

Abstract
Understanding “two-hit” experimental models is crucial for the rational development of therapies for hemorrhagic shock (HS). We modeled the clinical scenario of HS followed by polymicrobial sepsis (cecal ligation and puncture [CLP]) to investigate the molecular and functional alterations that occur within the gastrointestinal tract. Control, HS, CLP, simultaneous HS + CLP, and HS + delayed CLP by 24 h groups of Sprague-Dawley rats were studied for gastrointestinal transit and in vitro colonic circular muscle contractility to bethanechol. Reverse transcription–polymerase chain reaction quantified IL-6, IL-10, and heme oxygenase 1 messenger RNA expression in the isolated colonic muscularis 6 h after insult. Myeloperoxidase-positive neutrophils were quantified in colonic muscularis whole mounts. Mortality at 24 h was significantly increased in simultaneous mild HS + CLP (88%) over control, mild HS, CLP alone, or HS + delayed CLP. Cecal ligation and puncture significantly delayed transit compared with controls and HS alone. Hemorrhagic shock + delayed CLP animals had normal transit. Colonic contractions were suppressed by 50% after CLP compared with controls and HS. In contrast, HS + delayed CLP displayed control levels of contractile responses to bethanechol. Cecal ligation and puncture and simultaneous HS + CLP caused significant inflammatory messenger RNA induction of IL-6, iNOS, IL-10, and heme oxygenase 1 compared with control and HS, and these responses were significantly suppressed in HS + delayed CLP colonic muscularis extracts. Neutrophils were significantly recruited into the colonic muscularis following CLP after 24 h compared with control and HS. This recruitment was significantly less in the HS + delayed CLP animals. These data demonstrate the ability of mild HS to precondition the animal and protect it against a delayed, but not simultaneous, polymicrobial event.
Keywords: Gastrointestinal motility, hemorrhagic shock, sepsis, cecal ligation and puncture, IL-10
INTRODUCTION
Hemorrhagic shock (HS) with trauma and the subsequent development of multiple organ failure (MOF) is the leading cause of death for individuals aged 25 to 55 years, cutting lives short in their most productive years. The gut is often described as the motor of MOF because the loss of its integrity is a critical comorbidity factor for patients after HS, as well as trauma, surgery, sepsis, and burn injuries (1, 2). The intestine is highly sensitive to hemorrhage with major functional impairment of both the mucosa (3) and muscularis (4, 5). A major reason for this sensitivity is the disproportionate reduction in blood flow during circulatory shock caused by the preferential constriction of the splanchnic circulation to maintain core pressure. We and others have experimentally studied molecular and functional alterations to the gastrointestinal muscularis externa caused by HS. It has been shown that selective intestinal ischemia or systemic hypovolemic shock is associated with decreased gut motility that results in postinjury atony and functional ileus (5-8). The molecular basis for this dysfunction in gastrointestinal motility is an ischemic-induced inflammatory response within the intestinal muscularis externa that consists of the local activation of transcription factors and the upregulation of various cytokines and chemokines, in which the dense network of resident macrophages play a key role (6, 9). The final result of this hemorrhage-generated inflammatory milieu is the endothelial induction of intercellular adhesion molecule 1, the recruitment of leukocytes into the ischemic muscularis externa (7), and the elaboration of smooth muscle suppressive substances, such as NO via iNOS and prostanoids via COX-2 and reactive oxygen intermediates (5, 10).
Despite recent significant improvements in the clinical management of HS patients, the development of sepsis and ensuing MOF remains a leading cause of death in the surgical intensive care unit (11). The intestine and alterations in its barrier function are thought to play a pivotal role as an endogenous source of bacterial products and inflammatory mediators, which circulate systemically to the detriment of various organs, including the gut itself (12). Gastrointestinal ileus plays an important role in this process by limiting nutritional support, promoting bacterial overgrowth, and fostering mucosal barrier dysfunction.
We and others have shown that a single sublethal dose of endotoxin (LPS) disrupts in vivo and in vitro gastrointestinal motility. And this seems to be caused by the rapid activation of the dense network of resident macrophages that lie dormant within the intestinal muscularis (13-15), with the activated phagocytes exuding TNFα, IL-1β, IL-6, monocyte chemo-attractant protein 1, NO, and prostanoids, which cause ileus (14-19). We have also investigated the septic insult to the intestine using the more clinically relevant polymicrobial cecal ligation and puncture (CLP) model in rodents. Similar to LPS, CLP delayed gastrointestinal transit, decreased colonic contractile pressures, and suppressed circular muscle contractility of the jejunum and colon during a 4-day period. This functional deficit was also accompanied by the generation of a complex molecular inflammatory response and leukocyte sequestration into the muscularis externa by monocytes and neutrophils (20). Thus, the intestine is not only a source of bacteremia, but also an important target of bacterial products with major functional consequences to intestinal motility and the generation of cytokines, which likely participate in the development of MOF.
Clinically, patients seldom present with simple hemorrhage but most frequently experience multiple injurious events that occur in a typical sequential pattern. Therefore, it is vitally important to study the temporal interactions of various injurious events that simulate the clinical situation. Previously, various injurious interactions have been studied. An elegant “two-event” hypothesis has been put forth (21, 22), which postulates that an initial injury will prime the innate immune system such that a secondary event will synergistically interact with the aroused immune system leading to a rampant accentuated inflammatory response culminating in the devastating systemic inflammatory response syndrome (SIRS) (21, 22). Indeed, experimentalists have demonstrated that a priming event will result in exaggerated lung and liver injury to a subsequent insult (22). The specific priming injuries in two-hit models have included HS, superior mesenteric artery clamp, and burn, whereas the most common secondary injury consists of a septic challenge by systemic, intraperitoneal, or intratracheal endotoxin administration or CLP (23-27). This type of scenario is appropriately studied because clinically, an initial traumatic event is frequently followed by the development of an endogenous or exogenous infection.
However, the vulnerability window during which an individual is susceptible to the inflammatory synergy between two injurious events seems to be limited to around the first 36 to 72 h, at least for the lung (22, 27). It is currently unclear at which point and through which mechanisms the immune system of the body reverses gears and switches to a hypoinflammatory state called a compensatory anti-inflammatory responsive syndrome (28). Unfortunately, this secondary state of compromised host immunity predisposes critically ill patients to the development of late MOF to which most injured patients succumb (29).
