


Abstract
8-Oxoguanine-DNA glycosylase 1 (OGG1), with intrinsic AP lyase activity, is the major enzyme for repairing 7,8-dihydro-8-oxoguanine (8-oxoG), a critical mutagenic DNA lesion induced by reactive oxygen species. Human OGG1 excised the damaged base from an 8-oxoG·C-containing duplex oligo with a very low apparent kcat of 0.1 min–1 at 37°C and cleaved abasic (AP) sites at half the rate, thus leaving abasic sites as the major product. Excision of 8-oxoG by OGG1 alone did not follow Michaelis–Menten kinetics. However, in the presence of a comparable amount of human AP endonuclease (APE1) the specific activity of OGG1 was increased ∼5-fold and Michaelis–Menten kinetics were observed. Inactive APE1, at a higher molar ratio, and a bacterial APE (Nfo) similarly enhanced OGG1 activity. The affinity of OGG1 for its product AP·C pair (Kd ∼ 2.8 nM) was substantially higher than for its substrate 8-oxoG·C pair (Kd ∼ 23.4 nM) and the affinity for its final β-elimination product was much lower (Kd ∼ 233 nM). These data, as well as single burst kinetics studies, indicate that the enzyme remains tightly bound to its AP product following base excision and that APE1 prevents its reassociation with its product, thus enhancing OGG1 turnover. These results suggest coordinated functions of OGG1 and APE1, and possibly other enzymes, in the DNA base excision repair pathway.
INTRODUCTION
7,8-Dihydro-8-oxoguanine, or 8-oxoguanine (8-oxoG), is a major base damage induced in DNA by reactive oxygen species (ROS) and is often used as a key indicator of cellular oxidative stress (1). 8-OxoG is arguably the most important premutagenic lesion in DNA because of its tendency to mispair with A during DNA replication, leading to G→T transversion mutations (2,3). Such mutations may contribute to the loss of genomic integrity and cellular regulation associated with aging and a variety of disease states, including cancer (4–6). 8-OxoG, like other ROS-induced lesions, is repaired primarily via the DNA base excision repair pathway in which the first step is excision of the damaged base by a specific DNA glycosylase (7), namely 8-oxoguanine-DNA glycosylase (OGG), which has been identified in all organisms studied so far (7,8). The importance of 8-oxoG repair by OGGs is underscored by their functional conservation among bacteria and eukaryotes and by the structural conservation of these enzymes between yeast and mammals (9). All known OGGs are of the glycosylase/AP lyase type and excise 8-oxoG residues from DNA by attacking the N-glycosylic bond with an activated nucleophilic amino acid. In OGG1, the major eukaryotic species of OGG, and in other members of the endonuclease III (Nth) family of glycosylases/AP lyases, the nucleophile is a conserved lysine residue (10). Following base removal by these enzymes, a covalent Schiff base is formed between the enzyme and C1′ of the deoxyribose, which can be reduced by sodium borohydride to produce a stable ‘trapped complex’ (11). The Schiff base can either be hydrolyzed, freeing both the enzyme and intact AP site, or undergo a lyase reaction via β-elimination, resulting in cleavage of the DNA phosphate backbone at the abasic site with formation of 3′-phospho-α,β-unsaturated aldehyde (3′,4-hydroxy 2-pentenal) and 5′-phosphate termini (12–14).
The 3′-phospho-α,β-unsaturated aldehyde terminus produced by the β-elimination reaction cannot function as a primer for repair synthesis by DNA polymerases and must be removed by a 3′-phosphodiesterase activity, presumably that of the major AP endonuclease (APE1), to generate a 1 nt gap. DNA polymerases, including DNA polymerase β, then incorporate the correct base at the gapped site and the resulting nick is subsequently sealed by a DNA ligase to complete the repair process (15).
Earlier studies with bacterial and most eukaryotic DNA glycosylases/AP lyases showed that their AP lyase activity was comparable to the base excision activity, so that no free AP sites could be detected during the course of damaged base removal in an in vitro assay (16). This observation is consistent with the unified mechanism of DNA glycosylases/AP lyases which postulates coordinated base excision and β-elimination reactions as a result of Schiff base formation (17). Previous reports suggested that strand incision at AP sites opposite C by OGG1 was not coordinated with excision of 8-oxoG (18,19), indicating that OGG1 may be distinct from other characterized glycosylases/AP lyases in its inability to carry out concerted base excision and strand cleavage reactions. However, the kinetic mechanism of OGG1 was not investigated in earlier studies. Recently, a report on the catalytic mechanism of mouse Ogg1 (mOgg1) and an investigation of its substrate range was published (20). The low turnover reported for mOgg1 in that study was similar to earlier reports regarding human OGG1. It was suggested that the activity of mOgg1 may be affected by other repair enzymes, as has been reported for other DNA glycosylases (21,22), although no detailed kinetic studies have been reported. In this report we show that OGG1 is significantly stimulated by human APE1, an enzyme that acts subsequent to OGG1 in base excision repair. We have also provided the first direct evidence and detailed kinetics studies to elucidate the mechanism of OGG1 activation by APE1. Removal of 8-oxoG from DNA by OGG1 is rate limited by enzyme dissociation from its AP site product, for which APE1 competes, allowing OGG1 to turnover.
