

Abstract
The epithelial Na+ channel (ENaC) mediates the rate-limiting step in transepithelial Na+ transport in the distal segments of the nephron and in the lung. ENaC subunits are cleaved by proteases, resulting in channel activation due to the release of inhibitory tracts. Peptides derived from these tracts inhibit channel activity. The mechanism by which these intrinsic inhibitory tracts reduce channel activity is unknown, as are the sites where these tracts interact with other residues within the channel. We performed site-directed mutagenesis in large portions of the predicted periphery of the extracellular region of the α subunit and measured the effect of mutations on an 8-residue inhibitory tract-derived peptide. Our data show that the inhibitory peptide likely binds to specific residues within the finger and thumb domains of ENaC. Pairwise interactions between the peptide and the channel were identified by double mutant cycle experiments. Our data suggest that the inhibitory peptide has a specific peptide orientation within its binding site. Extended to the intrinsic inhibitory tract, our data suggest that proteases activate ENaC by removing residues that bind at the finger-thumb domain interface.
Keywords: Acid-sensing Ion Channels (ASIC), Allosteric Regulation, Epithelium, Protein Structure, Sodium Channels, ENaC, Peptide-binding Site
Introduction
The epithelial Na+ channel (ENaC)2 is expressed at the apical surface of Na+-transporting epithelia such as the distal nephron of the kidney, distal colon, and lung alveoli and airway. In conjunction with the Na+/K+-ATPase, ENaC transfers Na+ from the luminal to the interstitial space. This transfer is crucial in regulating blood pressure through its role in renal Na+ absorption and in regulating airway surface liquid volume and mucociliary clearance through its role in airway Na+ absorption. In accord with its role in these processes, improper ENaC function is implicated in several disorders. There is a growing body of evidence that enhanced ENaC activity in the airways of individuals with cystic fibrosis contributes to depletion of airway surface liquids resulting in poor mucociliary clearance (1–3). In the kidney, increased levels of aldosterone activate ENaC and increase the reabsorption of filtered Na+ (4). In both instances, increases in channel activity reflect, in part, enhanced channel proteolysis. Proteinuric states, characterized by excessive protein in the urine, are often accompanied by renal Na+ retention, volume expansion, and hypertension. Recent work indicates that volume expansion in proteinuric states reflects proteolytic activation of ENaC (5–7).
ENaC is a trimer composed of three homologous subunits, α, β, and γ (8, 9). ENaC subunits are members of the much larger ENaC/Degenerin family of ion channel proteins. These channels share a few salient features as follows: 1) most are gated by ligands and/or mechanical forces; 2) they are Na+-permeable and blocked by amiloride, a potassium-sparing diuretic; and 3) each subunit has two transmembrane helices (six transmembrane helices for the full channel), short intracellular N and C termini, and a large extracellular region comprised of several domains. Acid-sensing ion channels (ASIC) are also members of the ENaC/Degenerin family. The recently resolved structure of ASIC1 has provided important clues regarding the structural organization of ENaCs. Of note is that its extracellular region has well defined domains, termed finger, thumb, palm, knuckle, and β-ball.
ENaC α and γ subunits undergo a very unusual form of regulatory processing. Each subunit can be cleaved at two (or more) distinct extracellular sites resulting in the liberation of a small stretch of amino acids and an increase in channel activity (10, 11). For both subunits, we have shown that synthetic peptides corresponding to these released tracts, and subsets thereof, are inhibitory (12–14). We reasoned that the inhibitory peptides and proteolytically liberated fragments have similar binding sites and inhibitory mechanisms.
In an effort to elucidate the mechanism of proteolytic activation of ENaC, we functionally characterized the binding site for an α subunit-derived 8-residue inhibitory peptide. To map sites within α ENaC that interact with this peptide, we systematically mutated individual residues within several peripheral regions of the α subunit to Trp and measured the effect of these mutations on peptide-dependent channel inhibition. As some mutations may indirectly affect the ability of the peptide to inhibit the channel, we analyzed our data within a thermodynamic framework that allowed us to deduce the direct effects of mutations on peptide-dependent channel inhibition. Using these data, we performed double mutant cycle experiments to identify pairwise interactions. We found two residues that interact with a site toward the N terminus of the peptide and one residue that interacts with the C terminus of the peptide. Our results suggest that the peptide binds to both the finger and thumb domains, with the N terminus of the peptide binding at the finger-thumb interface. Because the peptide is an allosteric inhibitor of ENaC, these data provide support for the importance of the finger-thumb interface in the mechanism of ENaC gating.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis and Oocyte Expression
Mouse α, β, and γ ENaC subunit cDNAs in pBluescript SK− vector (Stratagene, La Jolla, CA) were used as templates to generate mutations using QuikChange II XL (Stratagene) following the manufacturer's instructions. Direct sequencing was used to confirm target mutations. cRNAs for wild-type and mutant α, β, and γ mouse ENaC subunits were synthesized with T3 mMessage mMachineTM (Ambion, Austin, TX) and purified using RNeasy® MinEluteTM cleanup kit (Qiagen, Valencia, CA). Stage V–VI Xenopus laevis oocytes were injected with 1 ng of cRNA of each ENaC subunit.