Previous two-hit studies with subsequent sepsis have mainly focused on lung and liver injury. In addition, mucosal injury is known to be limited by intestinal preconditioning before a subsequent severe prolonged period of ischemia (8, 30, 31). However, no two-hit studies have addressed the effects of HS followed by sepsis on inflammatory responses within the gastrointestinal muscularis and its motor function. Therefore, this study was designed to study molecular, cellular, and functional alterations that alter gastrointestinal motility in an experimental model that simulates the common clinical presentation of HS followed by the development of polymicrobial sepsis.
MATERIALS AND METHODS
Animals and experimental procedures
This study was approved by the University of Pittsburgh Institutional Animal Care and Use Committee and conforms to the National Institutes of Health guidelines for the care and use of laboratory animals. Male Sprague-Dawley (Harlan, Indianapolis, Ind) rats (250–300 g) were randomly placed in one of the following groups: control, sham HS, sham CLP, HS, CLP, HS + CLPimmediate and HS + CLP24 h (n = 6 for each group). The HS group was anesthetized by continuous isoflurane inhalation, and the left femoral artery was cannulated. Arterial blood pressure was monitored continuously. The HS protocol has been previously described (10, 32). After an initial bleed of 2.25 mL/100 g body weight over 10 min, blood was withdrawn or returned as needed to maintain a MAP of 40 mmHg. After 90 min of shock, the animals were resuscitated to a MAP of 80 mmHg by administration of the remaining shed blood plus two times the shed blood volume in Ringer’s lactate solution. Cecal ligation and puncture operative procedures were performed under sterile conditions such as previously described (20, 33). Cecal ligation and puncture animals underwent a small midline laparotomy; the cecum was eventrated and partially ligated using a 4-0 silk suture and punctured once in the middle region of the ligated cecum with a 21-gauge needle. Cecal ligation and puncture operative procedures were performed to induce polymicrobial enteric-derived peritonitis. Sham HS animals consisted of femoral cannulation without blood withdrawal for 90 min, and sham CLP consisted of laparotomy without cecal ligation or puncture.
Gastrointestinal motility measurements
Functional gastrointestinal motility (gastrointestinal transit and in vitro colonic circular smooth muscle contractility) was measured in the specified animal groups. In vivo gastrointestinal transit was measured 24 h after insult by orally administering fluorescein isothiocyanate–labeled Dextran (200 μL of 5 mg/mL stock solution, molecular weight = 70,000; Sigma, St Louis, Mo) to quantify gastrointestinal transit for a period of 90 min, as previously described (4, 34). Ninety minutes after oral administration, the entire gastrointestinal tract was harvested and divided into 15 equal pieces from the stomach to the distal colon. The contents were emptied into a vial with 2.0 mL of sterile water, mixed vigorously, and centrifuged (10,000 revolutions per minute for 10 min) to clarify. The fluorescent supernatant concentration of each bowel segment was quantified in duplicates in a multiwell fluorescence plate reader (GeminiXS, Molecular Devices, Sunnyvale, Calif; excitation 488 nm and emission 525 nm). A percentage transit distribution histogram was plotted for each gastrointestinal tract and a geometric center (GC) was calculated (GC = Σ [% of total fluorescent signal per segment × segment number]/100) (35).
Mechanical organ bath colonic circular muscle contractility was measured in the specified groups. Twenty-four hours after insult, the entire colon was harvested and placed in a cold preoxygenated Krebs-Ringer buffer (KRB) in a Sylgard glass dish. The mid colonic segment was opened along the mesentery, and the muscularis layer was separated from the mucosa using surgical techniques as previously described (5, 36). Parallel to the circular muscle layer, strips (1 mm × the circumference) were cut and placed in an organ bath containing warmed preoxygenated KRB solution (37°C). Mechanical activity was measured as previously described (6). In brief, one end of each strip was tied to a fixed post and the other was attached to an isometric force transducer (World Precision Instruments, Sarasota, Fla). Dose-response curves were generated by exposing the muscles to increasing concentrations of the muscarinic agonist bethanechol (0.3–300 μmol/L) diluted in KRB solution with 10-min wash intervals. Contractile activity was calculated as grams per square millimeter per second by converting weight and length of the strip to square millimeters of tissue.
Quantification of leukocytic infiltration
Histochemistry stainings were performed on full thickness muscularis whole mounts of the specified groups of animals, which were sacrificed as control, sham, 24 or 48 h after resuscitation. Segments of the mid colon were immersed in KRB in a Sylgard glass dish and opened along the mesenteric border. The mucosa and submucosa were removed, and the muscularis was fixed in 100% ethanol for 10 min. After washing with KRB, the tissue was treated with Hanker-Yates reagent (1 mg/mL, Sigma, St Louis, Mo) and hydrogen peroxide 3% (1 μL/mL) for detection of polymorphonu-clear neutrophils (PMNs) exhibiting myeloperoxidase (MPO) activity. Tissues were mounted on glass slides using Gel/Mount (Biomedia Corp, Foster City, Calif), coverslipped, and inspected by light microscopy (Nikon FXA, Fryer, Huntley, Ill) at a magnification of 200 times. Numbers of MPO-positive PMNs infiltrating the muscularis externa were determined from the mean counts collected from five optical fields in each whole mount.
SYBR green real-time reverse transcription–polymerase chain reaction of inflammatory gene expression
For molecular studies using quantitative reverse transcription–polymerase chain reaction (RT-PCR) on muscularis externa extracts, the specified groups were sacrificed 6 h after the experimental procedure: control, sham HS, sham CLP, HS, CLP, HS + CLPimmediate and HS + CLP24 h. As indicated, the HS with CLP animals were divided in two subgroups: (a) simultaneous CLP immediately after resuscitation and sacrifice after 6 h and (b) delayed CLP at 24 h after resuscitation and sacrifice after 6 h. Sham-hemorrhaged animals underwent cannulation and anesthesia for the identical period as HS animals, but were not bled. In addition, sham CLP animals had a midline laparotomy without CLP.
