SAS2, SAS4, and SAS5 have silencing phenotypes that overlap with those of cac1 and asf1 mutants (96, 97). We identified defects in histone H3-K56 acetylation in pol30 mutants using this approach and have described these findings separately (40). Below, we describe our analysis of acetylation of H3-K9 and H4-K16 in the pol30 mutants.
To determine whether pol30 mutants with defects in ASF1- and CAC1-dependent pathways were defective in acetylating Lys-9 on histone H3 or Lys-16 on histone H4, we conducted quantitative protein blot analyses of chromatin-associated histones isolated from POL30 and pol30 cells (Tables 3 and 4 and supplemental Fig. 4). These analyses indicated that H3-K9 was hypoacetylated in chromatin isolated from pol30-8 and pol30-6 mutants as well as from asf1Δ, cac1Δ, and asf1Δcac1Δ mutants (p = 0.061 for pol30-6, p = 0.018 for other mutants relative to wild type, n = 3). Differences in H3-K9ac levels in pol30-79 relative to POL30 cells varied greatly and may reflect semi-stable epigenetic differences in H3-K9ac from culture to culture. Consistent with this notion, H3 K9R mutants exist in multiple epigenetic states (see below and Fig. 4B). The reduction in H3-K9ac was severe in asf1Δ and cac1Δasf1Δ mutants, implying that Asf1p played a key role in incorporating H3-K9ac into chromatin (see also Refs. 83 and 85). H3-K9ac was also reduced in cdc44-5 mutants relative to wild-type cells. CDC44 encodes the largest subunit of the PCNA-loading complex, RF-C (99), which helps recruit Asf1p to the replication fork (41). In contrast, wild-type levels of H3-K9ac were observed in cells lacking SAS2, indicating that SAS-I was not required for acetylation of this residue (Table 3; see also Ref. 98). Together these results implied both CAF-1 and Asf1p contributed to incorporation of H3-K9ac into chromatin during replication-coupled processes. Quantitative Western blot analyses also revealed that chromatin-associated histone H4 was hypoacetylated at Lys-16 in all pol30 mutants relative to POL30 cells (p = 0.018, n = 3) and H4-K16ac levels were reduced the other mutants as well (Table 4). Thus, chromatin-associated histones were hypoacetylated at multiple residues in pol30 mutants (see also Ref. 40).
TABLE 3.
Acetylation of Lys on chromatin-associated histone H3
| Strain | Relative levels of H3-K9aca |
|---|---|
| POL30 | 100 |
| pol30-8 | 46 ± 2.8 |
| pol30-6 | 87 ± 19 |
| pol30-79 | 96 ± 50 |
| cac1Δ | 39 ± 3.3 |
| asf1Δ | 17 ± 2.0 |
| cac1Δasf1Δ | 14 ± 5.0 |
| cdc44-5 | 75 ± 18 |
| sas2Δ | 100 ± 22 |
a The level of chromatin-associated H3-K9ac relative to chromatin-associated H3 in each strain was determined by quantitative Western blot analysis as described under “Experimental Procedures” and was expressed as a percentage of that observed in POL30 cells. Data were calculated as (H3-K9ac/H3)mutant/(H3-K9ac/H3)POL30 × 100, where mutant = indicated strain, mean ± S.D., n = 3 to 6 (except n = 2 for sas2Δ).
TABLE 4.
Acetylation of Lys-16 on chromatin-associated histone H4
| Strain | Relative levels of H4-K16aca |
|---|---|
| POL30 | 100 |
| pol30-8 | 69 ± 9.5 |
| pol30-6 | 42 ± 3.4 |
| pol30-79 | 67 ± 13 |
| cac1Δ | 59 ± 12 |
| asf1Δ | 70 ± 13 |
| cac1Δasf1Δ | 66 ± 16 |
| cdc44-5 | 65 ± 24 |
| sas2Δ | 20 ± 3.1 |
a The level of chromatin-associated H4-K16ac relative to chromatin-associated H3 in each strain was determined by quantitative Western blot analysis as described under “Experimental Procedures” and was expressed as a percentage of that observed in POL30 cells. Data were calculated as [(H4-K16ac/H3)mutant/(H4-K16ac/H3)POL30] × 100, where mutant = indicated strain, mean ± S.D., n = 3 (except n = 2 for sas2Δ).
Rtt109p and SAS-I Interact with PCNA
To evaluate interactions between Pol30p and Rtt109p or the SAS-I complex in vivo, we turned to an approach based on FLIM-FRET between fusion proteins containing either CFP as the donor or YFP as the acceptor fluorophore. We assessed FRET donor lifetime changes of Pol30-CFPp in single living yeast cells also expressing Rtt109-YFPp, Sas4-YFPp, or Sas5-YFPp. Reduction in the CFP (donor) fluorescence lifetimes due to FRET with YFP was observed by recording the lifetime in yeast expressing Pol30-CFPp plus Rtt109-YFPp, Sas4-YFPp, or Sas5-YFPp relative to yeast expressing Pol30-CFPp only (Fig. 3A and supplemental Fig. 5A). No interactions were observed in cells expressing Pol30-CFPp plus the control protein Spc29-YFPp or the control protein Trx3-CFPp plus Rtt109-YFPp (supplemental Fig. 5A). A shift in peak position of lifetime distribution was observed in yeast expressing Pol30-CFPp plus Rtt109-YFPp, Sas4-YFPp, or Sas5-YFPp compared with Pol30-CFPp only (Fig. 3B and supplemental Fig. 5B). This shorter lifetime of the donor (Pol30-CFPp) in the presence of the acceptor fluorophore (Rtt109-YFPp, Sas4-YFPp, or Sas5-YFPp) indicates the close association of PCNA with Rtt109p and the SAS-I complex in vivo.
