

Abstract
Pancreatic cancer is a deadly disease that is virtually never cured. Understanding the chemoresistance intrinsic to this cancer will aid in developing new regimens. High expression of APE1/Ref-1, a DNA repair and redox signaling protein, is associated with resistance, poor outcome, and angiogenesis; little is known in pancreatic cancer. Immunostaining of adenocarcinoma shows greater APE1/Ref-1 expression than in normal pancreas tissue. A decrease in APE1/Ref-1 protein levels results in pancreatic cancer cell growth inhibition, increased apoptosis, and altered cell cycle progression. Endogenous cell cycle inhibitors increase when APE1/Ref-1 is reduced, demonstrating its importance to proliferation and growth of pancreatic cancer.
Keywords: Pancreatic cancer, APE1/Ref-1, siRNA, cell cycle
Introduction
Pancreatic cancer is a particularly egregious form of cancer with the worst 5-year survival rate of any cancer: <2% (1). Pancreatic cancers are hypoxic tumors that respond poorly to existing chemotherapeutic agents and radiation (2), underscoring the need to understand this type of cancer and develop new strategies to combat it.
Recently, reduction-oxidation (redox) signaling has been identified as an important system to study in pancreatic cancer (2, 3). We have been studying a DNA repair protein, apurinic/apyrimidinic endonuclease (APE1/Ref-1), that has a second major function in human tumors as a redox signaling protein (and goes by the alternate name of Ref-1). APE1/Ref-1 interacts with many transcription factors (HIF1-α, p53, AP1, NFκB CREB, and more) to facilitate their DNA binding via reduction of a cysteine residue (reviewed in Refs. 4, 5). In pancreatic cancer, several of the key transcription factors in controlling angiogenesis and growth such asNFκB, AP1, HIF-1, and STAT3 interact and/or are controlled by APE1/Ref-1's redox activity (5–7). The redox function of APE1/Ref-1 is emerging as having an important role in the tumor microenvironment, including pancreatic cancer cell growth, migration, and angiogenesis (3, 8, 9). In addition to its activity as a redox coactivator of transcription factors, the DNA repair function of APE1/Ref-1 is critical to genomic maintenance. Another well-demonstrated function of APE1/Ref-1 is the transcription cofactor activity, which is separate from its redox activity and has been shown to be essential for cell viability in mouse embryonic fibroblasts (MEFs) (10).
Many types of tumor cells demonstrate upregulation of APE1/Ref-1 protein levels (4) and then become hypersensitive to chemotherapeutic agents (especially DNA damaging agents) when APE1/Ref-1 protein levels are reduced (11–14). Treatment with gemcitabine, the foremost chemotherapeutic agent used in pancreatic cancer, increases protein and activity levels of APE1/Ref-1 in two human pancreatic cancer cell lines: Panc-1 and MiaPaCa-2. Upon treatment of Panc-1 cells with antisense oligonucleotides to APE1/Ref-1, Lau et al. observed a dramatic increase in sensitivity to gemcitabine (15). Furthermore, NFκB is known to be under redox control by APE1/Ref-1, and NFκB activity is a determinant of pancreatic cancer cells' response to gemcitabine. All these findings point to an important role of APE1/Ref-1 in pancreatic cancer pathophysiology.
A reduction of APE1/Ref-1 via siRNA in pancreatic tumor cells decreases the APE1/Ref-1 levels in both the mitochondria and nucleus. Similar to that seen in other cancer cell types, knocking down APE1/Ref-1 decreases pancreatic tumor cells' growth rate and colony-forming ability, and increases the percentage of cells remaining in G2/M and undergoing apoptosis.
Here we present new data characterizing the effect that reducing APE1/Ref-1 protein levels has on pancreatic cancer cell growth and cell cycle arrest. First we show that APE1/Ref-1 protein levels are elevated in patient samples of pancreatic adenocarcinoma and in peri-pancreatic metastases compared to normal pancreas cells. Second, APE1/Ref-1 siRNA not only reduces the amount of APE1/Ref-1 protein in the nuclear compartment but also in the mitochondria. Third, following synchronization, pancreatic cancer cells transfected with APE1/Ref-1 siRNA have delayed reentry into the cell cycle. This cell cycle arrest can be partially explained due to an increase in DNA damage as we observe an upregulation of H2AX-γ. However, reduced levels of APE1/Ref-1 did not increase ROS generation in Panc-1 or PaCa-2 cells. Finally, we molecularly characterized the cell cycle arrest by quantitating the changes of important proteins that regulate cell cycle. A decrease in APE1/Ref-1 levels causes increases in p21 and the inactive form of cdc-2, confirming cell cycle arrest. However, we observe no change in p27 expression. These data confirm that APE1/Ref-1 is important for the growth and proliferation of pancreatic cancer cells and that inhibitors of APE1/Ref-1 have therapeutic implications in the treatment of pancreatic cancer.
Materials and Methods
Tissue specimens and preparation
Pancreatic tissues were obtained from the Indiana University Simon Cancer Center Tissue Bank. All human tissue samples were collected according to a detailed IRB-approved protocol, informed patient consent, and HIPAA compliance protocol. Tissues were fixed overnight at room temperature in 10% NBF (neutral buffered formalin), then transferred through graded concentrations of alcohol to xylene inside a tissue processor. Tissues were embedded in paraffin before being microtomed into five-micron sections, mounted on positively charged slides and baked at 60°C.