Peptides
The peptides were synthesized and HPLC-purified by GenScript Corp. (Piscataway, NJ). All peptides were modified by N-terminal acetylation and C-terminal amidation.
Two-electrode Voltage Clamp
Electrophysiological measurements were performed 22–30 h after injection using a GeneClamp 500B voltage clamp amplifier (Axon Instruments, Foster City, CA), Clampex software (Axon Instruments), and a 20-μl oocyte recording chamber (Automate Scientific, San Francisco). Perfusion was controlled using a perfusion system employing a ValveLink8.2® controller and pinch valves (Automate Scientific). The standard bath solution was 110 mm NaCl, 2 mm KCl, 2 mm CaCl2, 10 mm HEPES, pH 7.4. Bath solutions with lower NaCl (1 mm NaCl) were made by substituting NaCl with N-methyl-d-glucamine in the standard bath solution. 10 mm stock solutions of peptides dissolved in water were used to add peptide to bath solutions, as indicated. The amiloride-sensitive component of the whole cell Na+ current was determined by perfusion with bath solution supplemented with 10 μm amiloride and was defined as the ENaC-mediated current.
Statistical Analysis
Significance comparisons between groups were performed with Student's t test, analysis of variance, or nonlinear mixed regression model analysis, as indicated.
Nonlinear Mixed Regression (NLMR) Model Analysis
To account for the dependence between repeated assessments of the same mutant under different experimental conditions, we used the nonlinear mixed effects R library nlme (15) to estimate the parameters of Equation 3. In the NLMR model formulation of this regression problem, the uncertainty concerning the value of the ιmax parameter (fixed effect) may be directly obtained from the output of the software. Because the kinetic constants appear as random effects, only their values (not the associated uncertainty) can be estimated through NLMR. To evaluate the uncertainty in these values and calculate the statistical significance of the former being different from the wild type, we utilized Markov Chain Monte Carlo methods. Briefly, the parameter estimates obtained by nlme were used to initialize the Gibbs sampler (a Markov Chain Monte Carlo algorithm), which was applied to the nonlinear regression problem of Equation 3. At convergence, the Markov Chain Monte Carlo algorithm provided simultaneous estimates for the model parameters and the associated uncertainties (S.E.). p values corresponding to the indicated hypotheses were calculated by Monte Carlo integration from the random samples obtained by the Gibbs sampler. These p values were adjusted by applying the Bonferonni correction, which in this case amounts to dividing the observed p value by the number of tests (number of different mutant types) examined. An adjusted value of p < 0.0001 was used to determine significance. For other multiple comparisons, one-way analysis of variance followed by a Student Newman-Keuls test and curve fitting to the Hill equation were performed using Igor Pro (Wavemetrics, Oswego, OR).
RESULTS
Influence of Channel Po on Apparent Peptide Affinity
We sought to identify α ENaC residues involved in α subunit-derived inhibitory peptide (P8; Ac-LPHPLQRL-amide; corresponding α ENaC residues 211–218) binding by disrupting binding through site-directed mutagenesis. However, we recently suggested that mutations (or any stimulus) that shift the equilibrium to favor an open channel state will also alter the apparent affinity of the channel for the inhibitory peptide, regardless of whether the mutation directly alters the peptide-binding site (12). This prediction is based on the notion that P8 is an allosteric inhibitor that thermodynamically stabilizes the closed state of the channel. As a consequence, factors that increase channel open probability (Po) are predicted to weaken apparent P8 affinity, as given by Equation 1,
where K′P is the apparent P8 affinity, and KCP is the dissociation constant for P8 bound to the ENaC closed state (see Ref. 12).
