

Abstract
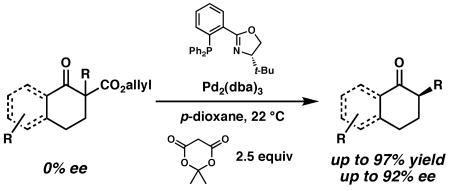
General homogeneous conditions for the palladium-catalyzed synthesis of carbonyl compounds with tertiary carbon stereocenters at the α-position are reported. The highly reactive catalyst tolerates a variety of substrate substitution and functionality, and generates enantioenriched cyclic ketones from racemic allyl β-ketoester starting materials.
Enantioselective protonation of achiral enolates or enol equivalents is an efficient route to access carbonyl compounds with tertiary carbon stereocenters at the α-position. Several distinct methods involving chiral proton sources or chiral catalysts have been developed for the enantioselective protonation of metal enolates.1,2 However, most of the reported methods are limited in substrate scope, few are catalytic, and together they do not provide a general solution for the enantioselective protonation of enolates.1d
Previously, we reported a series of catalytic enantioselective allylation reactions that deliver carbonyl compounds with adjacent quaternary stereocenters from various enolate precursors (Scheme 1).3,4 Crucial to the success of these transformations was the use of catalysts derived from Pd(0) and chiral phosphinooxazoline (PHOX) ligand 1.5,6 Based on kinetic and mechanistic studies carried out in our laboratories as well as computational studies performed in collaboration with the W. A. Goddard group at Caltech,7 we believe that in the course of the reaction a chiral Pd-enolate is generated in solution. We chose to explore a proton electrophile to take further advantage of this valuable intermediate for the preparation of tertiary stereocenters.8 As a result of these studies, we reported a highly enantioselective catalytic system for the decarboxylative protonation of racemic allyl β-ketoesters in the presence of Pd(OAc)2, (S)-t-Bu-PHOX (1), 4 Å molecular sieves (MS), and HCO2H (Scheme 1).9 Although this protocol is capable of generating cycloalkanones with excellent ee, each substrate required optimization of the amounts of 4 Å MS and HCO2H in order to suppress competitive allylation and maximize product ee. Moreover, the heterogeneous nature of the reaction hinders investigation of the mechanism of protonation. In response, we have sought substantially different protonation conditions to allow further development of a practical synthetic process. Herein, we report a highly enantioselective, general homogeneous catalytic system for the facile synthesis of tertiary stereocenters by protonation of ketone enolates.
Scheme 1.

Pd-Catalyzed Enantioselective Decarboxylative Allylation and Protonation of Racemic Allyl β-Ketoesters.
To achieve a homogeneous enantioselective protonation, the racemic allyl β-ketoester (±)-2 was exposed to Pd2(dba)3, (S)-t-Bu-PHOX (1), and a variety of achiral organic proton donors (Table 1). Gratifyingly, the use of dimethyl malonate did indeed lead to protonated product 3, although the ee was moderate (entry 1). Acetoacetic esters (entries 2 and 3) provided 3 in significantly higher ee than the malonate case, but at the expense of conversion. Acetylacetone derivatives (entries 4–6) were very reactive, with the more acidic analogues providing higher ee products. Noting that the more acidic compounds increased the rate of reaction dramatically, we chose next to explore other highly acidic organic proton donors. Ketosulfones (entries 7–9) led to protonated product 3 in very high ee, though conversion was sometimes slow. However, commercially available Meldrum's acid10 led to extremely rapid formation of 3 with good enantioselectivity (entry 10). Given the rapid reaction at 40 °C, we attempted the transformation at ambient temperature (23 °C) and observed a significant increase in product ee (entry 11). Further lowering the temperature required changing the solvent to THF, and although the ee was excellent, reactivity dropped sharply (entry 12). Notably, addition of MS to the reaction had a severe impact on the ee (entry 13).