FIGURE 3.

PCNA interacts with Rtt109p and SAS-I in vivo. Shown are confocal fluorescence lifetime images of CFP (A) and fluorescence lifetime distributions (B) in single living yeast cells expressing Pol30-CFPp (top), Pol30-CFPp plus Rtt109-YFPp (middle), and Pol30-CFPp plus Sas4-YFPp (bottom). Scale bar: 15 μm.
To confirm complex formation in the nuclei of single living cells, FRET efficiency between Pol30-CFPp and Rtt109-YFPp, Sas4-YFPp, or Sas5-YFPp was calculated by lifetimes obtained from the TCSPC decay histograms and fitted by a double exponential function. The lifetime of CFP, the calculated FRET efficiency, and the intermolecular distances are shown in Table 5 and supplemental Table 5. The FRET efficiency of Pol30-CFPp with Rtt109-YFPp, Sas4-YFPp, and Sas5-YFPp was ∼28.6, 25.7, and 23.1%, respectively, which corresponds to a Förster distance of 6.1, 6.2, and 6.4 nm, respectively (which is less than the diameter of a nucleosome), between these pairs of proteins in living cells.
TABLE 5.
PCNA interacts with Rtt109p and SAS-I by FLIM-FRET analysis
| Genotype | Lifetime | FRET efficiency | Distance |
|---|---|---|---|
| ns | nm | ||
| POL30-CFP | 1.456 ± 0.134 | ||
| POL30-CFP RTT109-YFP | 1.039 ± 0.161 | 0.286 | 6.055 |
| POL30-CFP SAS4-YFP | 1.012 ± 0.149 | 0.257 | 6.207 |
In contrast, no FRET was observed between Rtt109-YFPp and pol30-8- or pol30-6-CFPp; FRET interactions were also lost between SAS5-YFPp and pol30-8- or pol30-6-CFPp (Table 6 and Fig. 6). FRET between pol30-79-CFPp and Rtt109-YFPp or SAS5-YFPp could not be assessed, as POL30 plasmids could not be lost from strains co-expressing pol30-79-CFPp and Rtt109-YFPp or SAS5-YFPp during strain construction, indicating that these combinations may be lethal. These observations when combined support a model in which histone acetylation was coupled to PCNA and pol30 mutants with defects in ASF1- and CAC1-dependent pathways had reduced levels of chromatin assembly factor-dependent histone modifications, which, in turn, affected silencing.
TABLE 6.
pol30p mutants have defects in interacting with Rtt109p and SAS-I by FLIM-FRET analysis
| Genotype | Lifetime | FRET efficiency |
|---|---|---|
| ns | ||
| POL30-CFP | 1.39 ± 0.0680 | |
| POL30-CFP SAS5-YFP | 1.11 ± 0.0241 | 0.201 |
| pol30-6-CFP SAS5-YFP | 1.41 ± 0.0901 | |
| pol30-8-CFP SAS5-YFP | 1.43 ± 0.0351 | |
| pol30-79-CFP SAS5-YFP | NDa | ND |
| pol30-6-CFP RTT109-YFP | 1.40 ± 0.0532 | |
| pol30-8-CFP RTT109-YFP | 1.39 ± 0.0636 | |
| pol30-79-CFP RTT109-YFP | ND | ND |
a ND, not determined; lethal.
FIGURE 6.

pol30p mutants have defects in interacting with SAS-I and Rtt109p. Interactions between pol30-CFPp mutants and Sas5-YFPp (A) or Rtt109-YFPp (B) are shown: confocal fluorescence lifetime image of CFP (left) and fluorescence lifetime distribution (right) in yeast expressing the indicated proteins. Scale bar: 10 μm.
Altered Histone Modifications in pol30 Mutants Affect Silencing
To assess further the relationship among PCNA, histone acetylation, and silencing, we compared the silencing of pol30 and histone hypoacetylation mutants in two additional assays: silencing of an ADE2 reporter gene integrated at HMR and silencing of HMRae** (Figs. 4 and 5, Table 7, and supplemental Table 6). We generated HMR::ADE2 yeast expressing histone H3 or H4 mutants that mimicked the unacetylated forms of Lys-9 on H3 or Lys-16 on H4, H3 K9R and H4 K16R, and compared silencing in these mutants with pol30 mutants using a colony color assay. Cells expressing histone H3 K9R phenocopied the silencing defect of pol30-8 mutants at HMR::ADE2 (Fig. 4, A and B). Sectored colonies were observed in both histone H3 K9R and pol30-8 mutants, whereas red colonies were formed by wild-type cells. Similarly, sectored colonies were observed in pol30-8 cells expressing histone H3 K9R (Fig. 4C). As pol30-8 mutants primarily have silencing defects in a CAC1-dependent pathway (20, 38), we also examined the interactions between cac1Δ and histone H3 K9R at HMR::ADE2 (Fig. 4D). Colonies of cells lacking CAC1 and expressing wild-type histones H3 and H4 ranged from light pink to nearly white with occasional light pink and white sectors (Fig. 4D). Similar colony colors were observed in cac1Δ cells expressing histone H3 K9R. Double mutant analyses could not be conducted with H4 K16R and pol30 mutants, as colonies of HMR::ADE2 cells expressing histone H4 K16R were white and, therefore, derepressed (data not shown). In control experiments, ade2-1 strains containing a wild-type HMR locus plus pol30-8, cac1Δ, H3 K9R, or H4 K16R mutants all grew as red colonies (data not shown), confirming that these mutants did not disrupt the adenine metabolic pathway upstream of ADE2, which would have prevented the formation of a red pigment in a silencing-independent manner. The similar decrease of chromatin-associated H3-K9ac and the genetic epistasis among pol30-8, cac1, and H3 K9R suggest a network in which the PCNA-CAF-1 interaction ensures the deposition of H3-K9ac histones that contribute to silencing.