This idea was tested using the fact that high [Na+] decreases ENaC Po, a phenomenon known as Na+ self-inhibition (16). We measured the dose response of wild-type channels to P8 at three different external [Na+] (Fig. 1A). At low [Na+], where ENaC Po is high (16), the apparent P8 affinity was reduced as predicted by Equation 1. Increasing channel Po through the introduction of a point mutation also evokes the same response. Mutating the pore-lining residue βSer-518 to Lys gives an estimated Po of 0.97 (17) resulting in a greatly reduced P8 affinity (Fig. 1B). Similarly, ENaC channels lacking the γ inhibitory tract have a high Po and exhibit reduced efficacy of a peptide derived from the α subunit inhibitory tract (18).
FIGURE 1.

Effect of ENaC Po on P8 inhibition. A, effect of external [Na+] on P8 inhibition. Amiloride-sensitive currents were measured by two-electrode voltage clamp at −100 mV for 10 and 110 mm external Na+ and +40 mV for 1 mm external Na+ at the [P8] indicated. Values are mean ± S.D. (n = 6). Data were fit to the Hill equation. At 110 mm Na+, IC50 = 0.8 ± 0.1 μm; at 10 mm Na+, IC50 = 7 ± 1 μm; at 1 mm Na+, IC50 > 100 μm. B, effect of βS518K mutation on P8 inhibition. Effect of 10 μm P8 on amiloride-sensitive currents of oocytes injected with αβγ or αβS518Kγ ENaC were measured by two-electrode voltage clamp at −100 mV. Values are mean ± S.D. (n = 5). *, p < 0.001 versus wild-type by Student's t test.
To measure the effect of our mutations on P8 binding, we needed to determine whether mutations significantly enhanced channel Po, as this would indirectly reduce apparent P8 affinity. We recently observed a linear correlation between measurements of ENaC Po and Na+ self-inhibition (19), suggesting that measurements of Na+ self-inhibition could serve as a surrogate to estimate Po for each of our mutants. Na+ self-inhibition can be measured by rapidly replacing a 1 mm Na+ bath, where ENaC Po is high, with a 110 mm Na+ bath, where ENaC Po is lower (16). Acutely raising [Na+] immediately increases the driving force, resulting in a peak current (IPeak) that subsequently declines to a steady state current (ISS) as the channel arrives at a new equilibrium at a lower Po (19). With Na+ self-inhibition (ιNaSI) measured as ιNaSI = 100·(1 − ISS/IPeak), ENaC Po can be estimated using Equation 2,
based on our previous report describing the relationship between Na+ self-inhibition and channel Po (19). Equation 3 provides an estimate of the percent of current inhibition by P8 (ιP8) derived from our previously presented scheme (12) and Equation 2,
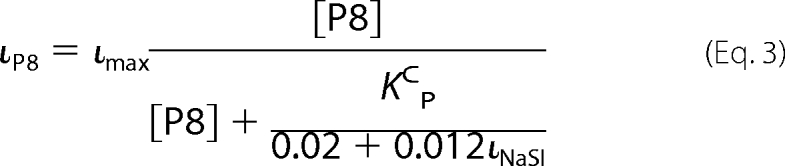 |
In Equation 3, ιmax represents maximal P8 inhibition as a percentage of total ENaC current. Therefore, we can estimate KCP for each species by measuring its P8 inhibition at a given [P8] and its Na+ self-inhibition prior to peptide addition using Equation 3, which has the form of the Michaelis-Menten equation.
Selection of Potential P8-binding Residues
The protease cleavage sites and excised inhibitory tract align within the finger domain of the channel. However, there is poor sequence identity between α ENaC and ASIC1 in their finger domains, including a 73-residue insert in α ENaC. As a result, the ASIC1 structure did not readily provide a predicted site of P8 interaction (Fig. 2A). To remedy this, we searched the RCSB protein data base and identified XC6422 from Xanthomonas campestris (PDB code 2FUK) as having two short interacting stretches of residues that share 70% sequence identity with α ENaC (Fig. 2B). One stretch corresponded to residues preceding and including P8, and the other stretch corresponded to the latter part of the finger including residues 287–294. Although XC6422 and α ENaC are not homologous, we hypothesized that a similar interaction might occur within α ENaC. Residues 287–294 in α ENaC roughly correspond to the α2-α3 loop in the late finger of ASIC1 (Fig. 2A). If P8 binds α ENaC near the α2-α3 loop, we hypothesized that nearby residues at the top of the thumb and the early finger may also be involved in P8 binding based on the ASIC1 structure. We tested our hypothesis by attempting to disrupt P8 binding through the introduction of bulky Trp residues at sites within three region as follows: 187–198 (early finger), 280–295 (α2-α3 loop; late finger), and 470–482 (top of the thumb). Based on these results, we extended our mutagenesis analyses to encompass residues 232–295 (late finger) and 466–487 (top of the thumb).