Immunostaining
Paraffin-embedded tissue sections were deparaffinized in running water. The tissues that were fixed in 10% NBF underwent antigen retrieval. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide (Sigma-Aldrich; St. Louis, MO, USA), and antigen retrieval was performed using DAKO Target Retrieval (citrate buffer pH 6.0). Immunostaining was performed using an APE1/Ref-1 antibody with a DAKO LSAB2 kit on a DAKO-automated immunostainer. Visual detection was performed using biotinylated secondary antibody (LSAB2), streptavidin-horseradish peroxidase complex, and DAB chromogen (DAKO). The slides were counterstained with hematoxylin, dehydrated through graded alcohols to xylene, then coverslipped with a permanent mounting medium. Control sections were treated with isotype control at the same concentration as primary antibody to verify the staining specificity. To assess APE1/Ref-1 staining, four independent pathologists evaluated the intensity and localization of the staining. Slides were scored as follows: percentage of cells staining, intensity of IHC stain (referred to as SI) (negative = 0, borderline/minimal = 1, moderate = 2, strong = 3), localization of stain in the cell (none, nuclear, cytoplasm, combination of cytoplasm/nuclear), and tumor distribution of the stain (homogenous, heterogeneous, focal, multifocal, and/or variable). p Values were calculated using Student's t-test.
Cell lines
Panc-1 and PaCa-2 were purchased from and authenticated by ATCC (Manassas, VA, USA). All cell lines were maintained at 37°C in 5% CO2 and grown in DMEM media (Invitrogen; Carlsbad, CA, USA) supplemented with 10% Cosmic Calf Serum (Hyclone; Logan, UT, USA) plus an antibiotic/antimycotic (Invitrogen).
Drugs
O6-benzylguaninewas purchased from Sigma Aldrich, dissolved in 100% DMSO, and stored as a 100-mM stock at −20°C.
Transfection of Panc-1 and PaCa-2 cells with APE1/Ref-1 and scrambled siRNA
Sequences of the double-stranded siRNAs are scrambled (5′ CCAUGAGGUCAGCAUGGUCUG 3′, 5′ GACCAUGCUGACCUCAUGGAA 3′) and APE1/Ref-1 (5′ GUCUG-GUACGACUGGAGU ACC 3′, 5′ UACUCCAGUCGUACCAGACCU 3′) as previously described (16–18). The 21-base sequence was subjected to a BLAST-search (NCBI) database of EST libraries to ensure that only one gene was targeted. Panc-1 and PaCa-2 cells were plated (1 × 104 and 8 × 103 cells/cm2, respectively) and allowed to attach overnight. The next day, oligofectamine reagent (Invitrogen) was used to transfect in the siRNA, following the manufacturer's protocol. Opti-MEM, siRNA, and oligofectamine were left on the cells for at least 6 hr; then regular DMEM media was added.
Western blot analysis
Cells were harvested, lysed in RIPA buffer (Santa Cruz Biotechnology; Santa Cruz, CA, USA), and protein was quantified and electrophoresed on a 12% SDS-polyacrylamide gel. The following antibodies were used at a dilution of 1:1,000: APE1/Ref-1 (Novus Biologicals; Littleton, CO, USA), Brahma-related gene 1(Brg-1), and mitochondrial cytochrome C oxidase II (mtCOXII) (Santa Cruz Biotechnology), PARP-1 (Cell Signaling Technology Inc.; Danvers, MA, USA), tubulin, and actin (Sigma Aldrich), p21 and p27 (Santa Cruz Biotechnology + 2% BSA), cdc-2 and phospho-cdc-2 (Cell Signaling Technology), and H2AX-γ (Upstate, Waltham, MD). Monoclonal APE/Ref-1 antibodies were produced in the Kelley laboratory and have been extensively used and characterized (19–23).
MTS assay for proliferation
Two thousand to 4,000 cells per 96-well plate were allowed to adhere overnight. Following transfection, MTS reagent was added, followed by optical density measurement at 490 nm. The values were standardized to wells containing media alone. To generate the doubling times, the curves in Figure 3(A) were fit using exponential regression (Microsoft Excel), and the R2 values were calculated. The curves for Panc-1 Scrambled, Panc-1 SiAPE1/Ref-1, PaCa-2 Scrambled, and PaCa-2 SiAPE1/Ref-1 had the following R2 values: 0.996, 0.978, 0.990, and 0.769, respectively.
Figure 3.
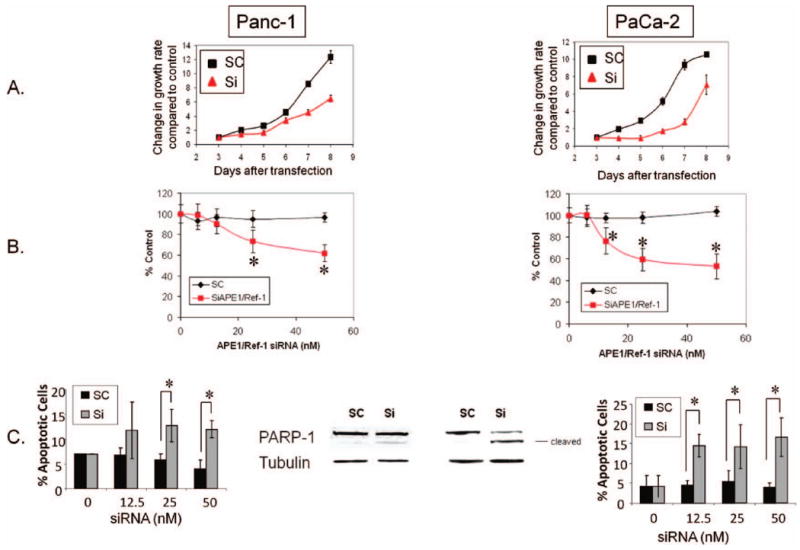
A reduction in APE1/Ref-1 protein reduces the growth rate and colony-forming ability of pancreatic cancer cell lines and increases the amount of apoptosis. (A) Growth rate of cells after exposure to 50 nM APE1/Ref-1 siRNA. (B) Colony-formation assays with each data point representing the mean ± SD from at least three separate experiments, with each experiment representing six replicate dishes per treatment. Solid squares depict siAPE/Ref-1-treated pancreatic cancer cells. *p < .005 using Student's t-test, comparing SC versus SiAPE1/Ref-1 at corresponding dose. (C) Bar graphs show average percentage of Annexin-positive cells (n = 4) ± SE. Middle panel: Representative Western blot probed with PARP-1 antibody on Day 3 after transfection with scrambled (SC) or APE1/Ref-1 siRNA (Si) at 50 nM.
