


Abstract
L1 elements are human transposons which replicate via an RNA intermediate. At least 15% of the human genome is composed of L1 sequence. An important initial step in the transposition reaction is nicking of the genomic DNA by L1 endonuclease (L1 EN). In vivo much of the genome exists in the form of chromatin or is undergoing biochemical transactions such as transcription, replication or repair, which may alter the accessibility of the L1 transposition machinery to DNA. To investigate this possibility we have examined the effect of substrate chromatinization on the ability of L1 EN to nick DNA. We find that DNA incorporated into nucleosomes is generally refractory to nicking by L1 EN. Interestingly, nicking of a minority of DNA sequences is enhanced when included in chromatin. Thus, dynamic epigenetic factors such as chromatinization are likely to influence the relatively permanent placement of L1 and other retroelements in the human genome.
INTRODUCTION
Transposons are segments of DNA capable of movement or replication within or between genomes. L1 elements are replicative human transposons whose mobilization involves a RNA intermediate (for a review see 1). Although most L1 elements lack intact 5′-ends (and are therefore not transpositionally competent), full-length elements (Fig. 1) encode two proteins essential for L1 retrotransposition (2). L1 ORF1 is a multimeric RNA-binding protein (3). L1 ORF2 contains both endonuclease and reverse transcriptase activities, as well as an uncharacterized region with homology to zinc finger domains (4,5).
Figure 1.

The human L1 retrotransposon. EN, endonuclease domain; RT, reverse transcriptase; ZN, putative zinc finger; vTSD, variable target site duplication. The 5′-UTR contains an internal promoter (black arrow), the 3′-UTR a poly(A) sequence.
As L1 endonuclease (L1 EN) is probably used to initiate the transposition reaction (5,6), the specificity of L1 EN influences the site of transposon insertion. We have recently shown that the specificity of this nuclease is similar to the sequence at the sites of L1 insertion in vivo and described the biochemical requirements of its nucleic acid recognition (5,7). Briefly, L1 EN is specific for DNA within a range of structural and sequence parameters, with minor groove width being of particular importance. The DNA sequence that best correlates with these requirements is TnAn, with nicking occurring preferentially at the TpA phosphodiester. L1 EN recognition of the 5′ (Tn) portion of this sequence is far more extensive and important for nicking than the rather minimally contacted 3′-half of the target DNA. Substitutions in this sequence which conserve the homopyrimidine or homopurine run are generally well tolerated. This experimental evidence has been corroborated by computer analysis of the sites of L1 and Alu element insertion, which suggested a nuclease specificity identical to that found in vitro for L1 EN (Pickeral et al., submitted for publication; 8). The macroscale distribution of retrotransposons in the genome, however, is likely to depend on the accessibility of the chromosome to the transposition machinery. Here we investigate the nucleolytic activity of L1 EN in the context of an environment which may contribute to chromosomal accessibility in vivo. We find that nucleosomal wrapping of DNA renders it a less efficiently nicked substrate, but when so wrapped some phosphodiesters at specific positions in the nucleosome are nicked at an increased rate.
MATERIALS AND METHODS
Protein and chromatin preparation
L1 endonuclease was prepared exactly as in Cost and Boeke (7). Briefly, the first 239 amino acids of L1.2 ORF2 were C-terminally fused to six consecutive histidine residues and purified by affinity and gel filtration chromatography. Nucleosome core particles were prepared with the same DNA sequences and in exactly the same manner as in Golding et al. (9). Briefly, a 209 bp portion of the Vk24 locus was amplified by PCR using 32P-labeled VkS (5′-tctcagaccggtttagtggcagtgggtcaggaac-3′) and Vk24PBSK (5′-attgggtaccgggccccccctcgaggtcg-3′) to create substrate 1. Substrate 2 was created by PCR amplification of 209 bp of the VkL8 locus with JH200-3 (5′-aacaatttcacacaggaaacagc-3′) and 32P-labeled JH200-12 (5′-aagttgctgcgattctcaccaat-3′). Chromatinized substrates were created by salt dialysis exchange of nucleosomes from chicken core particles, followed by sucrose gradient purification. Hydroxyl radical footprinting was done as in Golding et al. (9). A Molecular Dynamics PhosphorImager was used to collect the data and ImageQuant v.1.1 software was used for the purposes of quantitation; the scan is in the linear range. Scans with different exposure lengths were assembled with Adobe Photoshop 5.0.