FIGURE 2.

Predicted sites of P8 interaction with α ENaC. A, ribbon diagram of ASIC1 trimer. One subunit is highlighted, and the other two subunits are represented as transparent ribbons. Approximate locations of outer (Out) and inner (In) borders of the cell membrane are indicated. B, small sections of XC6422 from X. campestris (Protein Data Bank code 2FUK) were used to select potential P8-binding sites on α ENaC. Short stretches with high sequence identity between α ENaC and 2FUK were identified. In XC6422, these sequences interact (red). The 287–294-amino acid sequence of α ENaC corresponds to the α2-α3 loop of ASIC1. Based on the predicted placement of P8 corresponding residues 211–218 near the α2-α3 loop, and the proximity of the α2-α3 loop to the top of the thumb and the early finger within the resolved ASIC1 structure, we selected proximate tracts 187–199 (early finger), 280–295 (late finger), and 470–482 (top of the thumb) for initial experiments to probe P8 binding.
Mutations in the Finger and Thumb Domains Alter P8 Inhibition
Using Equation 3 above, we determined the underlying peptide-binding constant (KCP) of each mutant. To do this, we measured P8 (10 μm) inhibition (ιP8 = 100·(1 − IP8/ISS)) and Na+ self-inhibition (rapidly increasing [Na+] from 1 to 110 mm) of these mutants and of wild-type channels (Fig. 3, A and C). In addition, to fully characterize the relationship between P8 inhibition and Na+ self-inhibition for wild-type channels, we measured the P8 (10 μm) inhibition and Na+ self-inhibition of wild-type channels at 10 mm Na+ and P8 (10 μm) inhibition at 1 mm Na+ (Fig. 3B). To determine whether these mutations altered the peptide-binding constant, KCP, with respect to wild-type channels, we evaluated data for wild-type and mutant channels using NLMR models, which estimated the parameters of Equation 3 (Fig. 3C and supplemental Table S1). ιmax was a common parameter to the fits for all species, and KCP was an independent parameter for each species. For mutants where 10 μm P8 poorly inhibited the channel, we cannot readily distinguish between effects on KCP and ιmax in the absence of a dose-response curve. The fitted parameters for wild-type ENaC were similar to those determined from Fig. 1A in high [Na+]. Fig. 4A shows the average values of P8 inhibition and Na+ self-inhibition for each mutant overlaid with the fitted curve for wild-type ENaC. Mutants with peptide-binding constants similar to wild-type (Fig. 4A, black) had average P8 inhibition and Na+ self-inhibition values near the wild-type fitted curve. Mutants with weaker P8 affinity (Fig. 4A, red and blue) fell below the wild-type curve (p < 0.0001), and five of these mutants weakened KCP greater than 10-fold (blue; p < 0.0001).
FIGURE 3.

Nonlinear mixed regression model analyses of P8 inhibition of ENaC mutants. A, experimental protocol for measuring P8 inhibition with wild-type representative experiment shown. [P8] and [amiloride] (A) were 10 μm. cRNAs for wild-type and indicated mutant ENaC α subunits were mixed with wild-type β and γ ENaC cRNAs and injected into Xenopus oocytes at 1 ng/subunit/oocyte. The peak and steady state currents observed during the Na+ self-inhibition response are noted (IPeak and ISS), as are the currents following the addition of P8 and amiloride (IP8 and IAmil). B, P8 inhibition versus Na+ self-inhibition for individual oocytes expressing wild-type ENaC. Experiments were performed following the protocol in A (n = 104) or modified so that the 110 mm Na+ bath was instead 10 mm Na+ (n = 14) or 1 mm Na+ (n = 6). Na+ self-inhibition was reported as 100·(1 − ISS/IPeak). Data were fit to Equation 3. C, data for each mutant with fitted curves are shown. Each circle represents an individual experiment. Background shading of mutant labels indicate region of mutation as follows: early finger (white), late finger (gray), or top of thumb and wild type (black). Data were fit to Equation 3 by nonlinear mixed model analyses, with resulting parameters presented in supplemental Table S1.
FIGURE 4.

