

Abstract
A new method for the enantioselective synthesis of hexahydro-1H-benz[f]indoles is described. This copper-catalyzed enantioselective intramolecular alkene carboamination process can install vicinal tertiary and quaternary carbon stereocenters with high levels of diastereo- and enantioselectivity. The C-C bond-forming component of the reaction constitutes a C-H functionalization and no electronic activation of the aryl ring that undergoes addition is required. A known 5-HT1A receptor antagonist was synthesized efficiently using this method.
The intramolecular carboamination of alkenes is an attractive method for the synthesis of nitrogen heterocycles.1 This reaction has benefited particularly from methods involving palladium, copper and gold catalysis.2,3 In recent years, the catalytic asymmetric carboamination has been actively pursued. One approach involves the doubly intramolecular enantioselective carboamination wherein intramolecular alkene amination is followed by addition of the resulting reactive carbon intermediate to a π-bond tethered through the N-substituent. In this manner, Pd(II)2c and Cu(II)3a complexes have catalyzed formation of chiral bicyclic lactams and sultams, respectively.
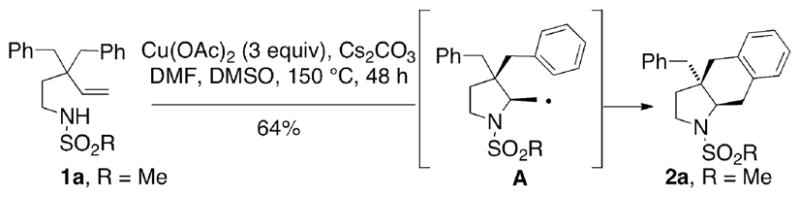
We desired to apply the copper-catalyzed carboamination reaction to the synthesis of other nitrogen heterocycles and believed that the proposed carbon radical intermediate,3c e.g., A (Scheme 1) could add to other nearby π-bonds.
Scheme 1.
Benz[f]indole via Carboamination Sequence
This reaction could in principle be (1) chemoselective, choosing radical addition either to an N-arylsulfonyl ring (as previously reported)3a or an aromatic ring at the allylic position (as in Scheme 1), (2) diastereoselective, choosing between formation of either a cis or trans ring fusion, and (3) enantioselective, catalyzed by a chiral copper(II)•ligand complex. The desired carboamination product 2 constitutes an otherwise difficult to access hexahydro-1H-benz[f]indole ring system and contains vicinal quaternary and tertiary carbon stereocenters. Some benz[f]indoles have demonstrated biological activity as dopamine antagonists4 and as anti-cancer agents.5 Surprisingly few methods for the synthesis of hexahydro-1H-benz[f]indoles have been reported.
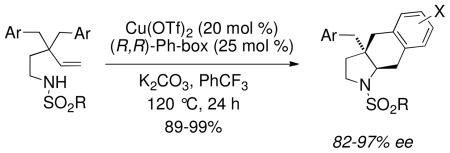
Our initial forays into the copper-promoted carboamination of N-mesyl substrate 1a were promising. Using 300 mol % of Cu(OAc)2, the cis-fused hexahydrobenz[f]indole 2a was obtained in 64% yield with >20:1 diastereoselectivity (Scheme 1).6 Encouraged by this result, we submitted 1a to catalytic, enantioselective conditions.3a To our delight, we obtained a 99% yield and 82% ee, with >20:1 diastereoselectivity using 20 mol % Cu(OTf)2, 25 mol % (R, R)-Ph-box, 300 mol % MnO2 at 120 °C in PhCF3 for 24 h (Figure 1, 2a).
Figure 1.

aEnantioselective Carboamination Scope. a20 mol % Cu(OTf)2 and 25 mol % (R,R)-Ph-Box were combined in PhCF3 (0.1 M w/r to 1) and heated at 60 °C for 2 h in a pressure tube, then 1, K2CO3 (100 mol %), MnO2 (300 mol %) were added and the reaction was heated at 120 °C for 24 h. Yield refers to product isolated from flash chromatography on SiO2. Enantioselectivity (%ee) was determined by chiral HPLC. bReaction run at 110 °C. cYield is for combined regioisomeric mixture, dr and %ee were the same for both isomers. SES = trimethylsilylethlysulfonyl, Bs = benzenesulfonyl, PMBS = 4-methoxybenzenesulfonyl, PCBS = 4-chlorobenzenesulfonyl, Ns = 4-nitrobenzenesulfonyl.
The trimethylsilylethylsulfonyl substrate 1b (R = SES) underwent the reaction with equal efficiency (Figure 1, 2b). Various arylsulfonyl substrates 1c-1g (R = Bs, Ts, PMBS, PCBS, Ns) provided hexahydro-1H-benz[f]indole adducts 2c-2g with even higher enantioselectivity (94–97% ee) and no trace of the sultam3a regioisomer. Substrates 1h-j with para aryl ring substitution (X = F, SMe, OMe) provided uniformly high yields and enantioselectivities. The meta-MeO substrate 3a gave the benz[f]indoles 4 as a 1.5:1 mixture of ortho and para regioisomers (with respect to aryl addition).3c
Interestingly, substrates with ortho substitution, 5a and 5b, gave regioisomers 6 and 7 where 7 is the result of a rearrangement where an aryl substituent has apparently shifted to the meta-position. This can be explained by a mechanism involving ipso-addition3d,7 followed by 1,2-alkyl shift (Scheme 2). A similar rearrangement occurred with the p-CF3-substituted substrate 1k, which gave a 3:1 mixture of products 2k and 8, the rearrangement product.
Scheme 2.