Nucleosomal nicking reactions
Nicking reactions were performed in 60 mM NaCl, 50 mM HEPES pH 7.5, 5 mM MgCl2 and ∼4% sucrose, with 1.75 µM L1 EN (and 2-fold dilutions thereof). Reactions proceeded for 0.5 h at 37°C, after which each was made 0.4 mg/ml proteinase K, 0.1% SDS and 50 mM EDTA and incubated at 55°C for 45 min. Reactions were then phenol/chloroform/isoamyl alcohol extracted and ethanol precipitated with glycogen as carrier, resuspended in 10 mM Tris pH 7.6, 1 mM EDTA, 50% formamide and electrophoresed through 8% acrylamide–8 M urea gels. Plasmid nicking reactions were performed identically but with the addition of 200 ng Bluescript DNA.
RESULTS
The accessibility of the eukaryotic genome to nuclear factors is regulated at several structural levels, the most fundamental being the presence or absence of a bound histone octamer. Nucleosomal incorporation of DNA can either prevent or facilitate access of DNA-binding proteins to the chromosome (10–12). To investigate the effect of chromatinization on the ability of L1 EN to nick DNA, several fragments of the Vk locus whose chromatin structure had been previously studied (9) were screened for the presence of L1 EN nicking sites. Two different 209 bp fragments of the immunoglobulin Vk locus which were nicked well by L1 EN were incorporated into nucleosomal monomers and challenged with varying amounts of L1 EN. Compared to free DNA, nicking of the histone-bound DNA was generally and substantially repressed (Fig. 2A and B). This repression was not limited to DNA in direct contact with the histone octamer, as nicking in the linker regions of both substrates was similarly repressed (e.g. nick 4, Fig. 2B). In contrast, several sites became more susceptible than free DNA to L1 EN when nucleosomal.
Figure 2.

L1 endonuclease nicking is repressed by chromatin. (A) Nicking on free and nucleosomal DNA. The boundary of the nucleosome is indicated by the bars to the right of the gels. Sites repressed when nucleosomal are numbered with black arrowheads; nucleosome-specific enhancements are annotated with letters and grey arrowheads. –, no L1 EN; G, guanosine-specific Maxam–Gilbert sequencing ladder; R, cleavage of the nucleosomal substrate with hydroxyl radicals. (B) As (A) but with substrate 2.
The DNA sequences of the sites nicked are shown in Figure 3. Sites which are repressed when chromatinized (#1–8) conform reasonably well to the L1 EN consensus. All repressed sites but one (#8) have a thymine at the critical –4 position relative to the nick. Sites which are enhanced when incorporated into nucleosomes do not appear to follow the L1 EN consensus. The magnitudes of the repression and enhancement at these sequences are quantitated in Figure 3. In general, the levels of nicking inhibition fall within a 5-fold range, with no apparent correlation between the magnitude of repression and the position of the DNA in the nucleosome. The range of values for enhancement is far greater. As quantitation of both enhanced and repressed sites was performed on lanes digested with identical amounts of L1 EN and only the 5′-most nick is detectable, relative over-digestion in the free DNA lanes results in nucleosome-mediated repression being somewhat underestimated and nucleosome-mediated enhancement conversely overestimated.
Figure 3.

Sites and quantitation of L1 EN nicking repression and enhancement. Listed are the sequences of the major L1 EN nicking sites from Figure 2A and B. Upper black arrowheads, nicked bond; lower grey arrowheads, nucleosome-specific enhancements. Bands from the lane containing the third EN concentration were used for quantitation. Background intensities at the appropriate position in lanes lacking EN were subtracted from the signal prior to comparison. When the background was equal to or greater than the signal the fold repression or enhancement was calculated without background subtraction and listed with a preceding >. The general consensus sequence for L1 EN nicking is TTTTAA, with nicking occurring at the TpA bond.
To reveal more fully the effect of chromatinization on L1 EN nicking at molecular resolution, traces of the nicking pattern of both free and nucleosome-incorporated DNA (from Fig. 2A) were aligned with the DNA sequence and hydroxyl radical footprint (Fig. 4). Two observations can be made: (i) most sites of repression occur at positions where the minor groove is maximally solvent exposed; (ii) sites of enhancement are always at or near minor groove exposure minima. The nicking and hydroxyl radical patterns of the substrate in Figure 2B were identically analyzed and yielded similar results (data not shown).
Figure 4.