Table 1. Screening of Achiral Proton Donorsa.
 | |||||
|---|---|---|---|---|---|
| entry | proton donor | time (h) | conversion (%)b | ee (%)c | |
| 1 | 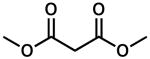 |
24 | 85 | 62 | |
| 2 | 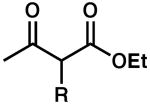 |
R = H | 24 | 50 | 80 |
| 3 | R = F | 24 | 55 | 89 | |
| 4 | 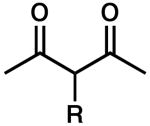 |
R = H | 24 | 95 | 50 |
| 5 | R = Ac | 2 | 100 | 74 | |
| 6 | R = Ms | 3 | 100 | 88 | |
| 7 | 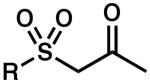 |
R = Me | 24 | 42 | 89 |
| 8 | R = Ph | 24 | 63 | 90 | |
| 9 | R = p-Tol | 5 | 100 | 84 | |
| 10 | 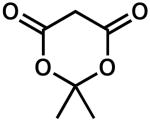 |
0.2 | 100 | 85 | |
| 11d | 0.5 | 100 | 90 | ||
| 12e | 24 | 50 | 95 | ||
| 13f | 1.5 | 100 | 43 | ||
Reactions performed with 0.1 mmol of (±)-2 at 0.033 M in p-dioxane.
Measured by 1H NMR spectroscopy.
Measured by chiral HLPC.
Reaction performed at 23 °C.
Reaction performed at 0 °C in THF solvent.
Reaction performed with 90 mg of 4 Å MS.
The combination of reactivity, selectivity, and availability prompted us to choose the Meldrum's acid scaffold for our further studies (Table 2). It was found that, in general, large substituents between the carbonyls of the proton source decrease the enantiopurity of 3, although smaller groups (e.g., CH3) are tolerated (entries 1–5). In contrast to the acyclic case, addition of a third resonance withdrawing group (e.g., acetyl) severely decreased reactivity and product ee (cf. Table 1, entry 9 and Table 2, entry 5). Electronic perturbation by substituting dimedone as the proton source led to only trace product in low ee (entry 6). Variation of the acetal portion with sterically diverse groups had a relatively small effect on ee (entries 7 and 8). Notably, simple spirocyclic derivatives of Meldrum's acid (entries 9 and 10) gave the highest ee, although conversion to product was sluggish. Larger bicyclic acetals gave mixed results (entries 11–13). Interestingly, the enantiopure menthone derived analogue performed equally well with either antipode of the chiral ligand, indicating that the catalyst directly controls the sense of enantioinduction (entries 12 and 13). None of the slight improvements observed with these various derivatives outweighed the convenience of the commercially available Meldrum's acid. Additionally, a screen of other chiral PHOX ligands revealed the superiority of the original (S)-t-Bu-PHOX (1).11
Table 2. Screening of Meldrum's Acid Derivativesa.
 | |||||
|---|---|---|---|---|---|
| entry | proton donor | time (h) | conversion (%)b | ee (%)c | |
| 1 | 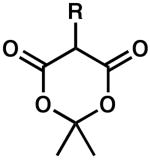 |
R = H | 0.5 | 100 | 90 |
| 2 | R = Me | 0.2 | 100 | 90 | |
| 3 | R = Ph | 24 | 86 | 68 | |
| 4 | R = Allyl | 1 | 100 | 67 | |
| 5 | R = Ac | 24 | 1 | 51 | |
| 6 | 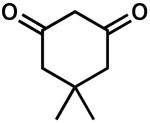 |
24 | 8 | 43 | |
| 7 | 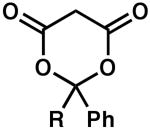 |
R = H | 5 | 100 | 85 |
| 8 | R = Me | 4 | 100 | 84 | |
| 9 | 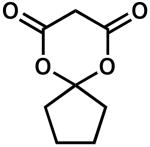 |
24 | 25 | 93 | |
| 10 | 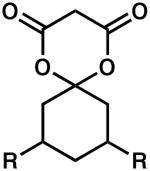 |
R = H | 24 | 86 | 91 |
| 11 | R = Me | 24 | 100 | 76 | |
| 12 | 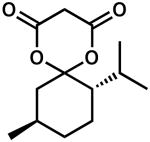 |
24 | 100 | 90 | |
| 13d | 24 | 100 | −90 | ||
Reactions performed with 0.1 mmol of (±)-2 at 0.033 M in p-dioxane.
Measured by 1H NMR spectroscopy.
Measured by chiral HLPC.
Reaction performed with (R)-t-Bu-PHOX (12.5 mol%).