MATERIALS AND METHODS
Expression and purification of recombinant OGG1 polypeptides
A human cDNA clone encoding the major 38 kDa isoform of OGG1, but with six additional amino acid residues inserted near the C-terminus of the polypeptide, was received from Drs A. P. Grollman and T. A. Rosenquist (13). When expressed in Escherichia coli the recombinant protein was completely insoluble. We then deleted the additional residues by site-directed mutagenesis in order to produce wild-type OGG1-1a containing 345 amino acid residues (23). The OGG1 coding sequence was then cloned into expression vector pET 28a (Novagen) by inserting the NdeI–EcoRI fragment in-frame after the coding sequence of an N-terminal leader peptide containing 20 amino acid residues, including six histidines (His tag) and a thrombin cleavage site. Missense mutations in the wild-type coding sequence were introduced using a Chameleon double-stranded mutagenesis kit (Stratagene) and confirmed by sequencing of the recombinant plasmid. His-tagged proteins encoded by these constructs were expressed in E.coli BL21 Codon Plus (Stratagene) overnight at 16°C. OGG1 expression was induced with 0.1 mM IPTG when the bacterial cultures reached an OD600 of 0.3–0.4. Cells were harvested by centrifugation and disrupted on ice with a Braun Sonic U sonicator at maximum power for 120 × 0.5 s pulses. Cell-free extracts were centrifuged for 15 min at 10 000 r.p.m. and the soluble fractions were applied to NTA (nickel–nitriloacetic acid)–agarose columns (Qiagen). Affinity purified His-tagged OGGs were eluted with 100 mM imidazole and, after dialysis against 250 mM NaCl, 20 mM Tris–HCl (pH 7.4) were digested with biotinylated thrombin (Novagen) at 16°C for 4 h, followed by batchwise treatment of the digested proteins with streptavidin–agarose (Novagen). Cleaved N-terminal His tag leader sequences and uncleaved enzymes were removed by batchwise treatment of the cleavage reaction mixture with NTA–agarose. Thrombin-cleaved proteins contained an additional Glu-Ser-His peptide at the N-terminus. Cleavage was confirmed by N-terminal peptide sequencing of purified proteins. The enzymes were dialyzed against 20 mM Tris–HCl (pH 7.4) 500 mM NaCl, 10% glycerol and stored at –80°C. Recombinant APE1 proteins and E.coli Nth were purified as described previously (24,25). Escherichia coli endonuclease IV (Nfo) and uracil DNA glycosylase (Udg) were purchased from Trevigen and New England Biolabs, respectively.
Preparation of DNA substrates
A 31mer oligonucleotide containing 8-oxoG at position 16 was purchased from Midland Certified Reagent Co., with the sequence 5′-GAA GAG AGA AAG AGA XAA GGA AAG AGA GAA-3′, where X denotes 8-oxoG. This oligo contains non-pairing bases and was chosen to minimize formation of intrastrand secondary structures and mispairing between identical strands during annealing. Oligonucleotides with the same sequence except for U or G at position X and their complementary oligonucleotide, with a C opposite X and an additional 5′-G residue, were purchased from Gibco BRL. All oligonucleotides used in this study were HPLC purified and the 8-oxoG-containing oligo was identified as a single species by mass spectrometry. Damage-containing oligos were annealed with a 1.2-fold excess of the complementary strand by heating to 95°C in 20 mM Tris–HCl (pH 7.4) 100 mM NaCl and then slow cooling to room temperature. Annealed duplexes were 3′-end-labeled with [α-32P]dCTP (Dupont) and Klenow exo-DNA polymerase (New England Biolabs). 5′-End-labeled substrates were prepared by labeling single-stranded damage-containing oligos with [γ-32P]ATP (Amersham) and T4 polynucleotide kinase (Pharmacia), prior to annealing with the complementary strand. Unincorporated radioactivity was removed using a Nick Spin G-50 column (Pharmacia). All annealed duplex substrates were >95% cleavable with the appropriate DNA glycosylases (data not shown). AP site-, β-elimination product- and 3′-OH product-containing oligos were prepared by incubating radiolabeled duplex U·C oligo with E.coli Udg, Udg together with E.coli Nth and Udg with APE1, respectively, followed by phenol/chloroform extraction and ethanol precipitation.
Enzyme cleavage assays
Enzyme assays were performed in 20 mM Tris–HCl (pH 7.4) 100 mM NaCl and 0.15 µg/µl bovine serum albumin (BSA) at 37°C in 20 µl reaction volumes. To measure AP lyase or AP endonuclease activities reactions were terminated by adding SDS and glycerol to 0.5 and 5%, respectively. Samples were loaded on 20% polyacrylamide gels containing 7 M urea without heating and electrophoresed in TBE buffer (89 mM Tris–borate, 2 mM EDTA, pH 8.3) at room temperature. Under these conditions the oligo duplexes used in these experiments were fully denatured and single strands containing thermolabile AP sites remained intact unless cleaved enzymatically. To measure glycosylase activity the reactions were terminated by adding SDS and piperidine to 0.5% and 200 mM, respectively, followed by heating at 95°C for 5 min in order to cleave DNA at AP sites, prior to loading on denaturing polyacrylamide gels. Following electrophoresis, the gels were analyzed in a PhosphorImager (Molecular Dynamics) and the radioactivity in DNA quantified using ImageQuant software (Molecular Dynamics). All quantitative experiments were performed in triplicate and standard deviations are included in the results, unless indicated otherwise.
Electrophoretic mobility shift assays
Gel shift experiments were performed in 20 mM Tris–HCl (pH 7.4) 100 mM NaCl, 0.15 µg/µl BSA and 15% glycerol in 20 µl reaction volumes. Reactions were incubated for 10 min at room temperature and bound complexes were separated from the free substrate by non-denaturing electrophoresis in 4% polyacrylamide gels containing 7 mM Tris–HCl (pH 7.9) 3 mM sodium acetate and 1 mM EDTA. Gels were electrophoresed at 4°C for 30 min at 100 V (8 mA) and vacuum dried prior to PhosphorImager quantification of radioactivity. Gel shifts were performed in triplicate and Kdapp values are shown with standard deviations.
RESULTS
Optimization of reaction conditions for OGG1
The glycosylase activity of OGG1 with the 8-oxoG·C duplex oligo substrate was measured under varying conditions of ionic strength, pH and BSA concentration (data not shown). The optimal salt concentration for base removal by OGG1 was determined to be 100 mM NaCl. Substitution of NaCl with KCl had no effect on enzyme activity. The pH optimum was found to be 7.4 in 20 mM Tris–HCl. Tris buffer was preferred to 20 mM HEPES, with the latter being slightly inhibitory at pH 8.0. Addition of BSA (0.15 µg/µl) optimally stabilized OGG1 and the enzyme maintained a linear reaction rate for at least 4 h. These optimal conditions for human OGG1 are nearly identical to those reported recently for mOgg1 (20).