Colony formation assay
To evaluate cell survival after siRNA transfection, a colony formation assay was used as previously described (24). Briefly, exponentially growing cells were plated at varying densities 72 hr after transfection. After approximately 12 days, colonies were stained with methylene blue (0.1% w/v) and scored. Percentage survival was calculated based on the plating efficiency of the scrambled control cells.
Apoptosis assays via Alexa Fluor 488-conjugated Annexin-V (Annexin-V)/Propidium Iodide staining
Cells were plated and transfected as described above, and apoptosis was assayed 72 hr after transfection. Cells were trypsinized, pelleted, washed in ice-cold PBS, and resuspended in 1× Binding Buffer (10 mM Hepes/NaOH pH7.4, 140 mM NaCl, 2.5 mM CaCl2). Apoptosis was analyzed using the Alexa Fluor® 488 Annexin-V Vybrant® Apoptosis Assay Kit in combination with propidium iodide (PI) (Molecular Probes; Eugene, OR, USA) as previously described (18).
Cell cycle staining via BrdU
To stain the cells for DNA content and analyze the movement of cells through G0/G1, S and G2/M, cells were plated, allowed to attach overnight, and transfected as above. On Day 3, cells were processed according to the manufacturer's directions (BD Pharmingen; San Diego, CA, USA) and as previously described (18). For the BrdU staining with synchronized cells, PaCa-2 cells were synchronized with 100 μM O6-benzylguanine (BG) (Sigma Aldrich) for 16 hr prior to adding serum-containing media (24, 25). Although BG inactivates the DNA repair protein alkylguanine alkyltransferase (AGT or MGMT), treatment with this agent is nontoxic in the absence of DNA-damaging agents in Panc-1 and PaCa-2 cells, as well as mice and humans (26, 27). Serum-containing media prompted the cells to reenter the cell cycle, allowing us to monitor the cells movement into S phase and G2.
ROS measurement
Oxidant-sensitive probe dihydrorhodamine 123 (DHR) is cell permeable, and oxidized rhodamine123 is preferentially retained by the mitochondria. ROS oxidize the nonfluorescent DHR to the positively charged fluorescent rhodamine 123 and therefore is an effective probe to measure ROS production (3, 13, 28). The production of ROS was determined by detecting the fluorescent intensity of DHR (Molecular Probes, Invitrogen, Carlsbad, CA, USA). Panc-1 and PaCa-2 cells were transfected with siRNA as described above. As a positive control for ROS production, tert-Butyl hydroperoxide solution (TBHP, 1 mM, 30 min) was utilized. After washing with PBS, the cells were incubated with 1μM DHR in fresh PBS for 30 min. Excessive probe was washed off using PBS. Cells were harvested with trypsin, and ROS fluorescence of labeled cells was measured by using a Coulter EPICS XL flow cytometer (Coulter). An average of 10,000 cells from each sample was counted, and each experiment was done in triplicate.
Results
APE1/Ref-1 protein levels are elevated in pancreatic adenocarcinoma and in peri-pancreatic metastases compared to normal pancreas
We obtained pancreatic cancer patient samples to begin our studies of APE1/Ref-1 protein levels in human pancreatic cancer. A total of 17 pancreatic cases were evaluated for APE1/Ref-1 immunostaining. Normal pancreatic tissue has moderate nuclear staining (SI = 2) in ∼70% of the normal acinar epithelium [Figure 1(A)] (n = 5). Strong nuclear immunostaining (SI = 3) is seen in the tumor cell epithelia in all pancreatic adenocarcinomas cases examined [Figures 1(B) and (C)] (n = 12). Figure 1(C) illustrates the intense nuclear staining in the tumor cells. Likewise, the immunostaining seen in the metastatic tumors is similar to the primary tumor sites, but slightly stronger in intensity [Figure 1(D)]. A statistically higher percentage of adenocarcinoma cells stain positive for APE/Ref-1, as compared to normal pancreas (p < .0001). Elevated levels of APE1/Ref-1 are consistent with our hypothesis that APE1/Ref-1 is involved in the progression and maintenance of pancreatic cancer.
Figure 1.

APE1/Ref-1 levels are elevated in pancreatic adenocarcinoma. (A) Normal pancreatic tissue (20×). (B) Primary pancreatic adenocarcinoma (20×). (C) Primary pancreatic adenocarcinoma (200×). (D) Pancreatic metastasis into the regional lymph node (20×).