Colonic muscularis messenger RNA (mRNA) expressions were quantified for IL-6, iNOS, IL-10, and heme oxygenase 1 (HO-1) using SYBR green two-step real-time RT-PCR (PE Applied Biosystems, Foster City, Calif). Total mRNA extraction was performed as previously described using the guanidinium-thiocyanate phenol-chloroform extraction method (7, 14). The RNA pellets were resuspended in RNAsecure resuspension solution (Ambion Inc, Austin, Tex). After resuspension, a DNAse treatment was carried out (DNA-free reagent, Ambion Inc). Equal aliquots (20 μg) of total RNA from each sample, quantified by spectrophotometry, were then processed for complementary DNA synthesis. Primers were designed according to published sequences or using Primer Express software (PE Applied Biosystems) and purchased from Life Technologies (Rockville, Md). Glyceraldehyde 3-phosphate dehydrogenase was used as an endogenous control. The efficiency and equality of the real-time PCR primer pairs were determined by amplifying serial dilutions of colonic muscularis complementary DNA. For each target gene, different MgCl2 (2-5 μmol/L) concentrations were tested to optimize the PCR amplification. Agarose gel electrophoretic analysis was used to verify the presence of a single product and to ensure that the amplified product corresponded to size predicted for the amplicon. Each sample was estimated in triplicate. The PCR mixture was prepared using the SYBR Green PCR Core Reagents (PE Applied Biosystems). The PCR conditions on an ABI PRISM 7700 Sequence Detection System (PE Applied Biosystems) were as recommended by the manufacturer. Relative quantification was performed using the comparative threshold cycle method as described previously by Schmittgen et al. (37) (see also User Bulletin no. 2, PE Applied Biosystems).
Data analysis
Results are presented as mean ± SEM. The data were analyzed using Student t test or ANOVA. EZAnalyze was used for F test and Bonferroni post hoc group comparisons where appropriate, and values of P < 0.05 were considered significant.
RESULTS
Experimental mortality
Mild HS for 90 min did not result in any mortality. Polymicrobial sepsis modeled by the single injury of CLP resulted in a 7% mortality rate (1/14 rats). When mild HS and CLP were performed concurrently (HS + CLPimmediate), an 88% mortality rate was observed (7/8 rats). Clinically, HS is frequently followed by the development a septic event, which can be polymicrobial in nature. But in contrast to the previous high mortality rate, when HS preceded CLP by 24 h (HS + CLP24 h), no mortality was recorded within the following 24 h.
Gastrointestinal transit
The single effects of mild HS and CLP on gastrointestinal transit were determined and compared with control rats. As previously described, in control animals, the nonabsorbable fluorescein isothiocyanate–labeled Dextran moved to the distal regions of the small intestine 90 min after oral administration, as reflected in a calculated GC of 8.8 ± 0.17 from the gastrointestinal transit distribution histograms (Fig. 1A). Sham HS and sham CLP did not significantly alter the geometric center compared with naive controls. Mild HS for 90 min followed by resuscitation for 24 h or 48 h resulted in no significant alteration in gastrointestinal transit compared with control (GC = 9.3 ± 0.44 and 9.1 ± 0.61, respectively). In contrast, 24 h after CLP-induced sepsis, animals demonstrated a significant delay in transit with a large portion of the fluorescent marker never leaving the stomach, yielding a calculated GC of 5.5 ± 0.84. In the animals which simultaneously received HS and CLP, the mortality rate (88%) was so high after 24 h that gastrointestinal transit was not determined in this group. We next investigated the effects of mild HS followed 24 h later by CLP. As shown in Figure 1B, preconditioning of the animals with mild HS followed by CLP after 24 h significantly reversed the transit delayed caused by CLP alone (GC = 7.8 ± 0.28) (ANOVA with adjusted Bonferroni individual group analysis, P < 0.05). Sham HS or sham CLP did not alter gastrointestinal transit (data not shown).
Fig. 1. Fluorescein isothiocyanate–labeled Dextran transit distribution histograms (A) in percent along the gastrointestinal tract in controls, HS, CLP, and HS + delayed CLP after a resuscitation period of 24 h.
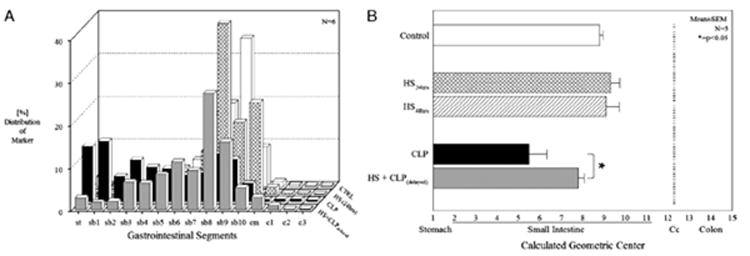
Controls and mild HS demonstrated a similar transit distribution pattern with a GC (B) of 8.81 ± 0.17 and 9.34 ± 0.44 with the transit marker being distributed in the terminal small bowel. Cecal ligation and puncture significantly delayed transit, which is reflected in the more orad distribution of the fluorescent Dextran to the proximal regions of the gastrointestinal tract (GC, 5.55 ± 0.84; *P < 0.025). After HS and delayed CLP, the transit distribution histogram and GC (7.80 ± 0.28) were similar to control values (n = 6 each group). c–colon; cm–cecum; sb–small bowel; st–stomach.
Intestinal circular muscle contractility
In vitro mechanical organ bath experiments were performed to determine the effect of HS and CLP, as single insults, and as a double-hit insult directly to the contractile activity of the colonic circular muscularis. The exposure of control muscle strips to bethanechol (0.3–300 μM) elicited a concentration-dependent increase in muscle contractility. Measurements of colonic circular muscle contractility 24 and 48 h after mild HS alone did not result in any significant change in the bethanechol-induced muscle contractile responses compared with controls (control, 4.8 ± 0.35 g/mm2 per second; 24 h, 4.9 ± 0.41 g/mm2 per second; 48 h, 4.80 ± 0.73 g/mm2 per second at 100 μM bethanechol; Fig. 2). Cecal ligation and puncture resulted in a significant suppression (50%) of colonic circular muscle contractility compared with controls (CLP, 2.4 ± 0.43 g/mm2 per second at 100 μM bethanechol, t test, P < 0.05). In addition, supporting the in vivo upper gastrointestinal transit data, colonic muscle contractility was significantly improved in the group that was subjected to HS followed by delayed CLP 24 h later, with contractions being measured an additional 24 h later (48 h after HS resuscitation; 4.90 ± 0.52 g/mm2 per second at 100 μM bethanechol, t test, P < 0.05). Colonic muscle activity could not be investigated in the simultaneous HS + CLP group because of the high mortality rate given by the Institutional Animal Care and Use Committee regulations. However, we assume that it would have been severely depressed compared with controls. Sham HS or sham CLP did not alter colonic circular muscle contractile responses to bethanechol (data not shown).
Fig. 2. The contractility of the colonic muscularis (grams per square millimeter per second) was measured in response to the cholinergic agonist bethanechol in concentrations of 0.3 to 300 μM.