OGG1 exhibits non-Michaelis–Menten kinetics
The steady-state rate of the glycosylase reaction of OGG1 with the 8-oxoG·C-containing duplex oligo substrate was measured over a wide range of substrate concentration (Fig. 1). The rate of base excision increased with increasing substrate concentration, but failed to approach a plateau at high concentrations. Linearity of the rate of product formation was determined at each substrate concentration to ensure that the steady-state rates of base excision by OGG1 were determined (data not shown).
Figure 1.

Kinetics of base excision by OGG1. OGG1 (1.25 nM) was incubated with varying concentration (3.125–100 nM) of the 8-oxoG·C-containing 32mer duplex oligo for 20 min at 37°C. Reactions were terminated by adding SDS and piperidine to 0.5% and 200 mM respectively, followed by heating at 95°C for 5 min. The products were analyzed as described in Materials and Methods.
Inhibition of OGG1 glycosylase activity by AP sites
OGG1 was incubated with a 20-fold molar excess of the 8-oxoG·C-containing duplex oligo in the presence of an additional 10-fold molar excess of oligo substrates containing an undamaged G·C pair, β-elimination product or intact AP site, which were added prior to OGG1 (Fig. 2). With excess 8-oxoG·C substrate, OGG1 excised 8-oxoG with an apparent kcat of 0.1 min–1. The presence of G·C pair- and β-elimination product-containing oligos at the start of reaction of OGG1 with the 8-oxoG·C substrate had no effect on the rate of base excision. In an identical reaction initiated in the presence of excess AP sites the rate of base excision was decreased ∼4-fold.
Figure 2.

Inhibition of OGG1 glycosylase activity by AP sites. OGG1 (25 nM) was incubated with 500 nM 8-oxoG·C oligo for 5 min at 37°C and the reaction terminated as described in Figure 1. Reaction 1, OGG1 only. Identical reactions were carried out by pre-mixing with 250 nM control G·C oligo (reaction 2), β-elimination product oligo (reaction 3) or AP site-containing oligo (reaction 4).
APE1 stimulates base excision by OGG1
OGG1 glycosylase activity was measured in the presence of a 20-fold excess of the 8-oxoG·C substrate (Fig. 3). Addition of human APE1 to the OGG1 reaction increased the rate of glycosylase-mediated base removal (Fig. 3). The effect of APE1 was significant, even when present at half the molar concentration of OGG1. When APE1 was present in 5- to 10-fold molar excess the stimulatory effect approached a maximum with a 5-fold increase in the rate of base removal. When incubated with the 8-oxoG·C substrate the preparation of APE1 used in these experiments did not show any contaminating glycosylase activity (data not shown). Similar stimulation of OGG1 in the presence of APE1 was also observed with other 8-oxoG-containing duplex oligo substrates (data not shown). Thus the activating effect of APE1 was independent of the substrate DNA sequence.
Figure 3.
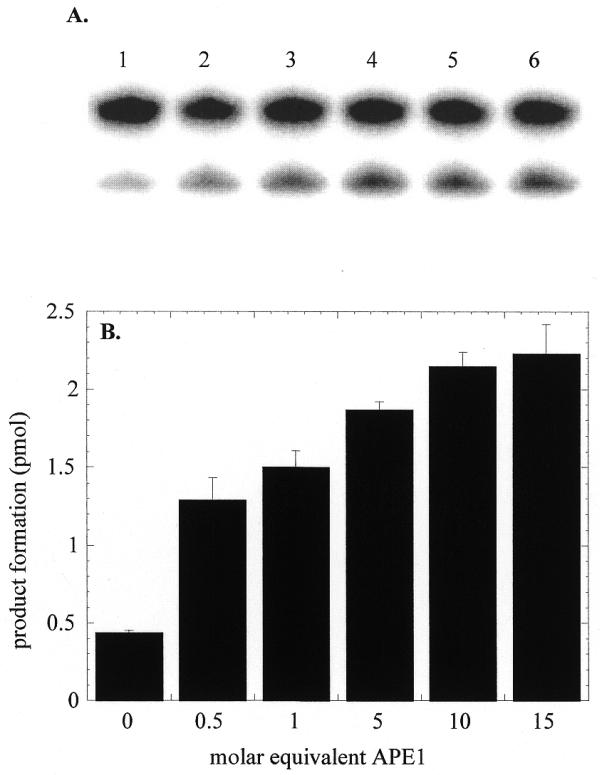
Stimulation of OGG1 glycosylase activity by APE1. OGG1 (25 nM) was incubated with the 8-oxoG·C oligo (500 nM) in the presence of varying amounts of APE1 (12.5–375 nM) and 1 mM MgCl2 for 5 min at 37°C. Termination of reactions and product analysis were performed as in Figure 1. Reaction 1, OGG1 only; reactions 2–6, OGG1 plus 12.5, 25, 125, 250 and 375 nM APE1. The actual data shown in (A) are presented as a bar graph in (B).
Product release by OGG1 is rate limiting
The rate of base excision by OGG1 was followed in single burst time–course reactions of the enzyme with a 20-fold excess of the 8-oxoG·C-containing duplex oligo (Fig. 4). OGG1 catalyzed one rapid reaction cycle, removing 8-oxoG with an apparent kcat of 0.5 min–1. Further reaction rounds proceeded at a constant velocity which was 5-fold lower than that of the initial burst. However, in the presence of a 5-fold molar excess of APE1 the initial burst velocity of the first cycle of base excision by OGG1 was unaffected and additional cycles progressed with an apparent kcat of 0.5 min–1, which was 5-fold higher than the steady-state kcat of OGG1 alone.
Figure 4.

Single burst kinetics of OGG1. OGG1 (25 nM) was reacted with 500 nM 8-oxoG·C substrate for 0.5–8 min. Reactions were terminated as described in Figure 1. Open circles, OGG1 alone; closed circles, OGG1 plus a 5-fold molar excess of APE1. Data points are means of three independent experiments.