HMRae** can be used as a model locus for analyzing mutations that lead to the relocalization of Sir proteins to inappropriate genomic loci via defects in histone modifications (40, 79, 96). Sir proteins are not normally present at HMRae** because neither the mutated E silencer, which has a point mutation at its Rap1p binding site and one base pair insertion at its Abf1p binding site (100), nor the I silencer is capable of recruiting Sir proteins to HMRae** (9, 40). This lack of Sir binding is the cause of the defect in silencing at this locus. In cells with hypoacetylated histones, however, Sir proteins become “mislocalized” to HMRae** via the presence of Sir binding sites on nucleosomes, thereby restoring silencing (e.g. see Refs. 40 and 79). Silencing at HMRae** is rescued in cells lacking ASF1or CAC1 and in pol30 mutants (19, 40, 96) (Fig. 5 and see below), implying that loss of chromatin assembly factor-dependent histone modifications had restored silencing in these strains. Consistent with this model, silencing defects at HMRae** are also partially suppressed in cells lacking SAS2, SAS4, or SAS5 or expressing histone H4 K16R as well as in cells lacking RTT109 or expressing H3 K56R (40, 96, 101–103) (Fig. 5). As Rtt109p acetylates Lys-9 in addition to Lys-56 on H3 (75, 84, 85, 104), we examined whether the loss of acetylation at Lys-9 on histone H3 could similarly suppress silencing defects at HMRae** and thereby restore mating in MATα cells. In patch mating assays, silencing was rescued at HMRae** in H3 K9R mutants (Fig. 5). In contrast, HMRae** was derepressed in cells expressing H3 K9Q mutants. HMRae** was also derepressed in H3 K9R,K56Q mutants, but HMRae** became silenced in H3 K9R,K56R mutants as well as in H3 K9Q,K56R mutants. Thus, although H3 K9R could promote silencing at HMRae**, the presence of a positive charge at residue 56 on H3 was more important than the charge status at residue 9 on H3 for silencing. In quantitative mating assays, H3 K9R mutants silenced HMRae** 6-fold more efficiently than did cells expressing wild-type histone H3 (p = 0.018, n = 3; Table 7).
We next tested for interactions between H3 K9R, H3 K14R and H4 K16R, as SAS-I readily acetylates both Lys-14 on H3 and Lys-16 on H4 but not Lys-9 on H3 in vitro (94). H3 K9R, H3 K14R, and H4 K16R mutants all restored silencing at HMRae** with similar efficiency, and combining H3 K14R with H4 K16R did not enhance silencing relative to either single mutant. In contrast, combining H3 K9R with H3 K14R and H4 K16R correlated with enhanced silencing at HMRae** (Table 7). Together, these results implied that loss of acetylation of Lys-9 on H3 restored silencing though a different pathway than did loss of acetylation of Lys-14 on H3 and Lys-16 on H4 and that H3-K14 and H4-K16 functioned through the same pathway. These pathways likely involve RTT109 and SAS2, respectively, as well as ASF1, CAF-1, RF-C, and PCNA (see Fig. 6). Global hypoacetylation of histones in the pol30 mutants from defects in replication-coupled chromatin assembly may have facilitated the mislocalization of Sir proteins to multiple genomic loci, thereby reducing the pools of Sir available for establishing silencing at chromosomal and extrachromosomal HMRs in earlier experiments (Table 2). Consistent with this notion, cells expressing histones H3 K9,14R H4 K16R double mutants had mild defects in silencing at native HM loci as measured by quantitative mating assays (Table 7; see also Ref. 62), and cells lacking acetylation of these residues as well as of H3-K56 have severe silencing defects at HML (105).
To confirm that more than one pathway functioning through POL30 was influencing silencing, we examined silencing at HMRae** in several pol30 mutants. pol30-79 plus pol30-6 and pol30-8 mutants have silencing defects that fall primarily in ASF1- or CAF-1-dependent pathways, respectively (20). pol30-79 (Fig. 5), pol30-6, and pol30-8 could each suppress the silencing defect at HMRae** (p = 0.018, n = 3 for each mutant), and pol30-42, which has the combined mutations of pol30-6 and pol30-8, further enhanced silencing (Fig. 5 and supplemental Table 6). Other pol30 mutants, which silence HMR::ADE2 efficiently (data not shown) and have no reported defects in ASF1- or CAC1-dependent pathways, could not rescue silencing at HMRae** (supplemental Table 6). Together, these results implied that pol30 mutants had restored silencing at HMRae** through multiple overlapping pathways involving CAC1 and ASF1, which modulate histone acetylation (see also Ref. 40).
DISCUSSION
In S. cerevisiae, silencing at HMR can be established during the passage through S phase, yet the initiation of DNA replication at silencers and the passage of a replication fork through HMR are not required for silencing (10–13). Despite this lack of requirement for DNA replication in establishing silencing, this study has indicated that pol30 mutants are defective in establishing silencing on both replicated and non-replicated HMR loci upon passage through S phase (Table 2). These observations imply that defects in a replication-coupled role of PCNA led to global changes in chromatin that were incompatible with the formation of silent chromatin or that PCNA contributed to silencing directly in a locus-specific, replication-independent manner. However, as Pol30p functions in replication- and repair-coupled processes and Pol30p did not preferentially mark HMR prior to establishing silencing (supplemental Fig. 1), we favor the former possibility.