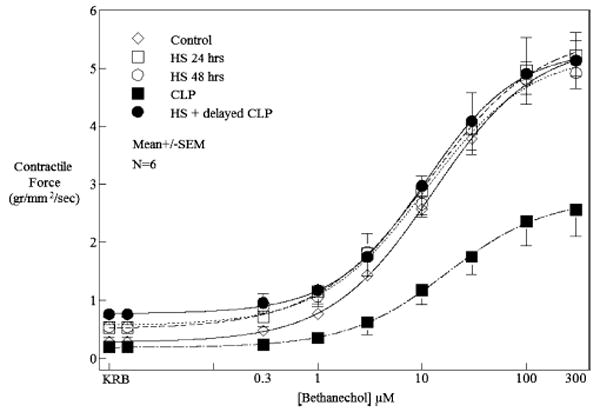
Twenty-four and 48 h after HS, contractility did not change compared with controls. Smooth muscle contractility of animals subjected to CLP was significantly decreased by approximately 50% compared with control animals at a representative bethanechol dose of 100 μM. Colon smooth muscle contractility was completely restored in HS and delayed CLP 24 h after resuscitation. Data shown represent mean contractility ± SEM at individual bethanechol doses (n = 5 each group).
Molecular inflammatory responses
The observed improvements in mortality and gastrointestinal motility in the 24-h preshocked intestine subjected to polymicrobial sepsis would predict that proinflammatory molecular signaling within the muscularis externa was moderated by the shock-induced ischemic preconditioning of the intestine. The underlying inflammatory molecular stimuli, which are known to participate in the inflammatory responses within the intestinal colonic muscularis (8), were investigated using SYBR green real-time RT-PCR analysis of the isolated colonic muscularis externa. IL-6 mRNA levels were investigated in seven groups of animals as a prototypical inflammatory cytokine within the colonic muscularis (control, sham HS, sham CLP, HS, CLP, simultaneous HS + CLP, and pre-HS + CLP). A 6-h time point was chosen for tissue harvest based on previously published data that showed this time to be maximal for inflammatory mediator expressions after CLP (9). As demonstrated in Figure 3A, no significant elevations in IL-6 mRNA were measured over control levels in the colonic muscularis specimens harvested 6 h after insult from sham HS, sham CLP, or mild HS groups of animals. However, both CLP alone and the simultaneous insult of HS + CLP resulted in greater than a 1,000-fold induction of IL-6 mRNA. In addition, as predicted by the improved motility, IL-6 mRNA induction was reduced by 60% in the pre-HS + CLP muscularis.
Fig. 3. Real-time RT-PCR for IL-6, iNOS, IL-10, and HO-1 mRNA expressions in the isolated colonic muscularis externa were measured 6 h after the designated insult and compared with controls.
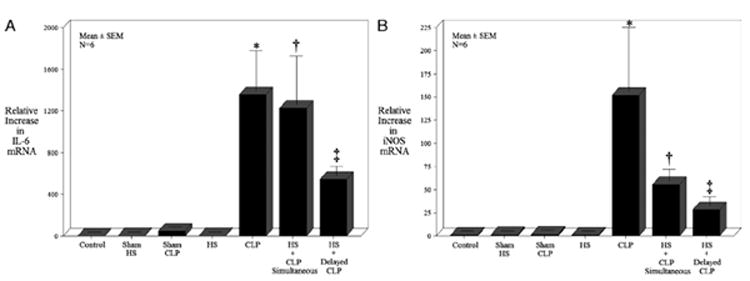
Inflammatory mediator expressions with the muscularis externa samples from controls, sham HS, sham CLP, HS, CLP, HS + CLP, and HS + delayed CLP were investigated. Data represent mean ± SEM for each group with F test analysis and Bonferroni post hoc group comparisons (*, †, and ‡P < 0.05 vs. controls; ‡P = 0.05 vs. CLP and HS + CLP; n = 5 each group).
Nitric oxide generated from iNOS has been shown to be important in colonic muscle dysfunction during inflammation (10), therefore, we quantified the expression of iNOS mRNA in the seven groups of animals. Similar to IL-6 expressions, no significant elevations in iNOS mRNA were measured over control levels in the colonic muscularis specimens harvested 6 h after insult from sham HS, sham CLP, or HS groups of animals. However, CLP alone resulted in 152-fold induction of iNOS correlating with the observed suppression in muscle function. Interestingly, the simultaneous insult of HS + CLP resulted in only a 55-fold increase over control, significantly less than that observed for CLP alone, although these animals were destined not to survive. The induction of iNOS mRNA was even less robust in the pre-HS + CLP animals, which demonstrated only a 28-fold increase compared with controls (Fig. 3B).
Our previous data suggest that the anti-inflammatory mediators IL-10 and HO-1 play a role in moderating colonic inflammation within the muscularis (11, 12). Therefore, we hypothesized that the pre-HS insult may function as an ischemic preconditioning of the colon, which initiates an early upregulation in IL-10 and HO-1. In contrast to the proinflammatory cytokines, the colonic muscularis anti-inflammatory mediators were exquisitely sensitive to experimental perturbation. As shown in Figure 3, C and D, the sham HS, sham CLP, and HS groups all demonstrated an induction of IL-10 and HO-1, with HS causing a 4-fold increase in both mediators over control. Cecal ligation and puncture alone and the simultaneous insults of HS + CLP each caused an approximate 65-fold induction of IL-10, which was significantly attenuated to only a 21-fold induction with a pre-HS stimulus 24 h before CLP. The HO-1 mRNA level was also significantly induced by CLP (20-fold) and synergistically induced by the simultaneous double hit (83-fold). The induction of HO-1 was again significantly reduced in the pre-HS + CLP tissues (8-fold) compared with both CLP alone and the simultaneous double-hit groups, with all tissues being harvested 6 h after the final experimental procedure.
Leukocytic infiltration
Twenty-four hours after HS, CLP and HS + CLPdelayed, the mid colonic muscularis was prepared in whole mounts to quantify PMN infiltrate into the muscularis after MPO histo-chemistry. In control, shams, and mild HS animals, MPO-positive cells were only occasionally detected (Fig. 4A–C, ). In contrast, CLP alone resulted in a significant recruitment of PMNs at 24 h into the colonic muscularis (CLP, 38 ± 2.8/cells at magnification ×200; Fig. 4D). Complimenting the above results, 24 h after HS + CLPdelayed, a significantly diminished number of PMNs were observed to be recruited into the colonic muscularis (20.2 ± 3.4) compared with CLP alone (ANOVA with adjusted Bonferroni individual group analysis, P < 0.05). A histogram of the quantified MPO-positive cells in the colon muscularis whole mounts is shown in Figure 5.