Stimulation of OGG1 base excision activity by mutant APE1 proteins and E.coli Nfo
Several enzymatically active mutants of APE1 were tested for their ability to stimulate OGG1 activity (Fig. 5). These include ND40, which has an N-terminal deletion of 40 amino acid residues, ND60-C-His with a deletion of 60 residues from the N-terminus and addition of six consecutive histidine residues at the C-terminus, and C65S, in which Cys65, implicated in the Ref-1 redox activity of APE1 (26), was substituted by Ser. The Ref-1 activity of APE1 was shown to be involved in reductive activation of redox-regulated transcription factors, such as c-Jun, and thus to stimulate their DNA binding activities in vitro (27). When incubated with a 5-fold molar excess of AP endonucleases in the presence of 1 mM MgCl2, OGG1 glycosylase activity was stimulated equally by all mutant APE1 proteins as well as by Nfo (Fig. 5, reactions 2–9). In reaction 7, 1 mM EDTA was present, effectively abolishing the AP endonuclease activity (28), but not the DNA binding capacity of APE1. Several catalytic mutants of APE1 have been reported, but were not used in this study because of their residual enzymatic activity and/or diminished DNA binding affinities (29). In the presence of EDTA inactive APE1 was not able to efficiently stimulate OGG1 turnover at equimolar concentration. However, if present in considerable excess (with 50-fold molar excess in reaction 8), inactive APE1 stimulated OGG1 similarly to the active enzyme.
Figure 5.

Stimulation of OGG1 glycosylase activity by mutant APE1 proteins and Nfo. OGG1 (25 nM) was incubated with 500 nM 8-oxoG·C substrate in the presence of AP endonucleases. Reactions 1 and 2, OGG1 alone. Reaction 1 was terminated with SDS and glycerol without heating. Reactions 2–9 were terminated as described in Figure 1. APE1 was added to reactions 3–8 as follows: 3, 125 nM ND40 APE1; 4, 125 nM ND60 APE1; 5, 125 nM C65S APE1; 6 and 7, 125 nM APE1; 8, 1.25 µM APE1. Reactions 1–6 and 9 contained 1 mM MgCl2. Reactions 7 and 8 contained 1 mM EDTA, reaction 9 125 nM Nfo.
APE1 inhibits sodium borohydride trapping of and AP lyase-mediated strand incision by OGG1
Sodium borohydride trapping of OGG1 with the 8-oxoG·C-containing duplex oligo was inhibited in the presence of APE1. When present at half the molar concentration of OGG1, APE1 decreased OGG1 trapping by ∼3-fold (Fig. 6A). The inhibition was APE1 dose dependent until a maximal decrease of 4- to 5-fold was approached when APE1 was used in 5-fold molar excess over OGG1.
Figure 6.

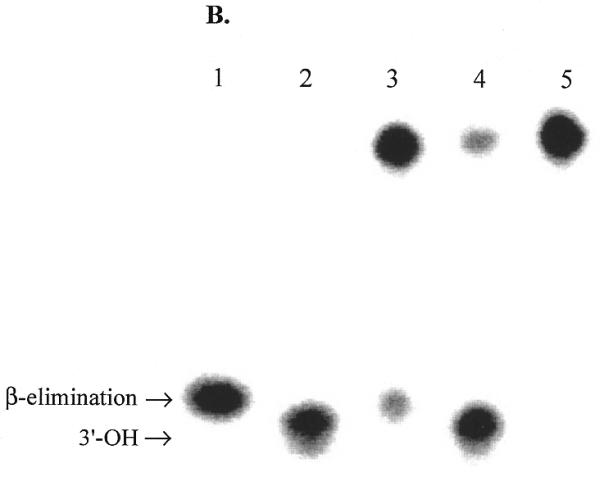
Effect of APE1 on sodium borohydride trapping and AP lyase activity of OGG1. OGG1 (5 nM) was incubated with 1 nM 8-oxoG·C-containing oligo substrate in the presence of 1 mM sodium borohydride with varying amounts of APE1 (2.5–25 nM) for 30 min at 37°C. (A) After termination of the reaction by adding 10 µl of 2× SDS sample buffer and heating for 5 min at 100°C, the trapped complexes were separated from free substrate by SDS–PAGE. (B) Analysis of 3′-termini generated by OGG1 and APE1. Lanes 1 and 2, mobility markers for the 3′-termini characteristic of the β-elimination product and AP site, respectively. Lane 1, 1 nM 5′-labeled U·C-containing duplex oligo was incubated at 37°C for 5 min with 1 µg E.coli Udg and 1 µg Nth; lane 2, 1 nM 5′-labeled U·C oligo was incubated with 1 µg E.coli Udg and 1.25 µM APE1; lanes 3–5, 1 nM 5′-labeled 8-oxoG·C-containing oligo was incubated for 5 min at 37°C with 30 nM OGG1 (lane 3), 10 nM OGG1 and 50 nM APE1 (lane 4) or 10 nM OGG1 and 500 nM APE1 (lane 5). An aliquot of 1 mM MgCl2 was present in lanes 1–4 and 1 mM EDTA in lane 5. All reactions were terminated with SDS (0.5%) and glycerol (5%) without heating.
The termini resulting from strand incision by OGG1 and by the combined action of OGG1 with APE1 were also examined (Fig. 6B). When presented with the 8-oxoG·C duplex oligo, OGG1 alone exclusively produced termini characteristic of a β-elimination product (lane 3). In the presence of both OGG1 and APE1, the overall strand incision was increased and all cleavage products had 3′-OH ends, indicative of the APE1 reaction product (lane 4). In the presence of OGG1, a 50-fold molar excess of APE1 and 1 mM EDTA no strand scission occurred, indicating that neither OGG1 nor APE1 cleaved the AP sites resulting from the glycosylase activity of OGG1 (lane 5).
Kinetics of OGG1, APE1 and E.coli Nfo
Steady-state velocities for OGG1 in the presence of a 5-fold molar excess of APE1 with the 8-oxoG·C substrate and for OGG1, APE1 and Nfo with an AP·C oligo substrate were plotted against substrate concentration (Fig. 7). Reciprocal plots of the data in Figure 7 were used to calculate kinetic constants presented in Table 1. In the presence of APE1, OGG1 showed normal single substrate kinetics and approached a maximum reaction velocity at ∼100 nM 8-oxoG·C-containing substrate (Fig. 7A). With the AP site-containing substrate OGG1 did not approach a maximal velocity and exhibited a biphasic enhancement of AP lyase activity at higher AP site concentration (25 nM in Fig. 7B). As in the case of OGG1 with the 8-oxoG·C substrate (Fig. 1), both APE1 and Nfo showed non-Michaelis–Menten kinetics with the AP site substrate, suggesting that these enzymes are inhibited by their products as well (Fig. 7C and D). APE1 cleaved the AP·C substrate with a kcat 20-fold higher than that of Nfo and 400-fold higher than that of OGG1 (Table 1). The kcat of OGG1 for the 8-oxoG·C substrate was increased ∼5-fold in the presence of a 5-fold molar excess of APE1 (Table 1).