Mutations at sites in the late finger and top of the thumb of α ENaC affect P8 inhibition. A, summary of the effects of mutations on P8 inhibition and Na+ self-inhibition. Each point represents the mean values of P8 and Na+ self-inhibition for an individual mutation. The fitted curve for wild-type ENaC (see Fig. 3B) is overlaid for comparison. Statistical analyses using NLMR were performed to determine whether mutants bound P8 with weaker affinity (red and blue) or 10-fold weaker affinity (blue) than wild-type. B, effect of Trp mutation at individual sites on P8 inhibition. Mutations in the latter half of the finger domain and the top of the thumb reduced P8 inhibition (10 μm P8). Two Cys (C) residues in the thumb that may be functionally important and three endogenous Trp (W) residues were not mutated. Values are mean ± S.D. (n = 5–12). Dashed line indicates wild-type value (77 ± 6%). Red and blue bars are as defined for A. p values were determined by nonlinear mixed regression model analyses (see Fig. 3) and are indicated here and in supplemental Table S1. Values of p < 0.0001 were considered significant.
Taking into account the effect of mutations on Po, we observed that mutations introduced into the late finger and the top of the thumb, but not the early finger, altered P8 inhibition (Fig. 4B). Mutations that reduced P8 inhibition at the top of the thumb occurred within a narrow range of positions (residues 470–473) and had modest effects on P8 inhibition. In contrast, mutations that reduced P8 inhibition were found throughout the late finger (residues 239–289) and had modest (Fig. 4B, red) to strong (blue) effects on P8 inhibition. These results suggest that P8 interacts primarily with residues in the late finger and with a limited number of residues at the top of the thumb.
Identifying Specific α ENaC-P8 Interactions with Double Mutant Cycle Analyses
Having identified sites that likely interact with P8, we sought to identify specific pairwise interactions between α ENaC and P8. We previously characterized the inhibition of ENaC by P8 analogues bearing substitutions throughout the peptide (13). Many of these substitutions reduced apparent peptide affinity. We reasoned that a channel mutation that reduced P8 binding may be reversed by a compensatory mutation in P8 akin to a double mutant cycle where the free energy of coupling, ΔΔGint, is non-zero (20). We therefore selected α ENaC mutants with reduced P8 affinity discovered in our Trp scan, and subsequently screened them against these peptides (Table 1). We identified four complementary peptide-channel pairs where a peptide mutation partially restored channel inhibition; both αD473W and αR289W with the mutant peptide LPKPLQRL and both αG252W and αQ254W with the mutant peptide LPHPLQRA (the underlined residue has been changed). αD473W is at the top of the thumb domain, and αR289W, αG252W, and αQ254W are in the latter half of the finger domain.
TABLE 1.
Screening data for mutant cycle analysis. Peptide inhibition of wild-type or α subunit mutant ENaC was determined at the indicated [peptide]. Mutant channel inhibition is expressed relative to wild-type channel inhibition for the same peptide. Values are expressed as mean (n = 2–3).
| Inhibition |
|||||||
|---|---|---|---|---|---|---|---|
| LPHPLQRL (10 μm) | APHPLQRL (30 μm) | LGHGLQRL (50 μm) | LPKPLQRL (50 μm) | LPHPLARL (10 μm) | LPHPLQAL (10 μm) | LPHPLQRA (50 μm) | |
| % | |||||||
| Wild-type | 77.22 | 52.19 | 51.22 | 42.24 | 51.22 | 49.64 | 41.82 |
| Inhmut/Inhwild type | |||||||
| G252W | 0.15 | 0.14 | 0.23 | 0.37 | |||
| Q254W | 0.39 | 0.34 | 0.30 | 0.70 | |||
| L255W | 0.46 | 0.20 | 0.14 | 0.21 | 0.19 | ||
| C256W | 0.49 | 0.40 | 0.25 | 0.17 | 0.50 | ||
| T267W | 0.11 | 0.13 | −0.02 | 0.11 | 0.02 | ||
| A274W | 0.52 | 0.41 | 0.56 | 0.52 | 0.37 | 0.36 | 0.31 |
| V275W | 0.43 | 0.35 | 0.41 | 0.31 | 0.21 | 0.23 | 0.18 |
| R289W | 0.29 | 0.21 | 0.32 | 0.48 | 0.20 | 0.20 | 0.11 |
| D473W | 0.76 | 0.73 | 0.67 | 1.02 | 0.54 | 0.48 | 0.48 |
If these sites are truly in physical contact, it is likely that P8 peptides with mutations at the third or eighth positions will have effects on the inhibition of mutant channels that are less than the additive effects of a mutant peptide on a wild-type channel and a wild-type peptide on a mutant channel. To investigate this further, we measured the affinity of several third position substituted 8-mers for αD473W and αR289W, as well as the affinity of several eighth position substituted peptides for αQ254W. In each case, we compared the results to the peptide binding affinity of wild-type ENaC (Fig. 5 and Table 2). Unfortunately, αG252W had a P8 affinity too weak for further study (data not shown). Although wild-type ENaC had 4-fold higher affinity for P8 than the αD473W mutant, these two channels had similar affinity for each of the peptides tested with third position substitutions. If ΔΔGint was zero (implying no coupling), we would have expected a 4-fold difference in affinity to be maintained for each of these peptides (i.e. the effects of peptide and channel mutations would be additive). We observed a similar effect for αR289W, where its 130-fold weaker affinity for P8 reduced to a 13-fold weaker affinity for each of the third position-substituted peptides tested. We also observed coupling between αQ254W and the eighth position of the peptide, so that a 10-fold difference in P8 affinity was eliminated for each of the eighth position-substituted peptides tested, as well as a 7-mer lacking a residue at this position. These results suggest that αAsp-473 and αArg-289 likely interact with the third position of P8 and that αGln-254 likely interacts with the eighth position of P8.