Fig. 4. Representative colonic muscularis whole mounts stained with Hanker Yates reagent for MPO-positive PMNs.
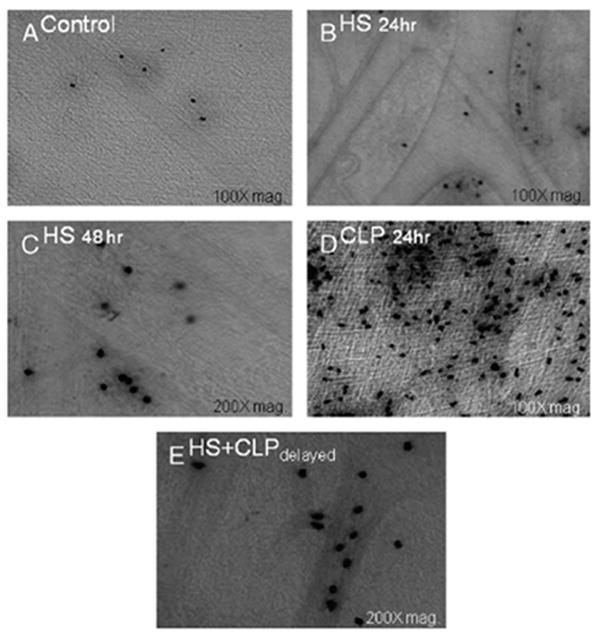
Few MPO-positive PMNs were seen in the control colonic whole mounts (A). Colonic muscularis whole mounts prepared 24 h (B) and 48 h (C) after HS showed an insignificant slight increase in PMN counts compared with control animals. Cecal ligation and puncture alone resulted in a massive recruitment of PMNs (D) compared with controls. Hemorrhagic shock and delayed CLP 24 h after resuscitation completely abolished the massive PMN infiltration compared with CLP (E).
Fig. 5. Histogram of MPO-positive neutrophil quantification within whole mounts of the colonic muscularis externa obtained from control, HS at 24 h, HS at 48 h, CLP, and HS + delayed CLP animals.
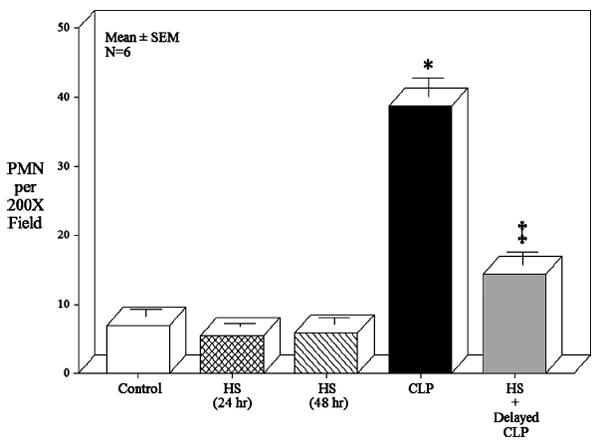
Five random fields of each colonic whole mount was observed under light microscopy and counted for numbers of MPO-stained PMNs at a magnification of 200 times. The mean ± SEM of each group was plotted and analyzed with an F test and Bonferroni post hoc group comparisons (* and ‡P < 0.05 vs. controls; ‡P = 0.05 vs. CLP, n = 5 each group).
DISCUSSION
The clinical scenario of HS followed by polymicrobial sepsis was simulated and investigated to shed light on molecular inflammatory events within the colonic muscularis and functional alterations in gastrointestinal transit and colonic muscle contractility that occur in this clinically relevant two-hit model.
It was previously proposed that the baneful SIRS occurs after a traumatic one hit, even in the absence of infection. Furthermore, in a two-hit model, a mild injury can create a priming inflammatory environment, which when compounded upon by a secondary, although otherwise innocuous, insult will also synergistically lead to severe end-organ injury and SIRS (21). This type of inflammatory synergy in a two-hit injury seems to have occurred in this study in the group of animals that received mild HS followed immediately by CLP, in which the polymicrobial-induced inflammatory response would have developed soon after shock (20). The mortality rate in this group of animals was nearly 90% compared with only minimal mortality in animals receiving a single injury.
However, published results investigating a priming phenomenon in various organs have been equivocal (8, 17, 38-40). The ostensible contradictions in these studies can probably be traced to differences in type, timing, and severity of the insults, as well as the particular experimental parameters being evaluated. All these distinctions probably have important implications in understanding the inflammatory response after trauma. In addition, the mechanism by which the so-called compensatory anti-inflammatory response syndrome might influence the inflammatory response is poorly understood (41). Evidence suggests that a time-dependent interaction exists between SIRS and compensatory anti-inflammatory response syndrome; a pro-inflammatory response may initially dominate but then later yield to a mounting anti-inflammatory response. Therefore, synergy in the two-hit hypothesis probably relies on the assumption that the immune system is in a hyperinflammatory state after the initial stimulus, which then increases the susceptibility of the patient to a secondary inflammatory injury.
Contrary to the two-hit synergistic effect and the high mortality rate recorded in the simultaneous HS + CLP model, no mortality was observed when CLP-induced sepsis was initiated 24 h after mild HS. Indeed, gastrointestinal transit measured 24 h after injury was significantly better in the combined HS delayed CLP animals compared with CLP alone, although HS by itself had no observable effect on transit at the 24- and 48-h periods. Similar results were obtained when recording isolated colonic circular muscle contractions 24 h after injury. Hemorrhagic shock alone did not alter contractions, but CLP alone caused a significant 50% depression in contractility. And as suggested by the upper gastrointestinal tract transit data, colonic contractility recorded from the HS with delayed CLP animals was improved over CLP alone.
The mild HS injury used in this study could be interpreted as a form of ischemic preconditioning. Recently, it has been shown that short periodic bursts of superior mesenteric artery occlusion can cause a prolonged ischemic preconditioning in the intestine, which results in improved function, decreased inflammation, and mucosal protection to a secondary prolonged period of ischemia (8). This is, of course, invoking the similar principle long known in the cardiac literature (42). Organ preconditioning has been broadly defined as a process whereby an antecedent event of transient ischemia, oxidative stress, temperature change, endotoxemia or drug administration bestows upon an organ or cell system a temporary tolerance to further insults (43). Indeed, we have previously demonstrated cross-tolerance in the intestine between a “tolerizing” dose of endotoxin and surgically induced postoperative ileus (17).