Figure 7.

Kinetics of OGG1, APE1 and Nfo. (A) OGG1 (1.25 nM) and APE1 (6.5 nM) were incubated with increasing amounts of the 8-oxoG·C-containing duplex oligo for 5 min at 37°C. (B) OGG1 (1.25 nM) was incubated with varying amounts (3.125–100 nM) of the AP·C-containing 32mer duplex oligo for 40 min at 37°C. Reactions were terminated by adding SDS and glycerol as in Figure 6B. (C) APE1 (0.5 pM) was incubated with increasing concentrations of the AP·C-containing oligo for 4 min at 37°C. (D) Escherichia coli Nfo (1.25 nM) was incubated with increasing concentrations of the AP·C-containing oligo for 4 min at 37°C. Substrates were in excess and reaction rates were in the linear range in all experiments. Data points are means of three independent experiments.
Table 1. Kinetic parameters of OGG1, APE1 and Nfo.
| Enzyme |
Substrate |
Activity |
Kmapp (nM) |
kcatapp ×103 (min–1) |
| OGG1 | 8-oxoG·C | Glycosylase | 9.9 ± 0.2 | 108 ± 7.1 |
| OGG1 + APE1 | 8-oxoG·C | Glycosylase | 8.9 ± 0.9 | 453 ± 16 |
| OGG1 | AP site | AP lyase | 7.2 ± 1.1 | 53.2 ± 7 |
| APE1 | AP site | AP endonuclease | 13.7 ± 1.2 | 21, 671 ± 2132 |
| Nfo | AP site | AP endonuclease | 21.2 ± 2.2 | 1062 ± 90 |
Values were calculated from Lineweaver–Burke plots of data presented in Figures 1 and 7.
Determination of dissociation constants for OGG1 and APE1
The relative binding affinities of OGG1 and APE1 for their substrates and products were determined by electrophoretic gel mobility shift assay (Table 2). All gel shift experiments were performed under conditions of enzyme inactivity. A mutant OGG1 protein (K249Q), in which the candidate active site Lys was substituted by Gln (9,30), was used to determine the binding affinity for various substrates. The K249Q mutant exhibits substrate binding specificity similar to that of the wild-type enzyme, but is catalytically inactive (9,30). Thus, this protein is suitable for quantifying the affinity of the enzyme for lesions containing 8-oxoG as well as for lesions resulting from subsequent steps in repair of 8-oxoG. Similar gel shift studies were reported for catalytically active mOgg1 (20). However, those results should be interpreted with caution because the observed complexes between the enzyme and various substrates may be affected by the generation of reaction products whose complexes with the enzyme could not be distinguished from the enzyme–substrate complex. The binding of APE1 to the AP site substrate and products was measured in the presence of 1 mM EDTA to ensure that the protein had no enzymatic activity. Our results show that OGG1 binds an AP site with significantly higher affinity than an 8-oxoG·C pair and interacts weakly with the duplex β-elimination product (Table 2). Interestingly, OGG1 had similar affinity for oligos containing a 3′-OH product at an APE1-generated strand break and an 8-oxoG·C pair. As expected, the affinity of APE1 for an AP site oligo was higher than that of OGG1. The β-elimination product was also bound more tightly by APE1 than by OGG1. APE1 showed similar affinity for an AP site and its 3′-OH product and had a comparatively low affinity for the β-elimination product (Table 2).
Table 2. Affinity of OGG1 and APE1 for repair substrates and products.
| Enzyme |
Substrate |
Kdapp (nM) |
| OGG1 | 8-oxoG·C | 23.4 ± 3.4 |
| OGG1 | AP site | 2.8 ± 0.2 |
| OGG1 | β-elimination | 223 ± 19 |
| OGG1 | 3′-OH | 20.7 ± 2.5 |
| APE1 | AP site | 1.7 ± 0.2 |
| APE1 | β-elimination | 72.7 ± 6.6 |
| APE1 | 3′-OH | 4.3 ± 0.5 |
All duplex oligonucleotides have identical sequences, except for the lesion opposite cytosine.
DISCUSSION
Although low turnover of mammalian OGG1 was recognized earlier (19,20), this report describes the first systematic investigation to elucidate the basis for the unusual kinetic properties of this enzyme. Our observations that human OGG1 did not exhibit Michaelis–Menten kinetics (reviewed in 31), excised 8-oxoG with a low apparent kcat (Table 1) and failed to approach a maximum reaction velocity (Vmax) in the presence of excess substrate suggested that the enzyme was inhibited by its own product. Our subsequent studies showing inhibition of OGG1 glycosylase activity in the presence of exogenous AP sites (Fig. 2) and the stimulation of its turnover in the presence of APE1 (Fig. 3), which has high affinity and activity for AP sites, supported this possibility. In single burst kinetics studies with OGG1 alone (Fig. 4) the enzyme completed one rapid reaction cycle, followed by significantly slower subsequent cycles, indicating that product release was rate limiting. APE1 relieved product inhibition and enhanced the specific activity of OGG1 5-fold (compare Figs 1 and 7A). In the presence of APE1 the steady-state and initial rates of base excision were about the same, suggesting that product dissociation of OGG1 is facilitated by APE1 (Fig. 4). This is consistent with the surprising observation that OGG1 has higher affinity for the AP·C product than for its 8-oxoG·C substrate (Table 2).