One replication-coupled way in which PCNA can influence silencing is through its role in directing the activity of chromatin assembly factors (20, 38). This role of PCNA connects DNA replication to nucleosome assembly and, hence, to the foundation upon which silenced chromatin is built as well as to chromatin at other loci across the genome. To understand how Pol30p influences chromatin structure and how chromatin structure impacts silencing, we analyzed the topology of HMR in pol30 mutants with silencing defects. In this analysis, we separately assessed the influence of pol30 mutants on chromatin structure in transcription-independent and Sir protein-dependent and -independent contexts. We reasoned that defects in packaging chromosomal DNA into nucleosomes after replication might lead to defects in silencing in the pol30 mutants if the density of nucleosomes at HMR was insufficient to permit Sir spreading. Supporting this notion, DNA sequences that exclude nucleosome formation can act as a barrier to Sir spreading (106), and as shown by ChIP analyses, the level of histone H3 at HMR and other genomic loci is reduced in cac1 mutants (76). As packaging DNA around nucleosomes induces negative supercoiling (107), gross defects in nucleosome density should have significantly altered the topology of HMR in pol30 mutants in our experiments. If such defects in packaging DNA also prevented Sir association at HMR, then Sir-dependent changes in topology also should have been perturbed. Instead, we observed similar topological patterns in pol30 versus POL30 cells at a modified HMR lacking the a1-a2 promoter region (supplemental Fig. 3). These findings revealed that mutant alleles of POL30 did not lead to severe defects in packaging promoter-independent DNA into nucleosomes. Thus, pol30 mutants likely compensated for defects in interacting with individual chromatin assembly factors by using alternative pathway(s) to ensure that the newly replicated DNA was packaged into chromatin. This compensation may have come at the cost, however, of incorporating inappropriately modified histones throughout the genome. We hypothesized that subtle pol30-dependent changes in the composition of chromatin, in turn, led to the defects in forming silent chromatin.
Under certain conditions, the rate of nucleosome assembly during DNA replication, rather than the final nucleosome density, could theoretically influence the establishment of silencing on a template that is replicated if a reduced rate of assembly resulted in the temporary loss of Sir protein binding sites on nucleosomes throughout HMR. Consistent with this model, loss of Asf1-dependent chromatin assembly in mammals results in delayed incorporation of histone H3 into chromatin (108, 109). If the rate of nucleosome assembly during DNA replication were significantly reduced in pol30 mutants versus POL30 cells, such a replication-coupled delay might lead to a more severe defect in establishing silencing on a replicated versus non-replicated HMR locus during S phase. Instead, we found that pol30-8 and pol30-79 mutants had similar defects in establishing silencing on both replicated and non-replicated templates (Table 2). Because other pathways can take over chromatin assembly in the absence of Asf1p function (108), and modifications like H3-K56ac may influence nucleosome turnover (109, 110), it is still an open question as to whether the rate of chromatin assembly contributes to defects in silencing.
Coupling PCNA and Chromatin Assembly to Histone Modifications
Our analyses demonstrated that pol30 mutants influenced histone modifications linked to chromatin assembly factors and that defects in these modifications altered silencing (Tables 3, 4, and 7, supplemental Fig. 4, Figs. 4 and 5, and Ref. 40). This study supports a central role for Pol30p in determining histone modifications in yeast.
Acetylation of Lys-9 on histone H3 is associated with newly synthesized histones (80, 81); this S phase-specific acetylation event likely depends on Asf1p and the histone acetyltransferases Rtt109p and Gcn5p (82–85) (see also Ref. 80). Our data imply that CAC1 also plays a role in enabling acetylation of this residue. Consistent with this notion, acetylation of Lys-9 on histone H3 and the silencing defects of pol30-8 and histone H3 K9R mutants correlate with a CAF-1-dependent pathway (Table 3 and Fig. 4) (20, 38). Our data also indicate that other histone modifications linked to CAC1 or ASF1 were perturbed in pol30 mutants including SAS-I-dependent acetylation of Lys-16 on histone H4 (Table 4 and supplemental Fig. 4) and Rtt109p-dependent acetylation of Lys-56 on histone H3 (40). These findings imply that PCNA can influence silencing by affecting the levels of multiple modifications on histones in yeast.
The work presented here supports a model in which the differing defects in silencing in the various pol30 mutants (19, 20, 38, 39), and likely other replication factors, are caused by defects in recruiting chromatin assembly factors to chromatin. This, in turn, leads to the incorporation of inappropriately modified histones throughout the genome. Hypoacetylated histones could help create high affinity binding sites for Sir proteins at multiple inappropriate loci across the genome (HMRae** being a model for one such locus) and result in an insufficient pool of Sir proteins being available for silent chromatin formation on replicated and non-replicated templates. Altered histone modifications at HMR initiated by errors in replication-coupled chromatin assembly during a previous S phase could also contribute to defects in establishing silencing. Future ChIP analyses will clarify how pol30 and other replication fork mutants affect histone modifications genome-wide.
At least two possible mechanisms could account for how POL30 affects histone modifications. First, in wild-type cells, newly synthesized histones could be post-translationally modified at a key residue(s) by a histone-modifying enzyme(s) prior to being loaded onto the chromosome during DNA replication, as has been suggested previously for H3-K9ac and -K56ac by Rtt109p (Refs. 75, 85, 87, and 111 and references therein). In this scenario, during replication, pol30 mutants would have been defective in recruiting the correct classes of chromatin assembly factors, which were bound to correctly modified histones. This defect would have led to the failure to load those histones onto the chromosome and to defects in silencing. The second possibility is that altered histone modifications in the pol30 mutants were caused by a defect in targeting a key enzyme(s) to chromatin during or shortly after nucleosome assembly, analogous to replication-coupled modifications observed in mammals (see the Introduction). Our demonstration, using single molecule methods, that Rtt109p and SAS-I interact with PCNA in vivo and that these interactions are defective in the pol30 mutants provides strong evidence for the second scenario but does not exclude the first possibility from occurring as well. Regardless of the mechanism(s) involved, the end result would be a genome with altered patterns of modifications on histones with the potential to negatively impact the regulation of individual genomic loci. Determining whether PCNA and chromatin assembly factors directly target histone acetyltransferases to the replication fork will help to clarify when and how these modifications occur. Currently, it also remains an open question as to whether or how specific chromatin assembly factors preferentially load different histones onto discrete regions of the genome during replication, but the targeted assembly of at least one variant, H2A.Z, is clearly influenced by modifications on histones H3 and H4 (112, 113). Further analysis of where specific histone modifications or, in more complex eukaryotes, histone variants linked to individual chromatin assembly factors are first observed throughout the genome upon DNA replication should begin to address this question (see (Refs. 114 and 115).