siRNA specific to APE1/Ref-1 effectively reduces the protein levels of APE1/Ref-1 in nuclei and mitochondria of human pancreatic cancer cells
To study the effects of APE1/Ref-1 expression on pancreatic cancer cell growth, we utilized siRNA to reduce protein expression. In Figure 2(A), Panc-1 and PaCa-2 human pancreatic cancer cells were treated with concentrations of APE1/Ref-1 siRNA ranging from 12.5 to 100 nM, resulting in a reduction in the amount of APE1/Ref-1 protein by >85% versus scrambled siRNA controls. As the amount of siRNA increases, APE1/Ref-1 protein levels decrease in a dose-dependent manner [Figure 2(A)]. Figure 2(B) shows representative Western blots demonstrating the expression of APE1/Ref-1 on Day 1–7 after transfection in both pancreatic cancer cell lines. Actin is utilized as a protein loading control. APE1/Ref-1 expression begins to return over time [see Figure 2(B), comparing Day 7 to Day 3]. Panc-1 cells demonstrate ∼85% reduction in APE1/Ref-1 protein on Day 3 and a 24% reduction on Day 7. The APE1/Ref-1 protein levels remain diminished longer in the PaCa-2 cells, as an 89% knockdown still exists on Day 3, and APE1/Ref-1 levels are still down by 75% on Day 7. A scrambled (SC) control siRNA is included in all panels to demonstrate the specificity of the APE1/Ref-1 siRNA as previously published (18).
Figure 2.

APE1/Ref-1 knockdown via siRNA results in a dose- and time-dependent decrease in APE1/Ref-1 protein in the mitochondria and nuclei. (A) Western blot of Panc-1 and PaCa-2 cells after transfection with APE1/Ref-1 siRNA. siRNA dose course on Day 3; oligofectamine (OF), scrambled (SC) and APE1/Ref-1 siRNA (Si). (B) Time course with siRNA 25 nM. (C) Representative Western blot probed for APE1/Ref-1 from mitochondria of PaCa-2 cells with quantitation (right). WCE = whole cell extract, Nuc/Cyto = Nuclei + cytoplasm, Mito = purified mitochondria.
APE1/Ref-1 has been found in both the nuclei and mitochondria of cancer cells (4, 29–30). Therefore, we also isolated mitochondria and found that APE1/Ref-1 protein levels are also reduced by 60% in the mitochondria of Panc-1 and PaCa-2 cells [Figure 2(B)]. To confirm this, we purified the mitochondria and probed the blots with mitochondrial-specific protein mtCOXII (mitochondrial cytochrome C oxidase II) (31). As a nuclear loading control, chromatin-associated protein Brg-1 (Brahma-related gene 1) was utilized. As expected, the mitochondrial preps expressed mtCOXII but not Brg-1. It is interesting to note that the size of APE1/Ref-1 is 37 kDa in both the nuclear and mitochondrial fractions. Previous literature reports that the mitochondrial APE1/Ref-1 protein is truncated (30); however, we found full-length APE1/Ref-1 protein in the mitochondria of Panc-1 and PaCa-2 cells. It is possible that the truncated form was below detection in our system.
APE1/Ref-1 siRNA effectively reduces the proliferation and colony-forming ability of human pancreatic cancer cells and increases apoptosis
As previously established in other cancer cell lines (18, 32, 33), APE1/Ref-1 knockdown reduces cell growth and decreases the ability of pancreatic cancer cells to form a colony, as shown in Figures 3(A) and (B). Figure 3(A) also shows that the reduction of APE1/Ref-1 protein in Panc-1 and PaCa-2 cells dramatically slows tumor cell growth rate following transfection. The cell population doubling time for the Panc-1 and PaCa-2 scrambled control cells is 33.4 hr and 30.6 hr, respectively; while the APE1/Ref-1 siRNA-treated cells double every 43.1 and 62.0 hr, respectively. To generate the doubling times, the curves in Figure 3(A) were fit using exponential regression, and the R2 values were all above 0.97 except for the PaCa-2 SiAPE1/Ref-1 treated cells (See Materials and Methods section). Thus, PaCa-2 cells that did not express high levels of APE1/Ref-1 did not enter exponential growth over the 7 days of posttransfection monitoring.
We utilized colony formation assays to assess the pancreatic cancer cell lines' ability to form a colony after knockdown of the APE1/Ref-1 protein. Both Panc-1 and PaCa-2 cells with reduced levels of APE1/Ref-1 demonstrate a statistically significant dose-dependent reduction in colony number (1.3- to 1.9-fold decrease) [Figure 3(B)], as well as a reduction in colony size. PaCa-2 cell colonies that did not express APE1/Ref-1 are more diffuse, while the Panc-1 colonies are smaller on average than the cells transfected with scrambled control (data not shown).
We next examined whether this reduced growth rate is due to an increase in apoptotic cell death. A reduction in APE1/Ref-1 increases the percentage of cells undergoing apoptosis which was seen clearly in siRNA-treated cells by Annexin-V staining at Day 3 [Figure 3(C)]. Confirming the increase in Annexin positivity, PARP-1 cleavage increases in pancreatic cancer cells that express reduced levels of APE1/Ref-1 protein [Figure 3C)]. In Panc-1 and PaCa-2 cells cells transfected with APE1/Ref-1 siRNA, the increase in PARP-1 cleavage is 2.4- and 5-fold greater, which is statistically significant (p < .05). Although the increase in apoptosis in both Panc-1 and PaCa-2 is 2- to 5-fold greater than the scrambled control, the majority of the cells (∼85%) are Annexin-negative, indicating that the differences in cell growth and colony formation are not entirely due to apoptosis. Therefore, we looked more closely at the effects of APE1/Ref-1 depletion on cell cycle.