These observations raised two important questions: (i) whether APE1 physically interacts with OGG1; (ii) whether APE1 activity is required for stimulation of the latter. In the presence of EDTA, APE1 was inactive and was able to stimulate OGG1 glycosylase activity <2-fold, compared to 5-fold stimulation when the enzyme was active (Fig. 5, reaction 7). This initially suggested that the AP endonuclease activity of APE1 is required for enhancement of OGG1 activity. However, we subsequently observed that the AP site binding affinity of APE1, rather than its activity, was essential for stimulation, because a 50-fold excess of APE1 in 1 mM EDTA was able to stimulate OGG1 to the same extent as the active enzyme (Fig. 5, reaction 8). Escherichia coli Nfo, an AP endonuclease structurally unrelated to APE1 (32,33), stimulated OGG1 at a similar level to APE1 (Fig. 5, reaction 9), indicating that a direct interaction between OGG1 and APEs is unlikely. Additionally, N-terminally truncated, C-terminally His-tagged and Ref-1 mutant APE1 proteins stimulated OGG1 turnover comparably to the wild-type enzyme (Fig. 5). Because the APE1 mutants used in this study are active but likely to have altered behavior in interacting with other proteins (27), these results also support our conclusion about the lack of a direct interaction between APE1 and OGG1. Independent experiments showing lack of co-immunoprecipitation of APE1 and OGG1 further support this conclusion (G.Roy and S.Mitra, unpublished results). However, we cannot completely exclude the possibility of some type of interaction between APE1 and OGG1 in the presence of AP site-containing DNA.
In an earlier study the inability of Nfo to stimulate human thymine-DNA glycosylase (TDG), which was readily stimulated by APE1, was reported. A direct interaction of TDG with APE1 but not with Nfo was suggested on the basis of these results (21). However, in that study the relative affinities of APE1 and TDG for an AP site were not determined. It should be noted that a 100-fold molar excess of APE1 was needed to stimulate TDG some 2-fold as shown in that report (21), while OGG1 activity in our study was enhanced 3- to 4-fold by only a 0.5-fold molar equivalence of APE1. It is, therefore, conceivable that TDG binds AP sites more tightly than APE1, and perhaps substantially more tightly than Nfo. Thus, the failure of Nfo to stimulate TDG may reflect disparities between the AP site affinities and enzymatic activities of APE1 and Nfo, rather than imply physical interaction between the two enzymes. We tested this possibility directly by measuring the reaction rates of APE1 and Nfo (Table 1). Both APEs cleaved AP sites more efficiently than OGG1, but the kcat of APE1 for the AP·C substrate was 20-fold higher than that of Nfo (Table 1). The affinity of Nfo for the AP site substrate could not be compared, as no catalytically inactive Nfo was available. Although OGG1 and APE1 demonstrated similar binding affinities for an AP site (Table 2), the rate of AP site cleavage by APE1 was 400-fold faster than that by OGG1 (Table 1). Thus, active APE1, even at sub-equimolar concentrations, was able to significantly enhance the removal of AP sites from the reaction and increase the overall rate of excision of oxidized guanine from DNA. Conceivably, any DNA-binding protein with specific affinity for an AP site would cause a similar stimulation of OGG1 glycosylase activity if present in sufficient excess. Additionally, considering the measurable affinity of OGG1 for the β-elimination product (Table 2), the 3′-phosphodiesterase activity of APE1 may also serve to free OGG1 molecules bound to this product. In any case, activation of OGG1 by APE1, observed in vitro, may have profound physiological relevance and is consistent with the ‘hand-off’ model recently proposed for the base excision repair pathway (34,35).
When the AP lyase activity of OGG1 was analyzed, all enzymatic strand cleavage products were found to result from β-elimination, as expected. With addition of APE1 to an OGG1 reaction with an 8-oxoG·C-containing oligo substrate all strand cleavage products had 3′-OH termini (Fig. 6B, lane 4). For this reason it was initially unclear whether APE1 stimulates the intrinsic AP lyase activity of OGG1, resulting in the formation of β-elimination products that are subsequently processed by APE1 to generate 3′-OH termini. Alternatively, the weak AP lyase activity of OGG1 could be overridden by the strong AP endonuclease activity of APE1, which will directly generate only 3′-OH ends from the uncleaved AP sites. A transient Schiff base is a necessary intermediate during initiation of the β-elimination reaction of AP lyases such as OGG1. Accordingly, any increase in the AP lyase activity of OGG1 would be expected to result in increased production of trapped complexes in the presence of sodium borohydride. In contrast, as shown in Figure 6A, addition of APE1 to an OGG1 reaction reduced sodium borohydride trapping ∼4-fold, indicating that, in the presence of APE1, the intrinsic AP lyase activity of OGG1 was largely superseded. Additionally, when a 50-fold molar excess of catalytically inactive APE1 was present OGG1 glycosylase activity was increased but no strand cleavage occurred (Fig. 5, reaction 8 and Fig. 6B, lane 5). This suggests that OGG1 dissociates from an AP site in the presence of APE1 prior to it proceeding to carry out the β-elimination reaction. These results, together with the identical initial velocities of OGG1 in the presence and absence of APE1 (Fig. 4), indicate that APE1 does not directly enhance catalysis by OGG1. Interestingly, both APE1 and Nfo failed to reach saturation with an AP site substrate (Fig. 7C and D, respectively), implicating product inhibition for these enzymes as well. APE1 and Nfo have both been shown to bind their cleaved AP site reaction products (29,36,37) and product inhibition of APE1 was proposed previously (29).
It was somewhat surprising that the maximal reaction velocity was not approached by OGG1 with increasing concentrations of an AP site substrate, although unlike the situation with the 8-oxoG·C substrate, the affinity of OGG1 for the β-elimination product of an AP site is relatively weak (Table 2). An apparently biphasic response was observed at higher AP site concentrations (25 nM), indicating that when AP sites are abundant OGG1 may undergo a second or distinct interaction with the AP site that enhances its AP lyase activity (Fig. 7B). This observation was highly reproducible and was confirmed by several experiments. It is possible that OGG1 may have dissimilar modes of AP site binding or multiple interactions with this substrate. The AP lyase activity of OGG1 may be dependent upon the conformation of the AP site (19), such that unproductive binding of the lesion may occur, although this remains to be established.