Replication and the Establishment of Epigenetic Processes
Other evidence for replication-coupled events influencing the establishment of silencing in yeast has come from studies of cells lacking SIR1. In a given population of cells lacking SIR1, individual cells will exist in two distinct transcriptional states at the HM loci that are mitotically stable (116, 117). When changes in the transcriptional state are monitored in the progeny of a single derepressed sir1Δ cell by pedigree analysis, a phenomenon known as the “grandmother effect” can be observed (117). In these switching events, all “granddaughter” cells of the original derepressed cell will switch to the silenced state during the same generation, suggesting that an event linked to DNA replication in an earlier cell cycle was inherited in the progeny, which permitted the establishment of silencing during the following cell cycle. It is tempting to surmise that heritable histone modification states regulated by DNA replication might influence the probability of establishing silencing and be responsible for this grandmother effect. Such an event could include a replication-coupled modification critical for silencing or a modification that must be erased in a replication-dependent manner for silencing, as has been proposed for the loss of methylation on histone H3 during silent chromatin formation (118). Consistent with this, pedigree analyses indicate loss of SET1- or DOT1-dependent methylation of histone H3-K4 or H3-K9, respectively, increases the probability of establishing silencing in a given cell division whereas loss of SAS2 decreases the likelihood that silencing will be established each cell cycle (119), analogous to our observations for pol30 mutants (Table 2).
Summary
Taken together, the results presented here imply that mutations in PCNA can lead to alterations in histone modifications that influence the formation of epigenetic processes. Although advances have recently been made in deciphering chromatin modifications involved in transcription and the enzymes responsible for those modifications, the role of DNA replication in defining histone modifications throughout the genome is still poorly understood. Future studies examining the relationship between histone-modifying enzymes and factors at the DNA replication fork will provide insights into how chromatin structures at individual loci are maintained across generations.
Supplementary Material
Acknowledgments
We thank Scott Briggs, Peter Burgers, Ann Ehrenhofer-Murray, Marc Gartenberg, Jasper Rine, Mitch Smith, Sharon Roth, and Arnold Stein for protocols, reagents, strains, and plasmids. We also thank Bruce Craig and Alexander Lipka for assistance with statistical analyses, Bo Yang for technical assistance, and Joe Ogas and Jasper Rine for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM55712 (to P. D. K.) and GM31105 (to J. Rine, with whom A. L. K. worked at the time). This work was also supported by Kimmel Scholar Award SKF-03-010 (to A. L. K.), a National Science Foundation award (to A. L. K.), a grant from the Purdue Office of the Vice President of Research (to A. L. K. and J. M. K. I.), and an American Cancer Society institutional research grant to the Purdue University Center for Cancer Research. Support for initial experiments was provided by the American Cancer Society (PF-01-126-01-MBC to A. L. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures,” “Results,” “References,” Tables 1–6, and Figs. 1–5.
- ORC
- origin recognition complex
- PCNA
- proliferating cell nuclear antigen
- CAF
- chromatin assembly factor
- Sir
- silent information regulator
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- FRT
- flippase recognition target.
REFERENCES
- 1.Rusché L. N., Kirchmaier A. L., Rine J. (2003) Annu. Rev. Biochem. 72, 481–516 [DOI] [PubMed] [Google Scholar]
- 2.Moretti P., Freeman K., Coodly L., Shore D. (1994) Genes Dev. 8, 2257–2269 [DOI] [PubMed] [Google Scholar]
- 3.Gardner K. A., Rine J., Fox C. A. (1999) Genetics 151, 31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Triolo T., Sternglanz R. (1996) Nature 381, 251–253 [DOI] [PubMed] [Google Scholar]
- 5.Luo K., Vega-Palas M. A., Grunstein M. (2002) Genes Dev. 16, 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bose M. E., McConnell K. H., Gardner-Aukema K. A., Müller U., Weinreich M., Keck J. L., Fox C. A. (2004) Mol. Cell. Biol. 24, 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoppe G. J., Tanny J. C., Rudner A. D., Gerber S. A., Danaie S., Gygi S. P., Moazed D. (2002) Mol. Cell. Biol. 22, 4167–4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudner A. D., Hall B. E., Ellenberger T., Moazed D. (2005) Mol. Cell. Biol. 25, 4514–4528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rusché L. N., Kirchmaier A. L., Rine J. (2002) Mol. Biol. Cell 13, 2207–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fox C. A., Ehrenhofer-Murray A. E., Loo S., Rine J. (1997) Science 276, 1547–1551 [DOI] [PubMed] [Google Scholar]