Reduced levels of APE1/Ref-1 result in cell cycle perturbation and G2 arrest
Using BrdU incorporation assays, we then investigated whether knockdown of APE1/Ref-1 changes the cell cycle profile. On Day 3 after transfection, the percentage of Panc-1 cells in G1 is significantly decreased, while the percentage of cells in G2/M increases by 32% in the APE1/Ref-1 siRNA-treated cells [Figure 4(A)]. The results are similar for PaCa-2 cells (data not shown). Oligofectamine treatment, utilized as a control, does not differ significantly from the scrambled control (data not shown). Furthermore, we synchronized PaCa-2 cells and monitored reentry into cell cycle when APE1/Ref-1 protein was reduced. At 8 hr after adding serum-containing media, 46% less cells have entered S phase (Figure 5), demonstrating that progression out of G1 is slower in the PaCa-2 cells that have reduced levels of APE1/Ref-1. These data suggest that knockdown of APE1/Ref-1 both delays and slows cell cycle progression in pancreatic cancer cells.
Figure 4.
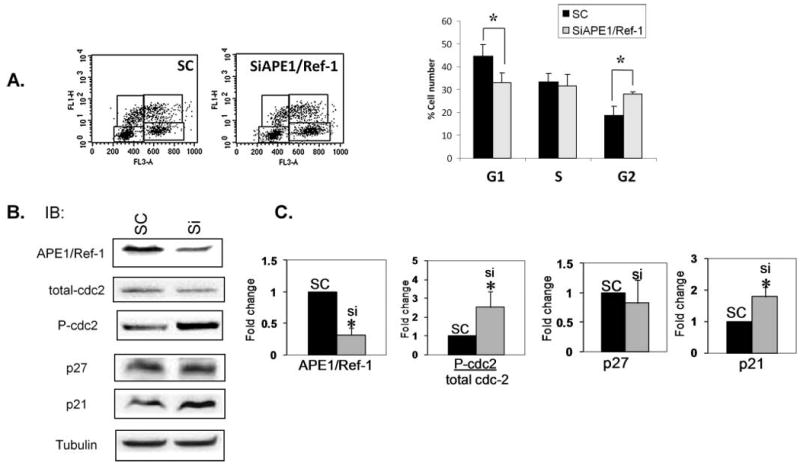
Reduced levels of APE1/Ref-1 alter cell cycle profile and levels of endogenous cell cycle proteins in asynchronous cells. Representative dot plots of BrdU incorporation assays in (A) asynchronous Panc-1 cells treated with SC and SiAPE1/Ref-1 (25 nM). Bar graphs demonstrate quantitation of ≥3 independent experiments with average ± SD. *p < .05 using Student's t-test, comparing SC vs SiAPE1/Ref-1. (B) Representative Western blot of cell cycle proteins including p21, p27, total cdc-2, and phosphorylated cdc-2. Tubulin is used as a loading control. (C) Quantitation of Western blot data for proteins shown in Panel B. Bar graphs demonstrate quantitation of ≥4 independent experiments with average ± SE. *p < .05 using Student's t-test, comparing SC versus SiAPE1/Ref-1.
Figure 5.

APE1/Ref-1 knockdown delays the reentry of cells into the cell cycle following synchronization. (A) Representative dot plots of synchronized PaCa-2 cells treated with SC and SiAPE1/Ref-1 (25 nM) stained with BrdU-FITC antibody and 7-AAD on Day 3 following transfection. (B) Bar graphs demonstrate quantitation of at least three independent experiments with average ± SD of SC control in black bars and SiAPE1/Ref-1 in gray bars. *p < .05 using student's t-test, comparing SC versus SiAPE1/Ref-1.
Decreased protein levels of APE1/Ref-1 cause increases in p21 and phosphorylated-cdc-2 levels, confirming G2/M arrest at a molecular level
We quantitated the levels of endogenous cell cycle inhibitors, p21 and p27 as well as monitoring the activity of the kinase responsible for the G2/M transition via phosphorylation of cdc-2 at tyr15 (34–36) (Figure 4). While we observe no change in p27 levels or total cdc-2, the levels of p21 increase 2-fold in cells that are not expressing high levels of APE1/Ref-1. The observed G2/M arrest is verified by the 2.5-fold increase in phosphorylated cdc-2 following APE1/Ref-1 siRNA treatment. Similar effects on the ratio of phospho-cdc-2 to total cdc-2 were observed in PaCa-2 cells (data not shown).
Reduced levels of APE1/Ref-1 did not increase ROS generation, but did increase overall levels of DNA damage
To investigate the effect of APE1/Ref-1 knockdown on the mechanism of cell cycle arrest we quantitated ROS levels and phosphorylation of H2AX (H2AX-γ) following APE1/Ref-1 silencing. By using oxidant-sensitive probe dihydrorhodamine 123 (DHR) analysis, we detected the ROS concentration in Panc-1 and PaCa-2 cells following knockdown of APE1/Ref-1 protein level. Panc-1 and PaCa-2 cells were treated with APE1/Ref-1 siRNA concentrations (50 nM), collected at various timepoints, and ROS was quantitated. As a positive control, cells were exposed to TBHP. Although ROS levels dramatically increased with TBHP in both cell lines, on Days 2 and 3 we did not see increased ROS generation in either cell line following knockdown of APE1/Ref-1 [Figures 6(A) and (B), and data not shown]. However, there is an 5-fold increase in the level of H2AX-γ when APE1/Ref-1 levels are knocked down in PaCa-2 cells [Figures 6(C) and (D)]. H2AX-γ was not detectable in the Panc-1 cells (data not shown).
Figure 6.