In this study, we also investigated molecular events that would begin to indicate the specific pathways that are involved in the protective response of mild HS to the subsequent development of a polymicrobial sepsis. We measured colonic muscularis IL-6 mRNA as a prototypical inflammatory marker within the injured muscularis externa because the detrimental effects of increased IL-6 on the bowel after sepsis (14) and HS (6, 44) have been demonstrated before, and because IL-6 is a consistent predictor of sepsis severity and mortality (45). Muscularis IL-6 mRNA was not significantly induced by either sham procedure or mild HS. However, a greater than 1,000-fold induction was measured in response to CLP alone and simultaneous HS + CLP. This massive induction, apparently caused primarily by CLP, was significantly inhibited by the 24-h pre-HS ischemic conditioning of the organ. Similarly, in a mild surgical stress and endotoxin double-hit model, Kamei et al. (46) found a significant reduction in mortality in the animals receiving both insults days apart with a corresponding inhibition in the endotoxin-induced increase in serum TNF-α and IL-6 levels.
Nitric oxide has been demonstrated to be a key inflammatory mediator, but it also plays a major role in regulating gastrointestinal motility via its production by nNOS in the enteric nervous system (47). Interestingly, iNOS from inflammatory cells within the muscularis externa has been demonstrated to play a significant role in the suppression of intestinal contractility after surgery, endotoxin, and CLP (16, 48). Here we show that the induction of colonic muscularis iNOS was down-regulated by mild shock regardless of the timing of the event because iNOS induction was inhibited significantly in both the immediate and delayed double-hit models compared with the induction by CLP alone.
IL-10 and HO-1 are known potent anti-inflammatory cytokines that are increased after CLP and HS (49). IL-10 is endogenously expressed in animals and patients with septic syndromes, and the concentrations of IL-10 in the blood and tissue compartments are often indicative of the magnitude and severity of the inflammatory injury (49). Therefore, as expected, the highest-injury group, the combined simultaneous HS + CLP animals, showed the largest increases in muscularis tissue IL-10 and HO-1 mRNA expression. Indeed, the intestine regarding these two anti-inflammatory mediators was very sensitive to injury-induced expression, as even sham animals were observed to have increased levels of expression. Mild shock resulted in a 4-fold increase in both mediators over control. A prolonged stable pre-elevation in IL-10 and HO-1 could be the anti-inflammatory mechanism that was associated with a decreased induction of IL-6 and iNOS, as well as HO-1 and IL-10 itself after the second hit of CLP. On the other hand, the decreased induction of the various cytokines could be caused by a generalized hyporesponsiveness to a second insult similar to that described before in macrophages/monocytes using a model of LPS preconditioning (50). The possible use of exogenous IL-10 or its preinduction as a treatment has been at times difficult to interpret clearly. Steinhauser et al. (51) noted that the endogenous production of IL-10 seemed to suppress the ability of the alveolar macrophages to subsequently kill bacteria. Interestingly, the preinduction of IL-10 and/or HO-1 by very low-dose carbon monoxide inhalation or other means has consistently been demonstrated to be protective in HS, acute lung injury, and endotoxemia (52, 53). However, the functional outcome of elevated IL-10 or HO-1 in various two-hit models is still unclear. But recently, Tamion et al. (54) indicated that intestinal I/R preconditioning decreased the inflammatory response to a secondary 1-h hemorrhage by inducing HO-1 overexpression. In addition, hemin-induced HO-1 mRNA was shown to attenuate HS-induced leukocyte adherence within the intestinal mesentery (55). Mechanistically, the preinduction of HO-1 and IL-10 by mild HS in this study could in part explain the observed decrease in leukocyte recruitment into the colonic muscularis in the HS + delayed CLP group of animals compared with CLP alone.
The onset of a secondary insult seems to be critical to the ultimate response of the organism, and the exact timing may be tissue specific (27). To our knowledge, the common clinical scenario of HS and a subsequent polymicrobial septic insult has not been previously modeled to study ileus. Depending upon various factors, modifications to immune and tissue function by a first hit could theoretically be either beneficial or harmful to the organism when it is exposed to a subsequent second insult.
In conclusion, in this two-hit experimental model of the clinical scenario of mild HS and sepsis, a time-dependent biphasic response to the second hit of bacteremia was measured in gastrointestinal motility, proinflammatory mediators, anti-inflammatory cytokines, and leukocyte recruitment. In this particular setting, in the short-term, mild HS augments mortality, injury, and the inflammatory response to bacteremia, and in the long-term, it blunts mortality, injury, and the inflammatory response to bacteremia. We believe that understanding the complex interaction of clinically relevant two-hit experimental models is crucial for the rational development of effective therapies for the leading cause of death for individuals during their most productive years of life.
Acknowledgments
This study was supported by the National Institutes of Health (grant no. R01-GM58241, R01-DK068610, and P50-GM53789).