FIGURE 5.

Pairwise interactions between P8 and α ENaC residues. Dose-response curves of various peptides with wild-type and mutant ENaCs as indicated (n = 5–8). Values are mean ± S.D. Data were fit to the Hill equation. Fitted curves for wild-type (dashed line) and mutant (solid line) channels are shown. IC50 values are in Table 2. Where inhibition was <70% at the maximally tested [peptide], maximum inhibition was fixed to 90% for curve fitting.
TABLE 2.
Dose-response curve parameters for double mutant cycle experiments
IC50 values were determined by fits of data in Fig. 3 to the Hill equation.
| Peptide | IC50 (μm) |
|||
|---|---|---|---|---|
| αβγ | αD473Wβγ | αR289Wβγ | αQ254Wβγ | |
| LPHPLQRL | 0.78 ± 0.09 | 3.1 ± 0.9 | 100 ± 10 | 9 ± 2 |
| LPYPLQRL | 2.5 ± 0.2 | 2.0 ± 0.7 | 32 ± 8 | |
| LPEPLQRL | 30 ± 5 | 39 ± 9 | 400 ± 20 | |
| LPKPLQRL | 64 ± 9 | 50 ± 10 | ||
| LPHPLQRA | 50 ± 30 | 50 ± 30 | ||
| LPHPLQRN | 56 ± 4 | 41 ± 6 | ||
| LPHPLQR_ | 130 ± 90 | 100 ± 70 | ||
Effects of ENaC Mutations on Na+ Self-inhibition
To control for the indirect effect on P8 inhibition by the effect of a mutation on channel Po, we measured the Na+ self-inhibition response of each mutant. Several mutants exhibited an altered Na+ self-inhibition response as compared with the wild-type response (Fig. 6). The sites of these mutations spanned a wide portion of the latter half of the finger and a narrow region of the top of the thumb. The sites at the top of the thumb correspond to residues in the first half of helix α5. Although α ENaC sequence identity to ASIC1 is poor in the finger domain, the sites in the late finger include residues between 264 and 286 that may correspond to helix α2, and indeed the observed effects on Na+ self-inhibition occurred at sites flanked by 2 or 3 residues where mutations had no effect. Projected onto a helical wheel (Fig. 6C), it becomes clear that mutations between 270 and 286 that affect Na+ self-inhibition lie on one face of a putative α-helix. We recently reported similar results for the γ subunit (21). The sites in the late finger between 236 and 256 correspond to no known structure, and the observed effects follow no discernable pattern.
FIGURE 6.

Mutations at sites in the late finger and top of the thumb of α ENaC affect Na+ self-inhibition. A, effect of Trp mutation at individual sites on Na+ self-inhibition, defined as 100·(1 − ISS/IPeak). Two Cys (C) residues in the thumb and three endogenous Trp (W) residues were not mutated. Values are mean ± S.D. (n = 5–12). Dashed line indicates wild-type value (37 ± 10%). p values versus wild-type channels were determined by analysis of variance with a Newman-Keuls post-hoc test. Values of p < 0.0001 were considered significant. B, helical wheel projection of residues 265–285. Mutations at residues that affected Na+ self-inhibition are gray. αTrp-278 was not mutated.
DISCUSSION
Proteolytic cleavage leading to channel activation is an unusual mechanism for channel regulation in biology. Although proteolytic cleavage has an important role in regulating ENaC, proteolysis may also regulate other members of the ENaC/Degenerin family (22). For ENaC, both the α and γ subunits undergo proteolysis in association with channel activation (10, 11, 14, 23). The activation of ENaC by proteolysis requires that a subunit be cleaved twice, releasing an intrinsic inhibitory tract. Peptides corresponding to the sequences of these excised tracts within the α and γ subunits inhibit ENaC currents (10, 12–14). These inhibitory tracts reside within the variable finger domains of these subunits and have no analogs within ASIC1 or other ENaC/Degenerin family members (8).