- 11.Kirchmaier A. L., Rine J. (2001) Science 291, 646–650 [DOI] [PubMed] [Google Scholar]
- 12.Li Y. C., Cheng T. H., Gartenberg M. R. (2001) Science 291, 650–653 [DOI] [PubMed] [Google Scholar]
- 13.Miller A. M., Nasmyth K. A. (1984) Nature 312, 247–251 [DOI] [PubMed] [Google Scholar]
- 14.Lau A., Blitzblau H., Bell S. P. (2002) Genes Dev. 16, 2935–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufman P. D., Kobayashi R., Stillman B. (1997) Genes Dev. 11, 345–357 [DOI] [PubMed] [Google Scholar]
- 16.Enomoto S., Berman J. (1998) Genes Dev. 12, 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith J. S., Caputo E., Boeke J. D. (1999) Mol. Cell. Biol. 19, 3184–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singer M. S., Kahana A., Wolf A. J., Meisinger L. L., Peterson S. E., Goggin C., Mahowald M., Gottschling D. E. (1998) Genetics 150, 613–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrenhofer-Murray A. E., Kamakaka R. T., Rine J. (1999) Genetics 153, 1171–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z., Shibahara K., Stillman B. (2000) Nature 408, 221–225 [DOI] [PubMed] [Google Scholar]
- 21.Foss M., McNally F. J., Laurenson P., Rine J. (1993) Science 262, 1838–1844 [DOI] [PubMed] [Google Scholar]
- 22.Prelich G., Kostura M., Marshak D. R., Mathews M. B., Stillman B. (1987) Nature 326, 471–475 [DOI] [PubMed] [Google Scholar]
- 23.Tan C. K., Castillo C., So A. G., Downey K. M. (1986) J. Biol. Chem. 261, 12310–12316 [PubMed] [Google Scholar]
- 24.Maga G., Hübscher U. (1995) Biochemistry 34, 891–901 [DOI] [PubMed] [Google Scholar]
- 25.Hübscher U., Maga G., Podust V. N. (1996) in DNA Replication in Eukaryotic Cells (DePamphilis M. L. ed) pp. 525–543, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26.Krishna T. S., Kong X. P., Gary S., Burgers P. M., Kuriyan J. (1994) Cell 79, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 27.Fien K., Stillman B. (1992) Mol. Cell. Biol. 12, 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho Y., Gruhler A., Heilbut A., Bader G. D., Moore L., Adams S. L., Millar A., Taylor P., Bennett K., Boutilier K., Yang L., Wolting C., Donaldson I., Schandorff S., Shewnarane J., Vo M., Taggart J., Goudreault M., Muskat B., Alfarano C., Dewar D., Lin Z., Michalickova K., Willems A. R., Sassi H., Nielsen P. A., Rasmussen K. J., Andersen J. R., Johansen L. E., Hansen L. H., Jespersen H., Podtelejnikov A., Nielsen E., Crawford J., Poulsen V., Sorensen B. D., Matthiesen J., Hendrickson R. C., Gleeson F., Pawson T., Moran M. F., Durocher D., Mann M., Hogue C. W., Figeys D., Tyers M. (2002) Nature 415, 180–183 [DOI] [PubMed] [Google Scholar]
- 29.Li X., Li J., Harrington J., Lieber M. R., Burgers P. M. (1995) J. Biol. Chem. 270, 22109–22112 [DOI] [PubMed] [Google Scholar]
- 30.Moldovan G. L., Pfander B., Jentsch S. (2007) Cell 129, 665–679 [DOI] [PubMed] [Google Scholar]
- 31.Burgers P. M. (2009) J. Biol. Chem. 284, 4041–4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krawitz D. C., Kama T., Kaufman P. D. (2002) Mol. Cell. Biol. 22, 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibahara K., Stillman B. (1999) Cell 96, 575–585 [DOI] [PubMed] [Google Scholar]
- 34.Moggs J. G., Grandi P., Quivy J. P., Jónsson Z. O., Hübscher U., Becker P. B., Almouzni G. (2000) Mol. Cell. Biol. 20, 1206–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamakaka R. T., Bulger M., Kaufman P. D., Stillman B., Kadonaga J. T. (1996) Mol. Cell. Biol. 16, 810–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaillard P. H., Martini E. M., Kaufman P. D., Stillman B., Moustacchi E., Almouzni G. (1996) Cell 86, 887–896 [DOI] [PubMed] [Google Scholar]
- 37.Mello J. A., Silljé H. H., Roche D. M., Kirschner D. B., Nigg E. A., Almouzni G. (2002) EMBO Rep. 3, 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharp J. A., Fouts E. T., Krawitz D. C., Kaufman P. D. (2001) Curr. Biol. 11, 463–473 [DOI] [PubMed] [Google Scholar]
- 39.Huang S., Zhou H., Katzmann D., Hochstrasser M., Atanasova E., Zhang Z. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13410–13415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller A., Yang B., Foster T., Kirchmaier A. L. (2008) Genetics 179, 793–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franco A. A., Lam W. M., Burgers P. M., Kaufman P. D. (2005) Genes Dev. 19, 1365–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tyler J. K., Collins K. A., Prasad-Sinha J., Amiott E., Bulger M., Harte P. J., Kobayashi R., Kadonaga J. T. (2001) Mol. Cell. Biol. 21, 6574–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyler J. K., Adams C. R., Chen S. R., Kobayashi R., Kamakaka R. T., Kadonaga J. T. (1999) Nature 402, 555–560 [DOI] [PubMed] [Google Scholar]
- 44.Groth A., Corpet A., Cook A. J., Roche D., Bartek J., Lukas J., Almouzni G. (2007) Science 318, 1928–1931 [DOI] [PubMed] [Google Scholar]