Binding studies with APE1 and a cleaved AP site substrate (Table 2) show that APE1 binds with high affinity to its own product, as does OGG1. Interaction of APE1 and β-polymerase was demonstrated previously (37) and moderate stimulation of APE1 in the presence of β-polymerase has been reported (29). It is thus likely that the optimum catalytic rate of individual base excision repair enzymes is achieved only in the presence of enzymes involved in successive steps of the repair process. We propose that sequential rounds of product inhibition of the glycosylase and AP endonuclease steps, and possibly other subsequent steps, may be characteristic features of the repair of 8-oxoG and other mutagenic base lesions in a wide range of organisms. The well-characterized glycosylases E.coli Fpg and MutY and human TDG and UNG1 have all been shown to bind AP sites with similar or higher affinity than for their respective substrates, although detailed kinetic studies of the effects of next-step repair enzymes on the activities of these glycosylases have not been reported (21,38–40).
APE1 is a ubiquitous multifunctional protein (41–43) and has been shown to be present at high concentration in HeLa cells (21,37), which is likely to significantly exceed that of OGG1. The poor activity of OGG1 towards AP sites, taken together with its ready dissociation from AP sites in the presence of sub-equimolar amounts of APE1, raises questions about the in vivo significance of the AP lyase activity of OGG1. One possible explanation is that during severe oxidative stress, when the 8-oxoG level in DNA is elevated, a concerted glycosylase/AP lyase reaction by OGG1 would create numerous single-strand breaks in the genome that could be lethal to cells. Thus, the poor AP lyase activity of OGG1 may be advantageous to cells and suggests a fundamental difference between the in vivo roles of OGG1 and other DNA glycosylases/AP lyases that do not leave intact AP site products. It may be beneficial if the strong binding of OGG1 to AP sites in vivo serves primarily to sequester these lesions from attack by other AP site-cleaving enzymes, e.g. Nth and DNA topoisomerases (44,45). The DNA strand breaks resulting from such attacks will prevent transcription and replication and could also trigger apoptosis. Thus OGG1 may provide a protective function until APE1 and other components required for subsequent steps of the base excision repair pathway are recruited. On the other hand, the observed enhancement of OGG1 AP lyase activity in the presence of high concentrations of AP site DNA could result in formation of numerous, localized single-strand breaks in DNA. In this situation it may be preferable for the cell to repair the damage via recombination, which requires single-strand breaks for initiation, rather than to repair individual AP sites. Alternatively, the increased number of single-strand breaks may function as a signal for the cell to undergo apoptosis and thus avoid mutations.
We showed earlier that oxidative stress increased APE1 mRNA and protein levels in human cells and APE1 was then translocated to the nucleus (46). An increase in cellular oxidative damage repair following APE1 induction and nuclear translocation in response to oxidative stress was observed (46). Stimulation of damaged base excision activity by APE1, as observed in this in vitro study, may also contribute to this increase. Elucidation of the physiological significance of OGG1 activation by APE1, in the context of complex signaling processes involved in recruitment of these and other necessary enzymes at the DNA lesion sites, will require further investigation.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Drs A. P. Grollman and T. A. Rosenquist (SUNY, Stony Brook, NY) for providing the human OGG1 cDNA clone. Dr T. G. Wood of the Recombinant DNA Laboratory (UTMB) assisted in plasmid construction. We thank Dr A. Kurosky, Director of the Protein Chemistry Laboratory (UTMB), for DNA and protein sequencing. We also thank Dr Y. W. Kow (Emory University) for critical reading of the manuscript and Drs R. S. Lloyd and D. W. Bolen (UTMB) for helpful discussions. This research was supported by US Public Health Service grants R01 CA81063, R01 ES08457 and AG10514.
References
- 1.Melvin T., Cunniffee,S.M.T., O’Neill,P., Parker,A.W. and Roldan-Arjona,T. (1998) Guanine is the target for direct ionisation of damage in DNA, as detected using excision enzymes. Nucleic Acids Res., 21, 4935–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klungland A., Rosewell,I., Hollenbach,S., Larsen,E., Daly,G., Epe,B., Seeberg,E., Lindahl,T. and Barnes,D.E. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl Acad. Sci. USA, 96, 13300–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minowa O., Arai,T., Hirano,M., Monden,Y., Nakai,S., Fukuda,M., Itoh,M., Takano,H., Hippou,Y., Aburatani,H. et al. (2000) Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl Acad. Sci. USA, 97, 4156–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiseman H., Kaur,H. and Halliwell,B. (1995) DNA damage and cancer: measurement and mechanism. Cancer Lett., 93, 113–120. [DOI] [PubMed] [Google Scholar]
- 5.Beckman K.B. and Ames,B.N. (1997) Oxidative decay of DNA. J. Biol. Chem., 272, 19633–19636. [DOI] [PubMed] [Google Scholar]
- 6.Lovell M.A., Xie,C. and Markesbery,W.R. (2000) Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res., 855, 116–123. [DOI] [PubMed] [Google Scholar]
- 7.Krokan H.E., Nilsen,H., Skorpen,F., Otterlei,M. and Slupphaug,G. (2000) Base excision repair of DNA in mammalian cells. FEBS Lett., 476, 73–77. [DOI] [PubMed] [Google Scholar]
- 8.Hazra T.K., Izumi,T., Maidt,L., Floyd,R.A. and Mitra,S. (1998) The presence of two distinct 8-oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucleic Acids Res., 26, 5116–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu R., Nash,H.M. and Verdine,G.L. (1997) A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol., 7, 397–407. [DOI] [PubMed] [Google Scholar]
- 10.Nash H.M., Bruner,S.D., Schärer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 11.Sun B., Latham,K.A., Dodson,M.L. and Lloyd,R.S. (1995) Studies on the catalytic mechanism of five DNA glycosylases. J. Biol. Chem., 270, 19501–19508. [DOI] [PubMed] [Google Scholar]
- 12.McCullough A.K., Dodson,M.L. and Lloyd,R.S. (1999) Initiation of base excision repair: glycosylase mechanisms and structures. Annu. Rev. Biochem., 68, 255–285. [DOI] [PubMed] [Google Scholar]