Addition and Rearrangement
Based upon these examples, it appears the propensity to undergo ipso rather than direct ortho substitution may be influenced by steric (in the case of ortho substituted) and electronic (in the case of the 4-CF3 substituent) factors. No regioisomers were observed in products 2a-j and direct substitution without going through an ipso intermediate is inferred for these compounds. Ortho substitution causes a decrease in available ortho additon sites while the the highly electron-withdrawing 4-CF3 group may influence the relative size of the orbital coefficients at the carbons that undergo addition of the electron-rich primary radical.
Transition state B and intermediate A can be used to rationalize the enantio- and diastereoselectivity of the reaction (Scheme 3). This model of enantioinduction is consistent with our other copper-catalyzed reactions where the N-substituent minimizes interaction with the closest bis(oxazoline) substituent.3a,8 The diastereoselectivity arises from the carbon radical of A adding to the aryl ring it is cis to. The structures of 2a, 6a, 7a and 8 were assigned by X-ray crystallography. The absolute and relative stereochemistry of the other adducts in Table 2 were assigned by NOE and by analogy (see Supporting Information).
Scheme 3.

Transition State Model for Enantioselectivity
We also explored the catalytic, diastereoselective carboamination reactions of the mono-benzylated alkenyl sulfonamides 9 (Scheme 4). It was necessary to use the N-mesyl derivatives of these substrates as the N-tosyl group competes successfully for addition of the carbon radical intermediate in these cases. The N-mesyl substrates 9a and 9b provided the trans-fused carboamination products 10 as the major diastereomers. Transition state C, which places the benzyl substituent in a pseudo-equatorial position, rationalizes the formation of the major diastereomers. It is interesting to note that the ortho-substituted 9b does not form products resulting from ipso substitution and rearrangement. This is likely because the formation of a trans-fused five-membered ring intermediate involved in this ipso substitution is more difficult to form than the transfused six-membered ring that would result from direct ortho addition that leads to the observed product. In the case of 5, a more favorable cis-fused spirocyclic intermediate (Scheme 2) can be formed.
Scheme 4.

Diastereoselective Carboaminations
Conversion of ortho-methoxy adduct 10b to the known 5-HT1A receptor antagonist4 11 was accomplished by removal of the mesyl group with Red-Al followed by N-alkylation (63%, two steps). Our synthesis of (±)−11 is 8 steps from γ-butyrolactone (24% overall yield) an improvement over its previous synthesis.4
In conclusion, this intramolecular alkene carboamination provides hexahydro-1H-benz[f]indoles in a concise and enantioselective manner. New vicinal quaternary and tertiary stereocenters can be formed in this reaction. The aromatic rings that undergo addition do not require any specific activating functionality and the C-C bond-forming step constitutes a C-H functionalization.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIGMS R01 078383) for support of this work and William W. Brennessel and the X-ray Crystallographic Facility at the University of Rochester for the X-ray structures of 2a, 6a, 7a and 8 and Dr. Cara L. Nygren and Dr. Stephan Scheins (SUNY-AB) for the X-ray structures of 10a and 10b.
Footnotes
Supporting Information Available Full experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews: Wolfe JP. Eur J Org Chem. 2007:571.Wolfe JP. Synlett. 2008:2913.Chemler SR. Org & Biomol Chem. 2009;7:3009. doi: 10.1039/B907743J.
- 2.Recent palladium-catalyzed carboaminations/carbonylations: Tsujihara T, Shinohara T, Takenaka K, Takizawa S, Onitsuka K, Hatanaka M, Sasai H. J Org Chem. 2009;74:9274. doi: 10.1021/jo901778a.Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157. doi: 10.1021/ja106989h.He W, Yip KT, Zhu NY, Yang D. Org Lett. 2009;11:5626. doi: 10.1021/ol902348t.Scarborough CC, Stahl SS. Org Lett. 2006;8:3251. doi: 10.1021/ol061057e.Houlden CE, Bailey CD, Ford JG, Gagne MR, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2008;130:10066. doi: 10.1021/ja803397y.Scarborough CC, Bergant A, Sazama GT, Guzei IA, Spencer LC, Stahl SS. Tetrahedron. 2009;65:5084. doi: 10.1016/j.tet.2009.04.072.Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J Am Chem Soc. 2009;131:9488. doi: 10.1021/ja9031659.A gold-catalyzed carboamination: Zhang G, Cui L, Wang Y, Zhang L. J Am Chem Soc. 2010;132:1474. doi: 10.1021/ja909555d.
- 3.Copper-catalyzed carboaminations: Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948. doi: 10.1021/ja0762240.Sherman ES, Chemler SR. Adv Synth & Catal. 2009;351:467. doi: 10.1002/adsc.200800705.Cu(II)-promoted carboaminations: Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896. doi: 10.1021/jo070321u.Fuller PH, Chemler SR. Org Lett. 2007;9:5477. doi: 10.1021/ol702401w.
- 4.Lin CH, Haadsma-Svensson SR, Phillips G, Lahti RA, McCall RB, Piercey MF, Schreur PJKD, Von Voigtlander PF, Smith MW, Chidester CG. J Med Chem. 1993;36:1069. doi: 10.1021/jm00060a015. [DOI] [PubMed] [Google Scholar]
- 5.Park HJ, Lee HJ, Min HY, Chung HJ, Suh ME, Park-Choo HY, Kim C, Kim HJ, Seo EK, Lee SK. Eur J Pharm. 2005;527:31. doi: 10.1016/j.ejphar.2005.10.009. and references therein. [DOI] [PubMed] [Google Scholar]
- 6.Manzoni MR. PhD Thesis. The University; Buffalo, SUNY: 2006. [Google Scholar]
- 7.Gonzales-Lopez de Turiso F, Curran DP. Org Lett. 2005;7:151. doi: 10.1021/ol0477226. [DOI] [PubMed] [Google Scholar]
- 8.Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.