In this study, we identified two distinct regions that are likely involved in the binding of an 8-residue peptide derived from the α subunit inhibitory tract. We note that mutations at these sites may have indirectly affected peptide binding through distortions of structure or access to the binding site, and we interpret our results mindful of this limitation. These regions encompass residues 470–473 in the thumb domain, and residues 239–289 in the finger domain. Neither region is contiguous with the α subunit inhibitory tract (residues 206–231) or residues corresponding to P8 (211–218). We also identified specific pairwise interactions between ENaC and P8. As Arg-289 and Asp-473 both interact with the third position of the peptide, our data suggest that Arg-289 and Asp-473 are in close proximity and that the N-terminal region of the peptide binds at a thumb-finger interface. As Arg-289 follows a stretch of residues that our Na+ self-inhibition data suggest is helical, our data suggest that a putative loop containing Arg-289 is in close proximity to the top of the thumb. This suggests that ENaC shares some common structural features with ASIC1 within the finger domain despite a lack of sequence identity in this region, as the α2-α3 loop within the finger domain of ASIC is also in close proximity to its thumb domain. We also found that Gln-254 interacts with the eighth position of P8. If the peptide assumes an extended conformation in the bound state, our data suggest that Gln-254 is not adjacent to the thumb-finger interface.
There is now strong evidence that P8 is an allosteric inhibitor of ENaC. First, we previously found that P8 binding is not voltage-dependent despite the fact that it is positively charged at pH 7.4, suggesting that P8 does not bind within the ion permeation pathway of the channel (14). Second, we have shown that apparent P8 affinity is influenced by channel open probability (Fig. 1, A and B). Third, we have identified mutations at residues in the finger and the thumb domains of α ENaC that attenuate P8 affinity, suggesting that P8 binds in the periphery of the channel and far from the pore. The finding that P8 is an allosteric inhibitor of ENaC leads to the conclusion that P8 inhibits the channel by preferentially stabilizing the closed state of the channel. Because P8 is derived from the furin-excised inhibitory tract, we suggest that the inhibitory tract functions largely through similar mechanisms. Based on this reasoning, we propose that furin activates ENaC through α subunit cleavage by removing an intrinsic allosteric inhibitor from a site partially defined by the finger-thumb interface.
In the course of this study, we measured the Na+ self-inhibition of α subunit Trp mutants within the finger and thumb domains. Interpretation of these results is complicated by the fact that we cannot readily distinguish between mutations that directly affect Na+ self-inhibition by altering Na+ binding and mutations that indirectly affect Na+ self-inhibition by altering the downstream allosteric machinery of the channel. For example, the βS518K pore mutant largely eliminates ENaC Na+ self-inhibition (21). If this was evidence for a Na+-binding site nearby, Na+ self-inhibition would be predicted to be voltage-dependent, contrary to published work (24). The simplest explanation is that βS518K stabilizes the open state of the channel relative to the closed state, raising the thermodynamic barrier to channel closure by Na+. Mindful of this limitation, our Na+ self-inhibition data provide evidence for an α-helix that encompasses residues 271–285.
The finger domains of members of the ENaC/Degenerin family of ion channels are hypervariable, which leads to the hypothesis that the finger domains of these proteins are functional modules. In the case of α ENaC, the finger domain appends protease sensitivity and possibly Na+ sensitivity as well. Given the modular finger hypothesis and the large number of sites in the finger domain where mutations altered Na+ self-inhibition, some of these sites may be directly involved in Na+ binding. Also, consideration of the P8-binding site within the finger and at the finger-thumb interface suggests that P8, and by extension the α-inhibitory tract, recruits much of the same allosteric machinery as used by Na+.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DK078734 (to O. B. K.), DK051391 (to T. R. K.), DK065161 (to T. R. K. and R. P. H.), and DK079307 (Pittsburgh Center for Kidney Research). This work was also supported by a scientist development grant from the American Heart Association and a grant from the PNC Charitable Trust Foundation (to M. G.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
- ENaC
- epithelial Na+ channel
- P8
- Ac-LPHPLQRL-amide
- ASIC1
- acid-sensing ion channel 1
- NLMR
- nonlinear mixed regression.