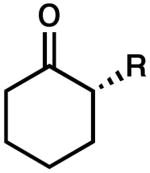
Encouraged by these results, the scope of this enantioselective protonation was explored (Table 3). The general set of reaction conditions employed in these studies were straightforward: racemic allyl β-ketoester was exposed to Pd2(dba)3 (5 mol%), (S)-t-Bu-PHOX (1, 12.5 mol%), and Meldrum's acid (2.5 equiv) in p-dioxane (0.033 M) at room temperature (22 °C). A variety of substituted cyclohexanone derivatives was prepared (entries 1–5). Especially noteworthy is the tolerance of a β-siloxy substituent (entry 5). The near quantitative yield and the absence of β-elimination products (i.e., enone) highlight the mild, neutral reaction conditions. 1-Tetralone derivatives (entries 6–8) were also accessible in high yield and ee. Finally, (S)-(+)-2-methyl-1-indanone (entry 9) was prepared in good yield, although the ee was moderate. When we attempted to carry out the reaction on larger scale, we observed a slight decrease in product ee. The origins of this decrease are unclear at this time.
Table 3. Enantioconvergent Decarboxylative Protonationsa.
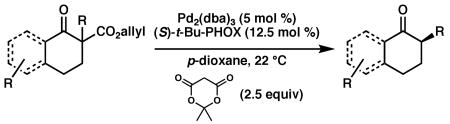 | |||||||
|---|---|---|---|---|---|---|---|
| entry | substrate | product | ee A (%) | time (h) | yield (%) | ee B (%) | |
| 1 | 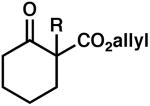 |
 |
R = Me | 88 | 0.5 | 99b | 86 (R) |
| 2 | R = Et | 92 | 0.5 | 99b | 89 (R) | ||
| 3 | R = Bn | 83 | 0.5 | 90 | 78 (S) | ||
| 4 | 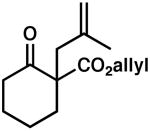 |
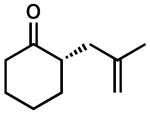 |
91 | 5 | 87 | 89 | |
| 5 | 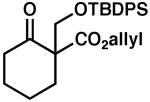 |
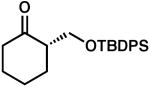 |
85 | 0.5 | 97 | 80 | |
| 6 | 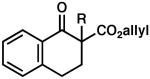 |
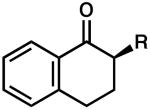 |
R = Me | 90 | 0.5 | 86 | 77 (S) |
| 7 | R = Allyl | 82 | 0.5 | 83 | 77 (R) | ||
| 8 | 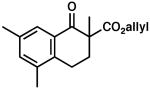 |
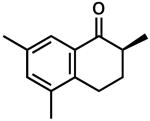 |
– | 0.5 | 77 | 77 | |
| 9 | 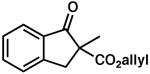 |
 |
– | 0.5 | 79 | 61 (S) | |
Reactions were performed on two different scales, A: 0.1 mmol of substrate, and B: 0.3 mmol of substrate, each at 0.033 M in p-dioxane. Column ee A reflects results for conditions A; time, isolated yield, and ee B are reported for conditions B. Isolated yields from conditions A were comparable to those with conditions B. The major enantiomer was the same under either set of conditions. Enantiomeric excess was measured following chromatography on silica gel; no erosion of ee was observed as a result of purification.
GC yield using tridecane as internal standard.
The absolute configuration of a number of products was established by a comparison of the observed sign of optical rotation to literature values (entries 1–3, 6, 7, and 9).11 Cyclohexanone substrates led to products in the opposite enantiomeric series compared to that of the tetralone and indanone cases (entries 1–3 vs. entries 6, 7, and 9). Identical behavior was observed when the reactions were performed with our heterogeneous Pd(OAc)2/HCO2H conditions,9 suggesting that the two enantioselective protonations proceed through very similar mechanisms.
Under our optimized reaction conditions, products arising from allylation of our postulated enolate intermediate were not observed. This contrasts with our previous work9 where it was necessary to supply an excess of acid (up to 8 equiv) to fully suppress competitive allylation. The only byproduct isolated under the homogeneous reaction conditions was 5,5-diallyl-2,2-dimethyl-1,3-dioxane-4,6-dione (4, Figure 1).12 Interestingly, we did not detect the mono-allylated Meldrum's acid as a byproduct.
Figure 1.