- 13.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Radicella J.P., Dherin,C., Desmaze,C., Fox,M.S. and Boiteux,S. (1997) Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 8010–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klungland A. and Lindahl,T. (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J., 16, 3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rabow L.E. and Kow,Y.W. (1997) Mechanism of action of base release by Escherichia coli Fpg protein: role of lysine 155 in catalysis. Biochemistry, 36, 5084–96. [DOI] [PubMed] [Google Scholar]
- 17.Dodson M.L., Michaels,M.L. and Lloyd,R.S. (1994) Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem., 269, 32709–32712. [PubMed] [Google Scholar]
- 18.Roldan-Arjona T., Wei,Y., Carter,K.C., Klungland,A., Anselmino,C., Wang,R., Augustus,M. and Lindahl,T. (1997) Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 8016–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjoras M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Rognes,T. and Seeberg,E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zharkov D.O., Rosenquist,T.A., Gerchman,S.E. and Grollman,A.P. (2000) Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem., 275, 28607–28617. [DOI] [PubMed] [Google Scholar]
- 21.Waters T.R., Gallinari,P., Jiricny,J. and Swann,P.F. (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem., 274, 67–74. [DOI] [PubMed] [Google Scholar]
- 22.Parikh S.S., Mol,C.D., Slupphaug,G., Bharati,S., Krokan,H.E. and Tainer,J.A. (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J., 17, 5214–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishioka K., Ohtsubo,T., Oda,H., Fujiwara,T., Kang,D., Sugimachi,K. and Nakebeppu,Y. (1999) Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell, 10, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Izumi T., Malecki,J., Chaudhry,M.A., Weinfeld,M., Hill,J.W., Lee,J.C. and Mitra,S. (1999) Intragenic suppression of an active site mutation in the human apurinic/apyrimidinic endonuclease. J. Mol. Biol., 287, 47–57. [DOI] [PubMed] [Google Scholar]
- 25.Asahara H., Wistort,P.M., Bank,J.F., Bakerian,R.H. and Cunningham,R.P. (1989) Purification and characterization of Escherichia coli endonuclease III from the cloned nth gene. Biochemistry, 28, 4444–4449. [DOI] [PubMed] [Google Scholar]
- 26.Walker L.J., Robson,C.N., Black,E., Gillespie,D. and Hickson,I.D. (1993) Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol., 13, 5370–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xanthoudakis S., Miao,G.G. and Curran,T. (1994) The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl Acad. Sci. USA, 91, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakabayashi H., Tsuji,T. and Seki,S. (1996) Purification and characterization of a 39kDa apurinic/apyrimidinic endonuclease from mouse ascites sarcoma cells. Acta Med. Okayama, 50, 131–137. [DOI] [PubMed] [Google Scholar]
- 29.Masuda Y., Bennett,R.A.O. and Demple,B. (1998) Dynamics of the interaction of human apurinic endonuclease (APE1) with its substrate and product. J. Biol. Chem., 273, 30352–30359. [DOI] [PubMed] [Google Scholar]
- 30.Guibourt N., Castaing,B., Van Der Kemp,P.A. and Boiteux,S. (2000) Catalytic and DNA binding properties of the Ogg1 protein of Saccharomyces cerevisiae: comparison between the wild type and the K241R and K241Q active-site mutant proteins. Biochemistry, 39, 1716–1724. [DOI] [PubMed] [Google Scholar]
- 31.Dixon M. and Webb,E.C. (1979) Enzymes, 3rd Edn. Academic Press, New York, NY, pp. 55–78.
- 32.Chan E. and Weiss,B. (1987) Endonuclease IV of Escherichia coli is induced by paraquat. Proc. Natl Acad. Sci. USA, 84, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Izumi T., Ishizaki,K., Ikenaga,M. and Yonei,S. (1992) A mutant Endonuclease IV of Escherichia coli loses the ability to repair lethal DNA damage induced by hydrogen peroxide but not that induced by methy methanesulfonate. J. Bacteriol ., 174, 7711–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- 35.Wilson S.H. and Kunkel,T.A. (2000) Passing the baton in base excision repair. Nature Struct. Biol., 7, 176–178. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi M., Lillis,R., Demple,B. and Takeshita,M. (1994) Interactions of Escherichia coli Endonuclease IV and Exonuclease III with abasic sites in DNA. J. Biol. Chem., 269, 21907–21914. [PubMed] [Google Scholar]
- 37.Bennett R.A.O., Wilson,D.M., Wong,D. and Demple,B. (1997) Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc. Natl Acad. Sci. USA, 94, 7166–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castaing B., Boiteux,S. and Zelwer,C. (1992) DNA containing a chemically reduced apurinic site is a high affinity ligand for the E.coli formamidopyrimidine-DNA glycosylase. Nucleic Acids Res., 20, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams S.D. and David,S.S. (1999) Formation of a Schiff base intermediate is not required for the adenine glycosylase activity of Escherichia coli MutY. Biochemistry, 38, 15417–15417. [DOI] [PubMed] [Google Scholar]
- 40.Bharati S., Krokan,H.E., Kristiansen,L., Otterlei,M. and Slupphaug,G. (1998) Human mitochondrial uracil-DNA glycosylase preform (UNG1) is processed to two forms one of which is resistant to inhibition by AP sites. Nucleic Acids Res., 26, 4953–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- 42.Xanthoudakis S., Smeyne,R.J., Wallace,J.D. and Curran,T. (1996) The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl Acad. Sci. USA, 93, 8919–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Izumi T., Henner,W.D. and Mitra,S. (1996) Negative regulation of the major human AP-endonuclease, a multifunctional protein. Biochemistry, 35, 14679–14683. [DOI] [PubMed] [Google Scholar]
- 44.Chaudhry M.A. and Weinfeld,M.J. (1995) The action of Escherichia coli endonuclease III on multiply damaged sites in DNA. J. Mol. Biol., 249, 914–922. [DOI] [PubMed] [Google Scholar]
- 45.Sabourin M. and Osheroff,N. (2000) Sensitivity of human type II topoisomerases to DNA damage: stimulation of enzyme-mediated DNA cleavage by abasic, oxidized and alkylated lesions. Nucleic Acids Res., 28, 1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramana C.V., Boldogh,I., Izumi,T. and Mitra,S. (1998) Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc. Natl Acad. Sci. USA, 95, 5061–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]