- 45.Henderson D. S., Banga S. S., Grigliatti T. A., Boyd J. B. (1994) EMBO J. 13, 1450–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chuang L. S., Ian H. I., Koh T. W., Ng H. H., Xu G., Li B. F. (1997) Science 277, 1996–2000 [DOI] [PubMed] [Google Scholar]
- 47.Li E., Beard C., Jaenisch R. (1993) Nature 366, 362–365 [DOI] [PubMed] [Google Scholar]
- 48.Estève P. O., Chin H. G., Smallwood A., Feehery G. R., Gangisetty O., Karpf A. R., Carey M. F., Pradhan S. (2006) Genes Dev. 20, 3089–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iida T., Suetake I., Tajima S., Morioka H., Ohta S., Obuse C., Tsurimoto T. (2002) Genes Cells 7, 997–1007 [DOI] [PubMed] [Google Scholar]
- 50.Milutinovic S., Zhuang Q., Szyf M. (2002) J. Biol. Chem. 277, 20974–20978 [DOI] [PubMed] [Google Scholar]
- 51.Fuks F., Burgers W. A., Brehm A., Hughes-Davies L., Kouzarides T. (2000) Nat. Genet. 24, 88–91 [DOI] [PubMed] [Google Scholar]
- 52.Sarraf S. A., Stancheva I. (2004) Mol. Cell 15, 595–605 [DOI] [PubMed] [Google Scholar]
- 53.Huen M. S., Sy S. M., van Deursen J. M., Chen J. (2008) J. Biol. Chem. 283, 11073–11077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schotta G., Lachner M., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T. (2004) Genes Dev. 18, 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams A., Gottschling D. E., Kaiser C. A., Stearns T. (1997) Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 56.Stearns T., Ma H., Botstein D. (1990) Methods Enzymol. 185, 280–297 [DOI] [PubMed] [Google Scholar]
- 57.Goldstein A. L., McCusker J. H. (1999) Yeast 15, 1541–1553 [DOI] [PubMed] [Google Scholar]
- 58.Wach A., Brachat A., Pöhlmann R., Philippsen P. (1994) Yeast 10, 1793–1808 [DOI] [PubMed] [Google Scholar]
- 59.Zhang W., Bone J. R., Edmondson D. G., Turner B. M., Roth S. Y. (1998) EMBO J. 17, 3155–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheff M. A., Thorn K. S. (2004) Yeast 21, 661–670 [DOI] [PubMed] [Google Scholar]
- 61.Kirchmaier A. L., Rine J. (2006) Mol. Cell. Biol. 26, 852–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang B., Kirchmaier A. L. (2006) Mol. Biol. Cell 17, 5287–5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Daganzo S. M., Erzberger J. P., Lam W. M., Skordalakes E., Zhang R., Franco A. A., Brill S. J., Adams P. D., Berger J. M., Kaufman P. D. (2003) Curr. Biol. 13, 2148–2158 [DOI] [PubMed] [Google Scholar]
- 64.Schmitt M. E., Brown T. A., Trumpower B. L. (1990) Nucleic Acids Res. 18, 3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vidi P. A., Chen J., Irudayaraj J. M., Watts V. J. (2008) FEBS Lett. 582, 3985–3990 [DOI] [PubMed] [Google Scholar]
- 66.Varghese L. T., Sinha R. K., Irudayaraj J. (2008) Anal. Chim. Acta 625, 103–109 [DOI] [PubMed] [Google Scholar]
- 67.Siegel R. M., Chan F. K., Zacharias D. A., Swofford R., Holmes K. L., Tsien R. Y., Lenardo M. J. (2000) Sci. STKE 2000, PL1. [DOI] [PubMed] [Google Scholar]
- 68.van Leeuwen F., Gottschling D. E. (2002) Methods Enzymol. 350, 165–186 [DOI] [PubMed] [Google Scholar]
- 69.Martins-Taylor K., Dula M. L., Holmes S. G. (2004) Genetics 168, 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matecic M., Martins-Taylor K., Hickman M., Tanny J., Moazed D., Holmes S. G. (2006) Genetics 173, 1939–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ayyagari R., Impellizzeri K. J., Yoder B. L., Gary S. L., Burgers P. M. (1995) Mol. Cell. Biol. 15, 4420–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eissenberg J. C., Ayyagari R., Gomes X. V., Burgers P. M. (1997) Mol. Cell. Biol. 17, 6367–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller A. M. (1984) EMBO J. 3, 1061–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Q., Fazly A. M., Zhou H., Huang S., Zhang Z., Stillman B. (2009) PLoS Genet. 5, e1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsubota T., Berndsen C. E., Erkmann J. A., Smith C. L., Yang L., Freitas M. A., Denu J. M., Kaufman P. D. (2007) Mol. Cell 25, 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tamburini B. A., Carson J. J., Linger J. G., Tyler J. K. (2006) Genetics 173, 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Leeuwen F., Gafken P. R., Gottschling D. E. (2002) Cell 109, 745–756 [DOI] [PubMed] [Google Scholar]
- 78.van Leeuwen F., Gottschling D. E. (2002) Curr. Opin. Cell Biol. 14, 756–762 [DOI] [PubMed] [Google Scholar]
- 79.Yang B., Britton J., Kirchmaier A. L. (2008) J. Mol. Biol. 381, 826–844 [DOI] [PubMed] [Google Scholar]
- 80.Kuo M. H., Brownell J. E., Sobel R. E., Ranalli T. A., Cook R. G., Edmondson D. G., Roth S. Y., Allis C. D. (1996) Nature 383, 269–272 [DOI] [PubMed] [Google Scholar]
- 81.Sobel R. E., Cook R. G., Perry C. A., Annunziato A. T., Allis C. D. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 1237–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adkins M. W., Carson J. J., English C. M., Ramey C. J., Tyler J. K. (2007) J. Biol. Chem. 282, 1334–1340 [DOI] [PubMed] [Google Scholar]
- 83.Adkins M. W., Tyler J. K. (2004) J. Biol. Chem. 279, 52069–52074 [DOI] [PubMed] [Google Scholar]