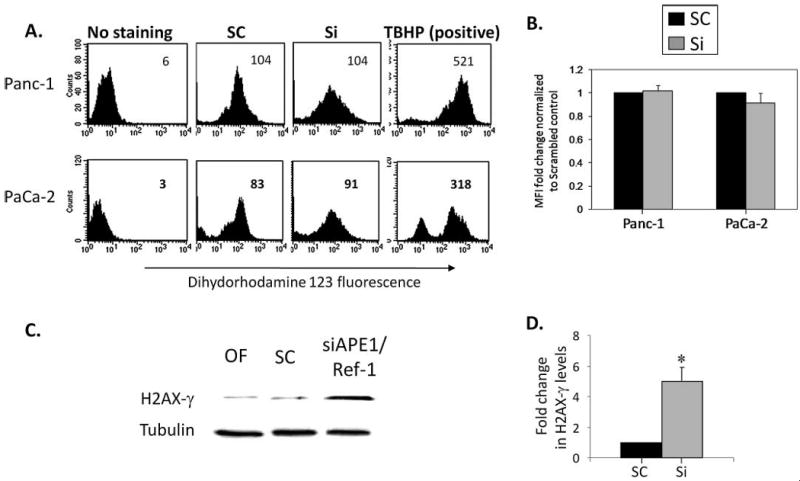
Knocking down APE1/Ref-1 increases DNA damage in pancreatic cancer cells but not the levels of ROS. (A) Knocking down of APE1/Ref-1 did not increase ROS generation in Panc-1 and PaCa-1 cell lines, but treatment with tert-butyl hydrogen peroxide (TBHP) did. The MFI (mean fluorescence intensity) of the oxidized form of dihydrorhodamine123 was analyzed by flow cytometry in APE1/Ref-1 siRNA (50 nM) transfected Panc-1 and PaCa-2 cells and analyzed on Day 3. These data are representative of three individual experiments and the averages are quantitated in Panel B. (C) Whole cell protein extracts from PaCa-2 cells were collected on Day 3 following transfection and analyzed for the presence of H2AX-γ by Western blot. A representative Western blot is presented, and the bar graph summarizes data from three experiments, with Tubulin used to normalize the amount of protein per lane. The amount of H2AX-γ is expressed relative to the respective scrambled controls in D. *p < .05 using student's t-test, comparing SC vs SiAPE1/Ref-1.
Discussion
Although alkylating chemotherapeutics are widely used cancer treatments, they have not been clinically effective in treating pancreatic cancer (37). Gemcitabine, the current standard for pancreatic cancer treatment, primarily demonstrates a palliative effect with isolated cases of long-term tumor regression (38). Success against this disease is still rare, and different treatment approaches are needed (1, 38–39). Although a previous report demonstrated that APE1/Ref-1 is elevated in pancreatic cancer cell lines as compared to a nontumorigenic pancreatic epithelial cell line (3), this is the first report to show that APE1/Ref-1 is elevated in patients with pancreatic adenocarcinoma and peri-pancreatic metastasis. This increase in APE1/Ref-1 expression could afford cellular protection against chemotherapeutic agents. In this report, we investigate APE1/Ref-1's importance in pancreatic cancer using siRNA for APE1/Ref-1. We demonstrate that targeting both the APE1/Ref-1 redox and DNA repair activities would dramatically increase chemotherapeutic cytotoxicity and could lead to eventual tumor regression.
Our goal is to understand the basic role that APE1/Ref-1 plays in the growth and/or resistance of pancreatic cancer cells to DNA-damaging agents and to assess whether APE1/Ref-1 would serve as a therapeutic target to potentiate the activity of current and novel agents in the treatment of pancreatic cancer. Knockdown of APE1/Ref-1 protein results in reduced proliferative capacity and colony-forming ability. Along with an increase in apoptosis are cell cycle effects that may be attributed to the DNA repair or the redox function of APE1/Ref-1 (32, 33, 40). Cell cycle effects are present in both synchronous and asynchronous cells, indicating that APE1/Ref-1 is important for transversing the cell cycle as well as moving cells from G1 to S phase. The cell lines utilized here are p53 mutant (41) and yet there is induction of p21, a well-established target of p53 transcriptional activity. We speculate that the induction of p21 may be due to p73 activity as p73 has been shown to induce p21 in colorectal cancer cell lines (42). Furthermore, in the paper by Grau et al., the authors demonstrate induction of p21 in pancreatic cancer cells following treatment with TGFβ indicating that TGFβ signaling may be regulating this response (43).
At least three possibilities exist to explain the cell cycle arrest: (a) accumulation of DNA damage supported by the H2AX-γ data in Figure 6(C); (b) lack of redox signaling; or (c) a combination of both functions. Quantitating DNA damage during specific phases of the cell cycle (44) in cells with reduced levels of APE1/Ref-1 protein will provide valuable information about the importance of the DNA repair function of APE1/Ref-1. Furthermore, we can analyze S-phase checkpoint proteins Chk1 and Cdc25A to investigate if replication errors are responsible for the observed cell cycle arrest (45). Although the levels of ROS did not increase following APE1/Ref-1 knockdown using the DHR123 staining, other reports demonstrate that APE1/Ref-1 protein levels do affect the generation of ROS (3, 5, 13). Further studies to quantitate long-term ROS generation via protein oxidation are underway to understand whether the increase in DNA damage observed with H2AX-γ antibody is due to accumulation of oxidative stress. To understand the mechanism by which APE1/Ref-1 affects cell cycle progression, we currently are conducting studies using small molecule inhibitors that affect only the DNA repair or redox function of APE1/Ref-1.
This is the first study in which APE1/Ref-1 protein levels have been quantitated in mitochondria following siRNA treatment. The mitochondrial genome is particularly susceptible to oxidative damage because mtDNA lacks protective histones and mtDNA repair systems that are less robust than systems protecting nuclear DNA (46). Perhaps APE1/Ref-1 knockdown compromises the DNA repair capacity of mitochondria. Studies are ongoing to (a) confirm an increase in mtDNA damage when APE1/Ref-1 protein is diminished via knockdown and to (b) assess mitochondrial function after knockdown.