ABBREVIATIONS
- CLP
cecal ligation and puncture
- GC
geometric center
- HO-1
heme oxygenase 1
- HS
hemorrhagic shock
- KRB
Krebs-Ringer buffer
- MOF
multiple organ failure
- MPO
myeloperoxidase
- PMN
polymorphonuclear neutrophils
- RT-PCR
reverse transcription–polymerase chain reaction
- SIRS
systemic inflammatory response syndrome
References
- 1.Marshall JC, Christou NV, Meakins JL. The gastrointestinal tract. The “undrained abscess” of multiple organ failure. Ann Surg. 1993;218:111–119. doi: 10.1097/00000658-199308000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deitch EA, Rutan R, Waymack JP. Trauma, shock, and gut translocation. New Horiz. 1996;4:289–299. [PubMed] [Google Scholar]
- 3.Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness [Review] Curr Opin Crit Care. 2003;9:143–151. doi: 10.1097/00075198-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Kallakuri S, Ascher E, Pagala M, Gade P, Hingorani A, Scheinman M, Mehraein K, Jacob T. Protective effect of glycine in mesenteric ischemia and reperfusion injury in a rat model. J Vasc Surg. 2003;38:1113–1120. doi: 10.1016/s0741-5214(03)00939-x. [DOI] [PubMed] [Google Scholar]
- 5.Hierholzer C, Kalff JC, Billiar TR, Bauer AJ, Tweardy DJ, Harbrecht BG. Induced nitric oxide promotes intestinal inflammation following hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2004;286:G225–G233. doi: 10.1152/ajpgi.00447.2002. [DOI] [PubMed] [Google Scholar]
- 6.Hierholzer C, Kalff JC, Chakraborty A, Watkins SC, Billiar TR, Bauer AJ, Tweardy DJ. Impaired gut contractility following hemorrhagic shock is accompanied by IL-6 and G-CSF production and neutrophil infiltration. Dig Dis Sci. 2001;46:230–241. doi: 10.1023/a:1005524021552. [DOI] [PubMed] [Google Scholar]
- 7.Kalff JC, Hierholzer C, Tsukada K, Billiar TR, Bauer AJ. Hemorrhagic shock results in intestinal muscularis intercellular adhesion molecule (ICAM-1) expression, neutrophil infiltration, and smooth muscle dysfunction. Arch Orthop Trauma Surg. 1999;119:89–93. doi: 10.1007/s004020050363. [DOI] [PubMed] [Google Scholar]
- 8.Moore-Olufemi SD, Kozar RA, Moore FA, Sato N, Hassoun HT, Cox CS, Jr, Kone BC. Ischemic preconditioning protects against gut dysfunction and mucosal injury after ischemia/reperfusion injury. Shock. 2005;23:258–263. [PubMed] [Google Scholar]
- 9.Zou L, Attuwaybi B, Kone BC. Effects of NF-kappa B inhibition on mesenteric ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2003;284:G713–G721. doi: 10.1152/ajpgi.00431.2002. [DOI] [PubMed] [Google Scholar]
- 10.Hierholzer C, Billiar TR. Molecular mechanisms in the early phase of hemorrhagic shock. Langenbecks Arch Surg. 2001;386:302–308. doi: 10.1007/s004230100242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarrar D, Chaudry IH, Wang P. Organ dysfunction following hemorrhage and sepsis: mechanisms and therapeutic approaches. Int J Mol Med. 1999;4:575–583. doi: 10.3892/ijmm.4.6.575. [DOI] [PubMed] [Google Scholar]
- 12.Grotz MR, Deitch EA, Ding J, Xu D, Huang Q, Regel G. Intestinal cytokine response after gut ischemia: role of gut barrier failure. Ann Surg. 1999;229:478–486. doi: 10.1097/00000658-199904000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cullen JJ, Mercer D, Hinkhouse M, Ephgrave KS, Conklin JL. Effects of endotoxin on regulation of intestinal smooth muscle nitric oxide synthase and intestinal transit. Surgery. 1999;125:339–344. [PubMed] [Google Scholar]
- 14.Eskandari MK, Kalff JC, Billiar TR, Lee KK, Bauer AJ. Lipopolysaccharide activates the muscularis macrophage network and suppresses circular smooth muscle activity. Am J Physiol. 1997;273:G727–G734. doi: 10.1152/ajpgi.1997.273.3.G727. [DOI] [PubMed] [Google Scholar]
- 15.Lodato RF, Khan AR, Zembowicz MJ, Weisbrodt NW, Pressley TA, Li YF, Lodato JA, Zembowicz A, Moody FG. Roles of IL-1 and TNF in the decreased ileal muscle contractility induced by lipopolysaccharide. Am J Physiol. 1999;276:G1356–G1362. doi: 10.1152/ajpgi.1999.276.6.G1356. [DOI] [PubMed] [Google Scholar]
- 16.Eskandari MK, Kalff JC, Billiar TR, Lee KK, Bauer AJ. LPS-induced muscularis macrophage nitric oxide suppresses rat jejunal circular muscle activity. Am J Physiol. 1999;277:G478–G486. doi: 10.1152/ajpgi.1999.277.2.G478. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz NT, Engel B, Eskandari MK, Kalff JC, Grandis JR, Bauer AJ. Lipopolysaccharide preconditioning and cross-tolerance: the induction of protective mechanisms for rat intestinal ileus. Gastroenterology. 2002;123:586–598. doi: 10.1053/gast.2002.34777. [DOI] [PubMed] [Google Scholar]
- 18.Wirthlin DJ, Cullen JJ, Spates ST, Conklin JL, Murray J, Caropreso DK, Ephgrave KS. Gastrointestinal transit during endotoxemia: the role of nitric oxide. J Surg Res. 1996;60:307–311. doi: 10.1006/jsre.1996.0048. [DOI] [PubMed] [Google Scholar]
- 19.Türler A, Schwarz NT, Turler E, Kalff JC, Bauer AJ. MCP-1 causes leukocyte recruitment and subsequently endotoxemic ileus in rat. Am J Physiol Gastrointest Liver Physiol. 2001;282:G145–G155. doi: 10.1152/ajpgi.00263.2001. [DOI] [PubMed] [Google Scholar]
- 20.Overhaus M, Togel S, Pezzone MA, Bauer AJ. Mechanisms of polymicrobial sepsis–induced ileus. Am J Physiol Gastrointest Liver Physiol. 2004;287:G685–G694. doi: 10.1152/ajpgi.00359.2003. [DOI] [PubMed] [Google Scholar]
- 21.Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am. 1995;75:257–277. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- 22.Moore EE, Moore FA, Harken AH, Johnson JL, Ciesla D, Banerjee A. The two-event construct of postinjury multiple organ failure. Shock. 2005;24(suppl 4):71–74. doi: 10.1097/01.shk.0000191336.01036.fe. [DOI] [PubMed] [Google Scholar]
- 23.Rizoli SB, Kapus A, Fan J, Li YH, Marshall JC, Rotstein OD. Immunomodulatory effects of hypertonic resuscitation on the development of lung inflammation following hemorrhagic shock. J Immunol. 1998;161:6288. [PubMed] [Google Scholar]
- 24.Schwacha MG. Macrophages and post-burn immune dysfunction. Burns. 2003;29:1–14. doi: 10.1016/s0305-4179(02)00187-0. [DOI] [PubMed] [Google Scholar]