REFERENCES
- 1.Rauh R., Diakov A., Tzschoppe A., Korbmacher J., Azad A. K., Cuppens H., Cassiman J. J., Dötsch J., Sticht H., Korbmacher C. (2010) J. Physiol. 588, 1211–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fajac I., Viel M., Gaitch N., Hubert D., Bienvenu T. (2009) Eur. Respir. J. 34, 772–773 [DOI] [PubMed] [Google Scholar]
- 3.Sheng S., Johnson J. P., Kleyman T. R. (2008) in The Kidney, Physiology and Pathophysiology (Alpern R. J., Hebert S. C. eds) 4th Ed., pp. 743–768, Elsevier Publishing, Philadelphia [Google Scholar]
- 4.Bhalla V., Hallows K. R. (2008) J. Am. Soc. Nephrol. 19, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 5.Kastner C., Pohl M., Sendeski M., Stange G., Wagner C. A., Jensen B., Patzak A., Bachmann S., Theilig F. (2009) Am. J. Physiol. Renal Physiol. 296, F902–F911 [DOI] [PubMed] [Google Scholar]
- 6.Passero C. J., Hughey R. P., Kleyman T. R. (2010) Curr. Opin. Nephrol. Hypertens. 19, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Passero C. J., Mueller G. M., Rondon-Berrios H., Tofovic S. P., Hughey R. P., Kleyman T. R. (2008) J. Biol. Chem. 283, 36586–36591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jasti J., Furukawa H., Gonzales E. B., Gouaux E. (2007) Nature 449, 316–323 [DOI] [PubMed] [Google Scholar]
- 9.Staruschenko A., Adams E., Booth R. E., Stockand J. D. (2005) Biophys. J. 88, 3966–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruns J. B., Carattino M. D., Sheng S., Maarouf A. B., Weisz O. A., Pilewski J. M., Hughey R. P., Kleyman T. R. (2007) J. Biol. Chem. 282, 6153–6160 [DOI] [PubMed] [Google Scholar]
- 11.Hughey R. P., Bruns J. B., Kinlough C. L., Harkleroad K. L., Tong Q., Carattino M. D., Johnson J. P., Stockand J. D., Kleyman T. R. (2004) J. Biol. Chem. 279, 18111–18114 [DOI] [PubMed] [Google Scholar]
- 12.Passero C. J., Carattino M. D., Kashlan O. B., Myerburg M. M., Hughey R. P., Kleyman T. R. (2010) Am. J. Physiol. Renal Physiol. 299, F854–F861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carattino M. D., Passero C. J., Steren C. A., Maarouf A. B., Pilewski J. M., Myerburg M. M., Hughey R. P., Kleyman T. R. (2008) Am. J. Physiol. Renal Physiol. 294, F47–F52 [DOI] [PubMed] [Google Scholar]
- 14.Carattino M. D., Sheng S., Bruns J. B., Pilewski J. M., Hughey R. P., Kleyman T. R. (2006) J. Biol. Chem. 281, 18901–18907 [DOI] [PubMed] [Google Scholar]
- 15.Pinheiro J. C., Bates D. M. (2000) in Mixed-Effects Models in S and S-PLUS (Chambers J., Eddy W., Härdle W., Sheather S., Tierney L. eds) pp. 271–414, Springer-Verlag, New York [Google Scholar]
- 16.Sheng S., Carattino M. D., Bruns J. B., Hughey R. P., Kleyman T. R. (2006) Am. J. Physiol. Renal Physiol. 290, F1488–F1496 [DOI] [PubMed] [Google Scholar]
- 17.Condliffe S. B., Zhang H., Frizzell R. A. (2004) J. Biol. Chem. 279, 10085–10092 [DOI] [PubMed] [Google Scholar]
- 18.Carattino M. D., Hughey R. P., Kleyman T. R. (2008) J. Biol. Chem. 283, 25290–25295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maarouf A. B., Sheng N., Chen J., Winarski K. L., Okumura S., Carattino M. D., Boyd C. R., Kleyman T. R., Sheng S. (2009) J. Biol. Chem. 284, 7756–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horovitz A. (1996) Fold. Des. 1, R121–R126 [DOI] [PubMed] [Google Scholar]
- 21.Winarski K. L., Sheng N., Chen J., Kleyman T. R., Sheng S. (2010) J. Biol. Chem. 285, 26088–26096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark E. B., Jovov B., Rooj A. K., Fuller C. M., Benos D. J. (2010) J. Biol. Chem. 285, 27130–27143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleyman T. R., Carattino M. D., Hughey R. P. (2009) J. Biol. Chem. 284, 20447–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bize V., Horisberger J. D. (2007) Am. J. Physiol. Renal Physiol. 293, F1137–F1146 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.