Diallyl Meldrum's Acid Byproduct of Protonation.
Since we have been unable to perform detailed mechanistic studies with our heteogeneous protonation system, we were eager to examine the mechanism with our new system. Kinetic studies of the homogeneous enantioselective protonation reaction were performed via 1H NMR spectroscopy in C6D6 (Figure 2).11 A plot of amount of allyl β-ketoester substrate 2 over time displayed zero-order decay, suggesting that ketoester 2 reacts very fast and generates an intermediate (possibly a Pd-carboxylate or Pd-enolate) that undergoes further reaction in a slower step (Scheme 2). Similar zero-order dependence in substrate was observed in the case of our related enantioselective allylation reaction,7 implying that the early stages of both processes are similar. While these results give some information regarding the first portion of the reaction pathway, further studies are required to elucidate the mechanism and origin of enantioselectivity.
Figure 2.
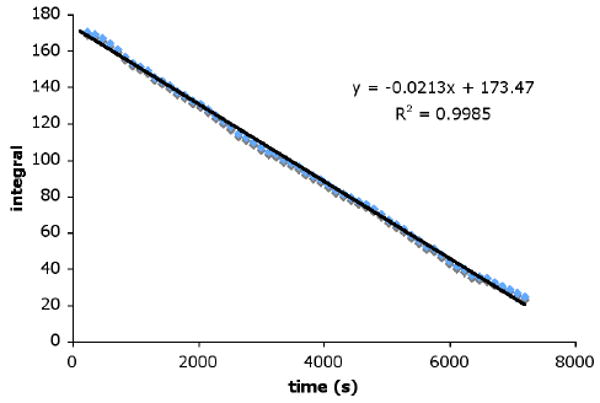
Plot of substrate 2 1H NMR integral vs. time.
Scheme 2.

Proposed Catalytic Cycle for Enantioselective Protonation.
In conclusion, we have developed a novel, homogeneous palladium-catalyzed system for the enantioconvergent decarboxylative protonation of racemic allyl β-ketoesters. The reaction tolerates a variety of substrate substitution and functionality and generates products with high enantiomeric excess. The method utilizes readily accessible racemic α-substituted allyl β-ketoesters and commercially available Meldrum's acid as the proton donor. One standard set of reaction conditions was amenable to the synthesis of a variety of α-tertiary cycloalkanones with high ee. The homogeneous conditions developed herein enabled preliminary kinetic experiments for investigation of the mechanism of this transformation. Additional explorations of the scope, mechanism, and application of this method are currently underway.
Supplementary Material
Acknowledgments
The authors thank Sankyo Co., Ltd. (financial support of TN), Eli Lilly (predoctoral fellowship to JTM), Amgen, and Bristol-Myers Squibb for generous funding.
Footnotes
Supporting Information Available Experimental details and NMR spectra of product compounds are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of enantioselective protonation, see:; (a) Duhamel L, Duhamel P, Plaquevent JC. Tetrahedron: Asymmetry. 2004;15:3653–3691. [Google Scholar]; (b) Eames J, Weerasooriya N. Tetrahedron: Asymmetry. 2001;12:1–24. [Google Scholar]; (c) Yanagisawa A, Yamamoto H. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 3. Springer; New York: 1999. pp. 1295–1306. [Google Scholar]; (d) Yanagisawa A, Yamamoto H. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Springer; New York: 2004. pp. 125–132. [Google Scholar]; (e) Fehr C. Angew Chem, Int Ed Engl. 1996;35:2566–2587. [Google Scholar]
- 2.For recent representative examples of enantioselective protonation, see:; (a) Donohoe TJ, Freestone GC, Headley CE, Rigby CL, Cousins RPC, Bhalay G. Org Lett. 2004;6:3055–3058. doi: 10.1021/ol049014q. [DOI] [PubMed] [Google Scholar]; (b) Liang G, Trauner D. J Am Chem Soc. 2004;126:9544–9545. doi: 10.1021/ja0476664. [DOI] [PubMed] [Google Scholar]; (c) Yanagisawa A, Touge T, Arai T. Angew Chem, Int Ed. 2005;44:1546–1548. doi: 10.1002/anie.200462325. [DOI] [PubMed] [Google Scholar]; (d) Sibi MP, Tatamidani H, Patil K. Org Lett. 2005;7:2571–2573. doi: 10.1021/ol050630b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schaefer C, Fu GC. Angew Chem, Int Ed. 2005;44:4606–4608. doi: 10.1002/anie.200501434. [DOI] [PubMed] [Google Scholar]; (f) Reynolds NT, Rovis T. J Am Chem Soc. 2005;127:16406–16407. doi: 10.1021/ja055918a. [DOI] [PubMed] [Google Scholar]; (g) Yamamoto H, Nakashima D. Synlett. 