- 25.Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock. 2003;19:358–365. doi: 10.1097/00024382-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 26.Ayala A, Chung CS, Lomas JL, Song GY, Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH, et al. Shock-induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Am J Pathol. 2002;161:2283–2294. doi: 10.1016/S0002-9440(10)64504-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lomas-Neira J, Chung CS, Grutkoski PS, Dunican A, Simms HH, Cioffi WG, Ayala A. Divergent roles of murine neutrophil chemokines in hemorrhage induced priming for acute lung injury. Cytokine. 2005;31:169–179. doi: 10.1016/j.cyto.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 29.Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma. 1996;40:501–510. doi: 10.1097/00005373-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Mallick IH, Yang W, Winslet MC, Seifalian AM. Ischaemic preconditioning improves microvascular perfusion and oxygenation following reperfusion injury of the intestine. Br J Surg. 2005;92:1169–1176. doi: 10.1002/bjs.4988. [DOI] [PubMed] [Google Scholar]
- 31.Wu B, Ootani A, Iwakiri R, Fujise T, Tsunada S, Toda S, Fujimoto K. Ischemic preconditioning attenuates ischemia-reperfusion–induced mucosal apoptosis by inhibiting the mitochondria-dependent pathway in rat small intestine. Am J Physiol Gastrointest Liver Physiol. 2004;286:G580–G587. doi: 10.1152/ajpgi.00335.2003. [DOI] [PubMed] [Google Scholar]
- 32.Hierholzer C, Kelly E, Tsukada K, Loeffert E, Watkins S, Billiar TR, Tweardy DJ. Hemorrhagic shock induces G-CSF expression in bronchial epithelium. Am J Physiol. 1997;273:L1058–L1064. doi: 10.1152/ajplung.1997.273.5.L1058. [DOI] [PubMed] [Google Scholar]
- 33.Overhaus M, Moore BA, Barbato JE, Behrendt FF, Doering JG, Bauer AJ. Biliverdin protects against polymicrobial sepsis by modulating inflammatory mediators. Am J Physiol Gastrointest Liver Physiol. 2006;290:G695–G703. doi: 10.1152/ajpgi.00152.2005. [DOI] [PubMed] [Google Scholar]
- 34.Kalff JC, Buchholz BM, Eskandari MK, Hierholzer C, Schraut WH, Simmons RL, Bauer AJ. Biphasic response to gut manipulation and temporal correlation of cellular infiltrates and muscle dysfunction in rat. Surgery. 1999;126:498–509. [PubMed] [Google Scholar]
- 35.Williams CL, Bihm CC, Rosenfeld GC, Burks TF. Morphine tolerance and dependence in the rat intestine in vivo. J Pharmacol Exp Ther. 1997;280:656–663. [PubMed] [Google Scholar]
- 36.Kalff JC, Schraut WH, Simmons RL, Bauer AJ. Surgical manipulation of the gut elicits an intestinal muscularis inflammatory response resulting in postsurgical ileus. Ann Surg. 1998;228:652–663. doi: 10.1097/00000658-199811000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription–polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 38.Schulman AM, Claridge JA, Ghezel-Ayagh A, Johnson O, III, Young JS. Differential local and systemic tumor necrosis factor-alpha responses to a second hit of lipopolysaccharide after hemorrhagic shock. J Trauma. 2003;55:298–307. doi: 10.1097/01.TA.0000028970.50515.A0. [DOI] [PubMed] [Google Scholar]
- 39.Patton JH, Jr, Lyden SP, Ragsdale DN, Croce MA, Fabian TC, Proctor KG. Granulocyte colony-stimulating factor improves host defense to resuscitated shock and polymicrobial sepsis without provoking generalized neutrophilmediated damage. J Trauma. 1998;44:750–758. doi: 10.1097/00005373-199805000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Fan J, Shek PN, Suntres ZE, Li YH, Oreopoulos GD, Rotstein OD. Liposomal antioxidants provide prolonged protection against acute respiratory distress syndrome. Surgery. 2000;128:332–338. doi: 10.1067/msy.2000.108060. [DOI] [PubMed] [Google Scholar]
- 41.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 42.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 43.Raeburn CD, Cleveland JC, Jr, Zimmerman MA, Harken AH. Organ preconditioning. Arch Surg. 2001;136:1263–1266. doi: 10.1001/archsurg.136.11.1263. [DOI] [PubMed] [Google Scholar]
- 44.Hierholzer C, Kalff JC, Audolfsson G, Billiar TR, Tweardy DJ, Bauer AJ. Molecular and functional contractile sequelae of rat intestinal ischemia/reperfusion injury. Transplantation. 1999;68:1244–1254. doi: 10.1097/00007890-199911150-00006. [DOI] [PubMed] [Google Scholar]
- 45.Damas P, Ledoux D, Nys M, Vrindts Y, De Groote D, Franchimont P, Lamy M. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann Surg. 1992;215:356–362. doi: 10.1097/00000658-199204000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamei K, Nimura Y, Nagino M, Aono K, Nakashima I. Surgical stress reduces mortality from endotoxin shock. Langenbecks Arch Surg. 2002;386:512–517. doi: 10.1007/s00423-001-0261-y. [DOI] [PubMed] [Google Scholar]
- 47.Stark ME, Bauer AJ, Sarr MG, Szurszewski JH. Nitric oxide mediates inhibitory nerve input in human and canine jejunum. Gastroenterology. 1993;104:398–409. doi: 10.1016/0016-5085(93)90407-4. [DOI] [PubMed] [Google Scholar]
- 48.Kalff JC, Schraut WH, Billiar TR, Simmons RL, Bauer AJ. Role of inducible nitric oxide synthase in postoperative intestinal smooth muscle dysfunction in rodents. Gastroenterology. 2000;118:316–327. doi: 10.1016/s0016-5085(00)70214-9. [DOI] [PubMed] [Google Scholar]
- 49.Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30:58–63. [PubMed] [Google Scholar]
- 50.Randow F, Syrbe U, Meisel C, Krausch D, Zuckermann H, Platzer C, Volk HD. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J Exp Med. 1995;181:1887–1892. doi: 10.1084/jem.181.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, Standiford TJ. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J Immunol. 1999;162:392–399. [PubMed] [Google Scholar]
- 52.Lee PJ, Alam J, Wiegand GW, Choi AM. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci U S A. 1996;93:10393–10398. doi: 10.1073/pnas.93.19.10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 54.Tamion F, Richard V, Lacoume Y, Thuillez C. Intestinal preconditioning prevents systemic inflammatory response in hemorrhagic shock. Role of HO-1. Am J Physiol Gastrointest Liver Physiol. 2002;283:G408–G414. doi: 10.1152/ajpgi.00348.2001. [DOI] [PubMed] [Google Scholar]
- 55.Moncure M, Chen L, Childs EW, Smalley D, Udobi KF, Cheung LY. Heme-oxygenase-1 mRNA expression affects hemorrhagic shock–induced leukocyte adherence. J Trauma. 2003;55:118–125. doi: 10.1097/01.TA.0000075333.04091.4A. [DOI] [PubMed] [Google Scholar]