Pancreatic cancer is a deadly disease that is virtually never cured (1). An APE1/Ref-1-targeted approach has the potential to augment current agents, especially gemcitabine, in the treatment of pancreatic cancer (15). Decreased APE1/Ref-1 levels may lead to an increased stress response, because removal of APE1/Ref-1's redox signaling affects downstream targets, including important proteins such as NFκB, AP-1, HIF-1α, p53, and others (47). We will continue to delineate the importance of the DNA repair and redox functions of APE1/Ref-1 in blocking survival and/or proliferation of pancreatic cancer cells. The recent findings indicating a growing role of APE1/Ref-1 in angiogenesis and the APE1/Ref-1 redox inhibitor as an antiangiogenic agent are areas that need additional study in pancreatic cancer (9). Finally, we will extrapolate the validity of this APE1/Ref-1-targeted approach to an in vivo therapeutic effect in pancreatic tumor xenografts or orthotopic models using small molecule inhibitors of APE1/Ref-1 such as E3330 (redox) (8) and methoxyamine (MX) (48) or CRT0044876 (7-nitroindole-2-carboxylic acid) (49). A better understanding of critical pathways and molecular mechanisms involved in pancreatic tumor development, progression, and resistance to traditional therapy is essential to having success against this deadly disease.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Moore MJ. Chemotherapy in pancreatic carcinoma. Current Oncol. 2003;5:s24–s26. [Google Scholar]
- 2.Duffy JP, Eibl G, Reber HA, Hines OJ. Influence of hypoxia and neoangiogenesis on the growth of pancreatic cancer. Mol Cancer. 2003;2:12. doi: 10.1186/1476-4598-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou GM, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther. 2008;7:2012–2021. doi: 10.1158/1535-7163.MCT-08-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 5.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie K, Wei D, Huang S. Transcriptional anti-angiogenesis therapy of human pancreatic cancer. Cytokine Growth Factor Rev. 2006;17:147–156. doi: 10.1016/j.cytogfr.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Ray S, Lee C, Hou T, Bhakat KK, Brasier AR. Regulation of signal transducer and activator of transcription 3 enhanceosome formation by apurinic/apyrimidinic endonuclease 1 in hepatic acute phase response. Mol Endocrinol. 2010;24:391–401. doi: 10.1210/me.2009-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, 2nd, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou GM, Karikari C, Kabe Y, Handa H, Anders RA, Maitra A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol. 2009;219:209–218. doi: 10.1002/jcp.21666. [DOI] [PubMed] [Google Scholar]
- 10.Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009;11:621–638. doi: 10.1089/ars.2008.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bapat A, Fishel M, Kelley MR. Going Ape as an approach to cancer therapeutics. Antioxid Redox Signal. 2009;11:651–668. doi: 10.1089/ars.2008.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Molecular Aspects of Medicine. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker LJ, Craig RB, Harris AL, Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau JP, Weatherdon KL, Skalski V, Hedley DW. Effects of gemcitabine on APE/ref-1 endonuclease activity in pancreatic cancer cells and the therapeutic potential of antisense oligonucleotides. Br J Cancer. 2004;91:1166–1173. doi: 10.1038/sj.bjc.6602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–686. [PubMed] [Google Scholar]
- 17.Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, Pommier Y, Lieberman J. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol. 2003;4:145–153. doi: 10.1038/ni885. [DOI] [PubMed] [Google Scholar]
- 18.Fishel ML, He Y, Reed AM, Chin-Sinex H, Hutchins GD, Mendonca MS, Kelley MR. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair (Amst) 2008;7:177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore DH, Michael H, Tritt R, Parsons SH, Kelley MR. Alterations in the expression of the DNA repair/redox enzyme APE/ref-1 in epithelial ovarian cancers. Clin Cancer Res. 2000;6:602–609. [PubMed] [Google Scholar]
- 20.Tell G, Pellizzari L, Pucillo C, Puglisi F, Cesselli D, Kelley MR, Di Loreto C, Damante G. TSH controls Ref-1 nuclear translocation in thyroid cells. J Mol Endocrinol. 2000;24:383–390. doi: 10.1677/jme.0.0240383. [DOI] [PubMed] [Google Scholar]
- 21.Hansen WK, Deutsch WA, Yacoub A, Xu Y, Williams DA, Kelley MR. Creation of a fully functional human chimeric DNA repair protein. Combining O6-methylguanine DNA methyltransferase (MGMT) and AP endonuclease (APE/redox effector factor 1 (Ref 1)) DNA repair proteins. J Biol Chem. 1998;273:756–762. doi: 10.1074/jbc.273.2.756. [DOI] [PubMed] [Google Scholar]
- 22.Robertson KA, Hill DP, Xu Y, Liu L, Van Epps S, Hockenbery DM, Park JR, Wilson TM, Kelley MR. Down-regulation of apurinic/apyrimidinic endonuclease expression is associated with the induction of apoptosis in differentiating myeloid leukemia cells. Cell Growth Differ. 1997;8:443–449. [PubMed] [Google Scholar]
- 23.Xu Y, Moore DH, Broshears J, Liu L, Wilson TM, Kelley MR. The apurinic/apyrimidinic endonuclease (APE/ref-1) DNA repair enzyme is elevated in premalignant and malignant cervical cancer. Anticancer Res. 1997;17:3713–3719. [PubMed] [Google Scholar]