Densitometric traces of hydroxyl radical and L1 EN nicking. Upper black arrowheads, sites of nicking repression; lower grey arrowheads, sites of nicking enhancement; light grey lines, divisions of 10 bp periodicity. The traces are from Figure 2A and correspond to dilution 3 (Free) and dilution 5 (Nucleosomal). These lanes were chosen for maximal clarity of presentation and are not intended to be quantitatively accurate.
To ensure that an unknown factor in the chromatin preparation was not inhibiting the nicking reaction in trans, the chromatinized fragment was mixed with naked supercoiled Bluescript DNA and incubated with L1 EN (Fig. 5). The plasmid was nicked as well in the presence of the chromatin-containing sucrose gradient fraction as in its absence. As the repression observed with the bound DNA occurs only in cis, we conclude that nucleosome-incorporated DNA is generally, though not absolutely, refractory to nicking by L1 EN.
Figure 5.
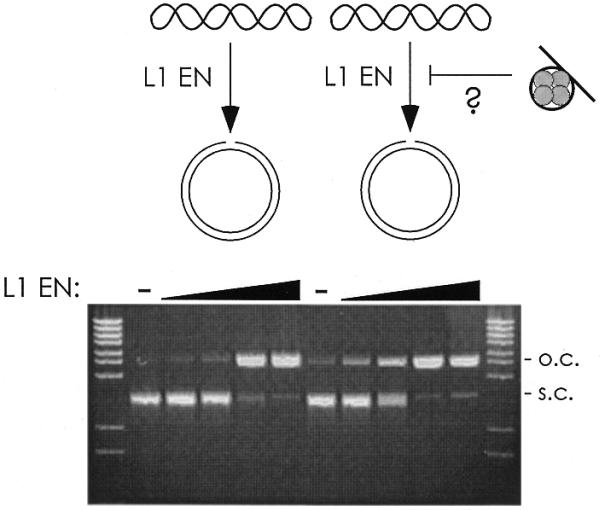
Nucleosome-mediated repression of L1 EN occurs only in cis. (A) Naked supercoiled Bluescript DNA was mixed with the chromatinized DNA and assayed for nicking. Lanes 2 and 7, no L1 EN; lanes 3–6 and 8–11, increasing 2-fold concentrations of L1 EN. s.c., supercoiled; o.c., open circle. DNA from the chromatin fragment is not visible as it has been electrophoresed off the gel in order to resolve the relatively large plasmids.
DISCUSSION
We have shown that chromatinization of DNA interferes with nicking by L1 EN. Akin to DNase I, L1 EN is unlikely to require access to both sides of the DNA helix to create a single nick (13). A simple inability to encircle its substrate is therefore unlikely to account for the observed inhibition. The general inability of L1 EN to nick nucleosomal DNA may result from the substantial distortion induced in the DNA helix by the histone octamer, but is also likely to result from repeated occlusion of the outward-facing minor groove by the tails of histones H2A and H2B as well as simple steric exclusion by the extended tails of histones H3 and H4 (14).
We observed repression of nicking activity on DNA very close to but not in direct contact with the histone octamer (e.g. nick 4, Fig. 2B). As this DNA is unlikely to be structurally perturbed, we suspect that the mechanism of this inhibition may be simple steric blockage of L1 EN. The translational position of this piece of DNA in the nucleosome has been mapped in detail (9). While the large majority of the nucleosomes have their DNA positioned as indicated by the black bar in Figure 2B, in a small subpopulation the histones are closer to the 5′-end of the DNA. This translational heterogeneity or similar heterogeneity present at the reaction temperature (15) may account for some portion of the observed repression. Nicking in extended full-length linker regions may be unaffected.
Our results stand in marked contrast to those observed with the retroviral integrases. Retroviral DNA integration using oligonucleotide viral end substrates is stimulated by the presence of histones on the target DNA, with retroviral integration occurring with 10–11 bp periodicity at major groove phosphodiester bonds on the non-nucleosomal faces of the DNA helix (10,12,16,17). L1 EN nicking, even at preferred sites with an exposed minor groove, is largely repressed when the DNA is nucleosomal. In this respect L1 EN activity behaves in a manner dissimilar to DNase I activity which, although repressed by chromatin along histone-protected lengths of the minor groove, is often unaffected or even stimulated by the incorporation of DNA into nucleosomes (16). Additionally, it is interesting to note that cleavage by the VDJ recombinase, consisting of RAG1 and RAG2, is also blocked by chromatinization, as these proteins can also function as a transposase (9,18).