- 84.Berndsen C. E., Tsubota T., Lindner S. E., Lee S., Holton J. M., Kaufman P. D., Keck J. L., Denu J. M. (2008) Nat. Struct. Mol. Biol. 15, 948–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fillingham J., Recht J., Silva A. C., Suter B., Emili A., Stagljar I., Krogan N. J., Allis C. D., Keogh M. C., Greenblatt J. F. (2008) Mol. Cell. Biol. 28, 4342–4353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Celic I., Masumoto H., Griffith W. P., Meluh P., Cotter R. J., Boeke J. D., Verreault A. (2006) Curr. Biol. 16, 1280–1289 [DOI] [PubMed] [Google Scholar]
- 87.Driscoll R., Hudson A., Jackson S. P. (2007) Science 315, 649–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Han J., Zhou H., Horazdovsky B., Zhang K., Xu R. M., Zhang Z. (2007) Science 315, 653–655 [DOI] [PubMed] [Google Scholar]
- 89.Maas N. L., Miller K. M., DeFazio L. G., Toczyski D. P. (2006) Mol. Cell 23, 109–119 [DOI] [PubMed] [Google Scholar]
- 90.Masumoto H., Hawke D., Kobayashi R., Verreault A. (2005) Nature 436, 294–298 [DOI] [PubMed] [Google Scholar]
- 91.Recht J., Tsubota T., Tanny J. C., Diaz R. L., Berger J. M., Zhang X., Garcia B. A., Shabanowitz J., Burlingame A. L., Hunt D. F., Kaufman P. D., Allis C. D. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 6988–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu E. Y., Bi X., Holland M. J., Gottschling D. E., Broach J. R. (2005) Mol. Cell. Biol. 25, 1846–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhou H., Madden B. J., Muddiman D. C., Zhang Z. (2006) Biochemistry 45, 2852–2861 [DOI] [PubMed] [Google Scholar]
- 94.Sutton A., Shia W. J., Band D., Kaufman P. D., Osada S., Workman J. L., Sternglanz R. (2003) J. Biol. Chem. 278, 16887–16892 [DOI] [PubMed] [Google Scholar]
- 95.Shia W. J., Osada S., Florens L., Swanson S. K., Washburn M. P., Workman J. L. (2005) J. Biol. Chem. 280, 11987–11994 [DOI] [PubMed] [Google Scholar]
- 96.Meijsing S. H., Ehrenhofer-Murray A. E. (2001) Genes Dev. 15, 3169–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Osada S., Sutton A., Muster N., Brown C. E., Yates J. R., 3rd, Sternglanz R., Workman J. L. (2001) Genes Dev. 15, 3155–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sutton A., Bucaria J., Osley M. A., Sternglanz R. (2001) Genetics 158, 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McAlear M. A., Tuffo K. M., Holm C. (1996) Genetics 142, 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kimmerly W., Buchman A., Kornberg R., Rine J. (1988) EMBO J. 7, 2241–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ehrenhofer-Murray A. E., Rivier D. H., Rine J. (1997) Genetics 145, 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu E. Y., Kim S., Replogle K., Rine J., Rivier D. H. (1999) Genetics 153, 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reifnyder C., Lowell J., Clarke A., Pillus L. (1997) Nat. Genet. 16, 109. [DOI] [PubMed] [Google Scholar]
- 104.Han J., Zhou H., Li Z., Xu R. M., Zhang Z. (2007) J. Biol. Chem. 282, 14158–14164 [DOI] [PubMed] [Google Scholar]
- 105.Yang B., Miller A., Kirchmaier A. L. (2008) Mol. Biol. Cell 19, 4993–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bi X., Yu Q., Sandmeier J. J., Zou Y. (2004) Mol. Cell. Biol. 24, 2118–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang J. C. (1982) Cell 29, 724–726 [DOI] [PubMed] [Google Scholar]
- 108.Galvani A., Courbeyrette R., Agez M., Ochsenbein F., Mann C., Thuret J. Y. (2008) Mol. Cell. Biol. 28, 3672–3685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaplan T., Liu C. L., Erkmann J. A., Holik J., Grunstein M., Kaufman P. D., Friedman N., Rando O. J. (2008) PLoS Genet. 4, e1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li Q., Zhou H., Wurtele H., Davies B., Horazdovsky B., Verreault A., Zhang Z. (2008) Cell 134, 244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Han J., Zhou H., Li Z., Xu R. M., Zhang Z. (2007) J. Biol. Chem. 282, 28587–28596 [DOI] [PubMed] [Google Scholar]
- 112.Raisner R. M., Hartley P. D., Meneghini M. D., Bao M. Z., Liu C. L., Schreiber S. L., Rando O. J., Madhani H. D. (2005) Cell 123, 233–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shia W. J., Li B., Workman J. L. (2006) Genes Dev. 20, 2507–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nakatani Y., Ray-Gallet D., Quivy J. P., Tagami H., Almouzni G. (2004) Cold Spring Harbor Symp. Quant. Biol. 69, 273–280 [DOI] [PubMed] [Google Scholar]
- 115.Loyola A., Bonaldi T., Roche D., Imhof A., Almouzni G. (2006) Mol. Cell 24, 309–316 [DOI] [PubMed] [Google Scholar]
- 116.Xu E. Y., Zawadzki K. A., Broach J. R. (2006) Mol. Cell 23, 219–229 [DOI] [PubMed] [Google Scholar]
- 117.Pillus L., Rine J. (1989) Cell 59, 637–647 [DOI] [PubMed] [Google Scholar]
- 118.Katan-Khaykovich Y., Struhl K. (2005) EMBO J. 24, 2138–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Osborne E. A., Dudoit S., Rine J. (2009) Nat. Genet. 41, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.