- 24.Fishel ML, Newell DR, Griffin RJ, Davison R, Wang LZ, Curtin NJ, Zuhowski EG, Kasza K, Egorin MJ, Moschel RC, Dolan ME. Effect of cell cycle inhibition on Cisplatin-induced cytotoxicity. J Pharmacol Exp Ther. 2005;312:206–213. doi: 10.1124/jpet.104.073924. [DOI] [PubMed] [Google Scholar]
- 25.Arris CE, Boyle FT, Calvert AH, Curtin NJ, Endicott JA, Garman EF, Gibson AE, Golding BT, Grant S, Griffin RJ, Jewsbury P, Johnson LN, Lawrie AM, Newell DR, Noble ME, Sausville EA, Schultz R, Yu W. Identification of novel purine and pyrimidine cyclin-dependent kinase inhibitors with distinctmolecular interactions and tumor cell growth inhibition profiles. J Med Chem. 2000;43:2797–2804. doi: 10.1021/jm990628o. [DOI] [PubMed] [Google Scholar]
- 26.Dolan ME, Chae MY, Pegg AE, Mullen JH, Friedman HS, Moschel RC. Metabolism of O6-benzylguanine, an inactivator of O6-alkylguanine-DNA alkyltransferase. Cancer Res. 1994;54:5123–5130. [PubMed] [Google Scholar]
- 27.Dolan ME, Roy SK, Fasanmade AA, Paras PR, Schilsky RL, Ratain MJ. O6-benzylguanine in humans: metabolic pharmacokinetic and pharmacodynamic findings. J Clin Oncol. 1998;16:1803–1810. doi: 10.1200/JCO.1998.16.5.1803. [DOI] [PubMed] [Google Scholar]
- 28.Szabados E, Fischer GM, Gallyas F, Jr, Kispal G, Sumegi B. Enhanced ADP-ribosylation and its diminution by lipoamide after ischemia-reperfusion in perfused rat heart. Free Radic Biol Med. 1999;27:1103–1113. doi: 10.1016/s0891-5849(99)00151-3. [DOI] [PubMed] [Google Scholar]
- 29.Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 30.Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 32.Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 33.Vascotto C, Cesaratto L, Zeef LA, Deganuto M, D'Ambrosio C, Scaloni A, Romanello M, Damante G, Taglialatela G, Delneri D, Kelley MR, Mitra S, Quadrifoglio F, Tell G. Genomewide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics. 2009;9:1058–1074. doi: 10.1002/pmic.200800638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow JP, Siu WY, Ho HT, Ma KH, Ho CC, Poon RY. Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. J Biol Chem. 2003;278:40815–40828. doi: 10.1074/jbc.M306683200. [DOI] [PubMed] [Google Scholar]
- 35.Dekoj T, Lee S, Desai S, Trevino J, Babcock TA, Helton WS, Espat NJ. G2/M cell-cycle arrest and apoptosis by n-3 fatty acids in a pancreatic cancer model. J Surg Res. 2007;139:106–112. doi: 10.1016/j.jss.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 36.Ujiki MB, Ding XZ, Salabat MR, Bentrem DJ, Golkar L, Milam B, Talamonti MS, Bell RH, Jr, Iwamura T, Adrian TE. Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol Cancer. 2006;5:76. doi: 10.1186/1476-4598-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore MJ, Feld R, Hedley D, Oza A, Siu LL. A phase II study of temozolomide in advanced untreated pancreatic cancer. Invest New Drugs. 1998;16:77–79. doi: 10.1023/a:1006043332368. [DOI] [PubMed] [Google Scholar]
- 38.Burris HA., 3rd Recent updates on the role of chemotherapy in pancreatic cancer. Semin Oncol. 2005;32:1–3. doi: 10.1053/j.seminoncol.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 39.Kern S, Tempero M, Conley B, et al. Pancreatic Cancer: An Agenda for Action. Bethesda, MD: National Cancer Institute; 2001. [Google Scholar]
- 40.Yang S, Misner B, Chiu R, Meyskens FL., Jr Redox effector factor-1, combined with reactive oxygen species, plays an important role in the transformation of JB6 cells. Carcinogenesis. 2007;28:2382–2390. doi: 10.1093/carcin/bgm128. [DOI] [PubMed] [Google Scholar]
- 41.Mohiuddin M, Chendil D, Dey S, Alcock RA, Regine W, Mohiuddin M, Ahmed MM. Influence of p53 status on radiation and 5-flourouracil synergy in pancreatic cancer cells. Anticancer Res. 2002;22:825–830. [PubMed] [Google Scholar]
- 42.Yu J, Zhang L, Hwang PM, Rago C, Kinzler KW, Vogelstein B. Identification and classification of p53-regulated genes. Proc Natl Acad Sci USA. 1999;96:14517–14522. doi: 10.1073/pnas.96.25.14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grau AM, Zhang L, Wang W, Ruan S, Evans DB, Abbruzzese JL, Zhang W, Chiao PJ. Induction of p21waf1 expression and growth inhibition by transforming growth factor beta involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res. 1997;57:3929–3934. [PubMed] [Google Scholar]
- 44.Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007;6:1239–1248. doi: 10.1158/1535-7163.MCT-06-0633. [DOI] [PubMed] [Google Scholar]
- 45.Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 46.Marcelino LA, Thilly WG. Mitochondrial mutagenesis in human cells and tissues. Mutat Res. 1999;434:177–203. doi: 10.1016/s0921-8777(99)00028-2. [DOI] [PubMed] [Google Scholar]
- 47.Luo M, He H, Kelley MR, Georgiadis M. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu L, Gerson SL. Therapeutic impact ofmethoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr Opin Investig Drugs. 2004;5:623–627. [PubMed] [Google Scholar]
- 49.Madhusudan S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, Talbot DC, Hammonds T, Freemont PA, Sternberg MJ, Dianov GL, Hickson ID. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005;33:4711–4724. doi: 10.1093/nar/gki781. [DOI] [PMC free article] [PubMed] [Google Scholar]