The observed pattern of repression and enhancements may seem surprising at first, as repression was often found at minor groove maxima and enhancement at the minima. We believe that these observations can be nicely explained by the distorted geometry of DNA when in contact with a nucleosome and principally by the likely separate and asymmetric location of the L1 EN catalytic site relative to its DNA-binding loops. Nicking by L1 EN requires extensive interaction with four or more bases 5′ of the nicked bond. The nucleotide residues 3′ of the nick contribute little to the nicking specificity (7). As most of the L1 EN nicking sites on free DNA map to positions of minor groove solvent exposure when incorporated into nucleosomes, cleavage by L1 EN would require binding and recognition of DNA 4–5 bp 5′ (at the minor groove minima, the place of contact with the histones) (Fig. 6B). Conversely, recognition of the DNA 5′ of sites of enhanced cleavage is unimpeded by histone contacts. Envisioning L1 EN catalytic site access to the nicked bond is more difficult, but may depend on the specific shape of L1 EN, distortion of DNA on the nucleosome surface induced by L1 EN or perturbation of the nucleosome phasing prompted by L1 EN binding (Fig. 6C).
Figure 6.
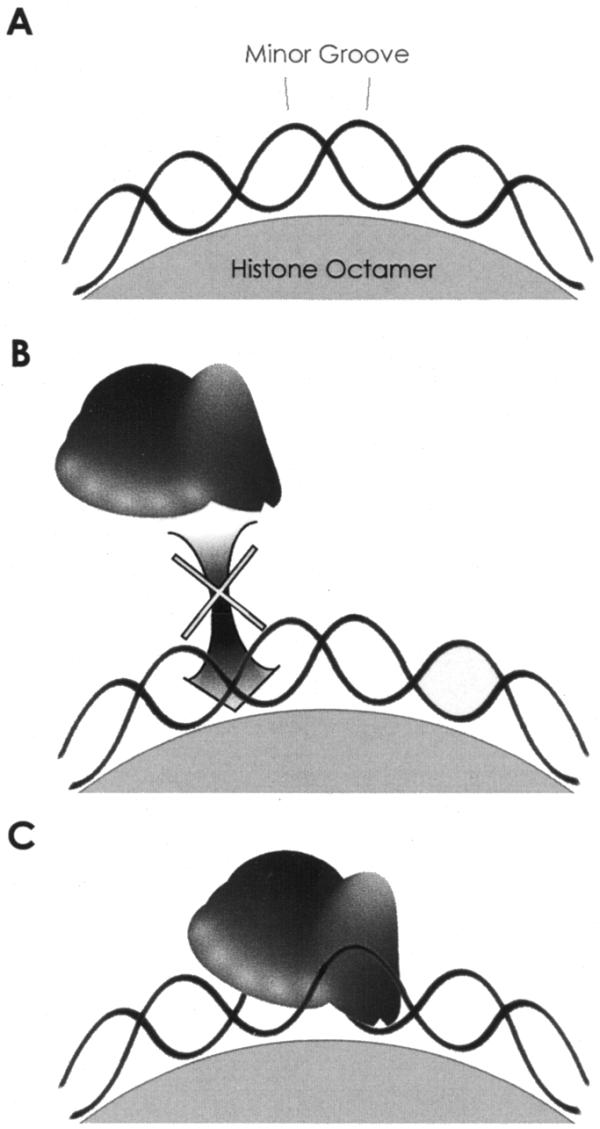
A model for L1 EN nicking of nucleosomal DNA. (A) DNA wrapped around the histone octamer. (B) Repression of L1 EN. Nicking at the center exposed minor groove requires extensive minor groove recognition up to 4–5 bp 5′, an interaction which is blocked by the histone octamer. (C) Enhancement of nicking. Recognition of the minor groove is unimpeded by the octamer. Either the active site of L1 EN is flexible enough to access the relatively protected cleaved phosphodiester or L1 EN binding initiates a change in the rotational position of the DNA within the nucleosome (not shown).
It is unclear what level of nicking activity is required by L1 for transposon insertion in vivo. The resistance of chromatin to L1 integration is not likely to be absolute, as nucleosome-mediated inhibition is overcome at high enzyme concentrations. The specificity of L1 EN on naked DNA largely mirrors the specificity of in vivo L1 transposon insertions, but a subset of L1 integration sites deviate from the consensus (5,7,8). Given the nucleosome-specific nicking detected here, we suggest that some of these apparently anomalous insertions may result from nicking at non-consensus sites distorted on the nucleosome face into a structural context favorable for nicking.