2006:151–152. [Google Scholar]; (h) Mitsuhashi K, Ito R, Arai T, Yanagisawa A. Org Lett. 2006;8:1721–1724. doi: 10.1021/ol0603007. [DOI] [PubMed] [Google Scholar]; (i) Frost CG, Penrose SD, Lambshead K, Raithby PR, Warren JE, Gleave R. Org Lett. 2007;9:2119–2122. doi: 10.1021/ol070603g. [DOI] [PubMed] [Google Scholar]; (j) Amere M, Lasne MC, Rouden J. Org Lett. 2007;9:2621–2624. doi: 10.1021/ol070712v. [DOI] [PubMed] [Google Scholar]; (k) Poisson T, Dalla V, Marsais F, Dupas G, Oudeyer S, Levacher V. Angew Chem, Int Ed. 2007;46:7090–7093. doi: 10.1002/anie.200701683. [DOI] [PubMed] [Google Scholar]; (l) Fehr C. Angew Chem, Int Ed. 2007;46:7119–7121. doi: 10.1002/anie.200701428. [DOI] [PubMed] [Google Scholar]
- 3.(a) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 4.For a review of these enantioselective allylation reactions developed by our lab and others, see: Mohr JT, Stoltz BM. Chem Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183., and references therein.
- 5.For the development of phosphinooxazoline ligands, see: Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336–345. doi: 10.1021/ar9900865.Williams JMJ. Synlett. 1996:705–710., and references therein.
- 6.We have recently reported a convenient general synthetic route to PHOX ligands see:; Tani K, Behenna DC, McFadden RM, Stoltz BM. Org Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]
- 7.Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J Am Chem Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]
- 8.To our knowledge, there are three other groups that have reported related Pd-catalyzed systems that produce similar enantioenriched products.; (a) Henin F, Muzart J. Tetrahedron: Asymmetry. 1992;3:1161–1164. [Google Scholar]; (b) Aboulhoda SJ, Henin F, Muzart J, Thorey C, Behnen W, Martens J, Mehler T. Tetrahedron: Asymmetry. 1994;5:1321–1326. [Google Scholar]; (c) Abouldoda SJ, Letinois S, Wilken J, Reiners I, Henin F, Martnes J, Muzart J. Tetrahedron: Asymmetry. 1995;6:1865–1868. [Google Scholar]; (d) Baur MA, Riahi A, Henin F, Muzart J. Tetrahedron: Asymmetry. 2003;14:2755–2761. [Google Scholar]; (e) Sugiura M, Nakai T. Angew Chem, Int Ed Engl. 1997;36:2366–2368. [Google Scholar]; (f) Hamashima Y, Somei H, Shimura Y, Tamura T, Sodeoka M. Org Lett. 2004;6:1861–1864. doi: 10.1021/ol0493711. [DOI] [PubMed] [Google Scholar]
- 9.Mohr JT, Nishimata T, Behenna DC, Stoltz BM. J Am Chem Soc. 2006;128:11348–11349. doi: 10.1021/ja063335a. [DOI] [PubMed] [Google Scholar]
- 10.(a) Meldrum AN. J Chem Soc. 1908;93:598–601. For the initial synthesis of this compound, see: [Google Scholar]; (b) Davidson D, Bernhard SA. J Am Chem Soc. 1948;70:3426–3428. doi: 10.1021/ja01190a060. For the correct structural assignment, see: [DOI] [PubMed] [Google Scholar]; (c) Bonifácia VDB. Synlett. 2004:1649–1650. For a highlight of recent chemistry based on Meldrum's acid, see: [Google Scholar]
- 11.See the Supporting Information for details.
- 12.(a) Bouillon G, Schank K. Chem Ber. 1980;113:2630–2635. [Google Scholar]; (b) Kayaki Y, Koda T, Ikariya T. J Org Chem. 2004;69:2595–2597. doi: 10.1021/jo030370g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.