We have previously suggested that L1 EN, like DNase I, may be useful in investigating questions regarding DNA and/or chromatin structure (7). On free DNA L1 EN nicks at kinkable regions of DNA present between regions of very stiff DNA structure. While it may be coincidental that positions of L1 EN nicking and minor groove maxima correlate (Fig. 4), it may be the case that the DNA structural features recognized by L1 EN are the same as those sensed by nucleosomes when searching for the rotational position of lowest free energy. As the curvature of DNA on the nucleosome is discontinuous (14), deformable positions may be favored for particularly large bending and therefore rotation to the outside of the nucleosome particle. It will be interesting to see whether nicking by L1 EN proves generally predictive of this phasing.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Nurjana Bachman and all members of the Boeke laboratory for helpful discussions and an anonymous reviewer of this paper for helpful criticism. This work was supported in part by NIH training grant 5T32CA09139-24 (G.J.C.), NIH grant CA16519 (J.D.B.) and NIH grant AI40227 (A.G. and M.S.S.).
References
- 1.Kazazian H.H. and Moran,J.V. (1998) The impact of L1 retrotransposons on the human genome. Nature Genet., 19, 19–24. [DOI] [PubMed] [Google Scholar]
- 2.Moran J.V., Holmes,S.E., Naas,T.P., DeBerardinis,R.J., Boeke,J.D. and Kazazian,H.H.,Jr (1996) High frequency retrotransposition in cultured mammalian cells. Cell, 87, 917–927. [DOI] [PubMed] [Google Scholar]
- 3.Hohjoh H. and Singer,M.F. (1996) Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J., 15, 630–639. [PMC free article] [PubMed] [Google Scholar]
- 4.Mathias S.L., Scott,A.F., Kazazian,H.H.,Jr, Boeke,J.D. and Gabriel,A. (1991) Reverse transcriptase encoded by a human retrotransposon. Science, 254, 1808–1810. [DOI] [PubMed] [Google Scholar]
- 5.Feng Q., Moran,J., Kazazian,H. and Boeke,J.D. (1996) Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell, 87, 905–916. [DOI] [PubMed] [Google Scholar]
- 6.Luan D.D., Korman,M.H., Jakubczak,J.L. and Eickbush,T.H. (1993) Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell, 72, 595–605. [DOI] [PubMed] [Google Scholar]
- 7.Cost G.J. and Boeke,J.D. (1998) Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry, 37, 18081–18093. [DOI] [PubMed] [Google Scholar]
- 8.Jurka J. (1997) Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl Acad. Sci. USA, 94, 1872–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golding A., Chandler,S., Ballestar,E., Wolffe,A.P. and Schlissel,M.S. (1999) Nucleosome structure completely inhibits in vitro cleavage by the V(D)J recombinase. EMBO J., 18, 3712–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pryciak P.M. and Varmus,H.E. (1992) Nucleosomes, DNA-binding proteins and DNA sequence modulate retroviral integration target site selection. Cell, 69, 769–780. [DOI] [PubMed] [Google Scholar]
- 11.Owen-Hughes T. and Workman,J.L. (1994) Experimental analysis of chromatin function in transcription control. Crit. Rev. Eukaryot. Gene Expr., 4, 403–441. [PubMed] [Google Scholar]
- 12.Pruss D., Bushman,F.D. and Wolffe,A.P. (1994) Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core. Proc. Natl Acad. Sci. USA, 91, 5913–5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sück D., Lahm,A. and Oefner,C. (1988) Structure refined to 2Å of a nicked DNA octanucleotide complex with DNase I. Nature, 332, 464–468. [DOI] [PubMed] [Google Scholar]
- 14.Luger K., Mader,A.W., Richmond,R.K., Sargent,D.F. and Richmond,T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 15.Godde J.S. and Wolffe,A.P. (1995) Disruption of reconstituted nucleosomes. The effect of particle concentration, MgCl2 and KCl concentration, the histone tails and temperature. J. Biol. Chem., 270, 27399–27402. [DOI] [PubMed] [Google Scholar]
- 16.Pruss D., Reeves,R., Bushman,F.D. and Wolffe,A.P. (1994) The influence of DNA and nucleosome structure on integration events directed by HIV integrase. J. Biol. Chem., 269, 25031–25041. [PubMed] [Google Scholar]
- 17.Pryciak P.M., Sil,A. and Varmus,H.E. (1992) Retroviral integration into minichromosomes in vitro. EMBO J., 11, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiom K., Melek,M. and Gellert,M. (1998) DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell, 94, 463–470. [DOI] [PubMed] [Google Scholar]