

Abstract
Glomerular disease can be triggered or exacerbated by microbes that activate the immune system by Toll-like receptor (TLR) ligation. TLR9 activation promotes host defenses through the enhancement of innate and adaptive immune responses that facilitate the recruitment of leukocytes to areas of inflammation. We defined the role of TLR9 in experimental crescentic glomerulonephritis. Wild-type mice administered a TLR9 ligand and sheep anti-mouse glomerular basement membrane antibody developed histological injury with impaired renal function, which was attenuated in TLR9 knockout mice. Consistent with enhanced renal injury, wild-type mice exhibited enhanced T helper 1 and T helper 17 cellular immune responses. Kidney mRNA expression of inflammatory cytokines and chemokines as well as leukocyte recruitment were increased in wild-type mice. The use of bone marrow chimeric mice demonstrated that while both bone marrow and tissue cell TLR9 are required for maximal injury, bone marrow TLR9 is more important. Administration of a TLR9 inhibitor before sheep anti-mouse glomerular basement membrane globulin in wild-type mice attenuated cellular nephritogenic immunity that resulted in decreased renal injury. Administration of the inhibitor 7 days after disease initiation decreased glomerular leukocyte recruitment as well as renal injury. These results define the role of TLR9 in experimental crescentic glomerulonephritis and identify therapeutic potential for TLR9 inhibitors in attenuating renal injury, decreasing cellular nephritogenic immunity early in disease, and decreasing kidney effector responses later.
The generation of autoinflammatory responses with subsequent organ injury is not well understood. Little is known about the context in which humans develop autoimmunity and autoinflammatory responses, while attempts at replication of these diseases in experimental animal models are challenging and often unrewarding. The discovery of Toll-like receptors (TLRs), which recognize molecular signatures from infectious agents or endogenous ligands, has provided insights into the development and pathogenesis of autoinflammation and organ injury. TLRs that evolved to protect host from infections can be activated by self molecules. Thus while signaling via TLRs promotes protective innate and adaptive immunity, excess responses may also promote intense organ inflammation and injury.1 This is best established for the endosomally (intracellularly) located TLRs, which detect nucleic acids. Inappropriate activation of TLRs 7, 8, and 9 facilitates the development of heightened cellular immunity, autoantibodies, and organ inflammation.2,3 Hypomethylated DNA from bacterial or viral microbes ligate TLR9, which activates dendritic cells, triggering inflammatory responses and promoting Th1-polarized adaptive immune responses.4,5 The immunostimulatory effects of TLR9 can be reproduced by synthetic oligodeoxynucleotides (ODNs), which contain unmethylated deoxycytidyl-deoxyguanosine (CpG) motifs, CpG-ODN.6
Glomerulonephritis (GN) is a common cause of end stage renal failure, and crescentic GN represents the most severely injurious form. CD4+ T helper (Th) cells are crucial for the development of crescentic GN. CD4+ Th cells are polarized into subsets dependant on their cytokine production. For Th1 cells, the signature cytokine produced is interferon-γ (IFN-γ), for Th2 cells interleukin (IL)-4 and IL-17 for the Th17 cell subset. Evidence from human and experimental studies suggests glomerular crescent formation is driven by Th1 dependant nephritogenic immune responses that direct cell mediated effectors inducing glomerular injury.7,8 Although Th1 driven nephritogenic immune responses induce severe experimental crescentic GN, Th2 predominant responses are less severe, and Th2 associated cytokines, IL-4 and IL-10, can attenuate injurious Th1 directed glomerular injury.9 Recent evidence suggests that Th17 cells are also required for full disease expression in experimental anti-glomerular basement membrane (GBM) GN.10 While T cells drive systemic and local immune responses, macrophages are the key effector cells of glomerular injury in this model.11 Furthermore, macrophage depletion effectively halts the progression of crescentic GN.12
TLRs have been implicated in the initiation and disease progression of several forms of human and experimental kidney disease, recently reviewed.13 In murine experimental crescentic GN, ligation of extracellular TLRs (TLR2/4) enhance renal injury,14,15,16,17 whereas in experimental lupus, ligation of TLR9 enhances lupus nephritis,18,19 although these results were not confirmed in TLR deficient mice. Surprisingly lupus prone (MRL/Mplpr/lpr) TLR9−/− mice suffer increased mortality and renal injury compared with control (MRL/Mplpr/lpr) mice, suggesting endogenous TLR9 has immune regulatory function.20 These results highlight the importance of studying the effects of TLR9 in wild-type (WT) and TLR9−/− mice to ascertain function for TLR9 in disease pathogenesis. The role of TLR9 in rapidly progressive immune-mediated crescentic GN, the most severe acute form of GN, has not been studied. Clinically, this would be of interest because of the known correlation between infection and GN.
Since current treatments of crescentic GN are associated with considerable morbidity and mortality, the use of better targeted therapies is desirable. Although suppressive ODNs, which were relatively nonspecific, showed promise in treating experimental arthritis21 and GN in lupus prone mice,22 recently more specific TLR9 inhibitors have been developed. These TLR9 inhibitors contain repeating guanine (G-G-G-G) motifs and successfully limit inflammatory cytokine production in mice and humans,23 but have not been studied in (immune mediated) kidney disease.
In this series of experiments, we defined a role for TLR9 (using WT and TLR9−/− mice) in enhancing renal injury in experimental crescentic GN. TLR9 was required for development of autoinflammatory responses and full expression of kidney injury. We found that TLR9 ligation induced Th1 and Th17 systemic nephritogenic responses and increased the recruitment of glomerular cellular effectors, which resulted in enhanced GN with impaired renal function. Subsequently, we demonstrated successful attenuation of renal injury after administration of a TLR9 inhibitor; the inhibitor suppressed Th1 and Th17 nephritogenic immune responses if administered pre-emptively and decreased glomerular effector T cell and macrophage recruitment when administered after disease initiation.
Materials and Methods
Experimental Design
TLR9−/− mice on a C57BL/6 background5 were bred at Monash Medical Centre (Melbourne, Australia), whereas mice WT (C57BL/6 [CD45.2] and for some chimeric studies congenic CD45.1 mice) were obtained from Monash Animal Services (Melbourne, Australia). All mice were housed in specific pathogen-free conditions, in microisolators at Monash Medical Centre. Sheep anti-mouse GBM antibody was generated as previously described.9 Nonaccelerated autologous phase anti-GBM GN was induced in age-matched, 8- to 10-week-old, male mice after the i.p. administration of CpG-ODN (80 μg) in 200 μl of PBS, followed 2 hours later by the i.p. injection of 30 mg of sheep anti-mouse GBM antibody. All mice, WT and TLR9−/− mice, weighed 27 ± 2 g. For the inhibition studies, mice were injected i.p. with the TLR9 inhibitor, IRS869 (80 μg). IRS869 is a regulatory DNA sequence with TLR9 specificity23 or PBS control 2 hours before the administration of CpG-ODN and then twice a week at the same dose. Mice were sacrificed after 21 days. Studies were performed in accordance with National Health and Medical Research Council of Australia guidelines and approved by the Monash University Animal Ethics Committee. For all studies the number of mice was ≥ eight mice per group; individual numbers are listed in the figure legends. Bone marrow (BM) chimeric mice were generated as previously described.24 Flow cytometry demonstrated >90% reconstitution. Results are expressed as mean ± SEM. For statistical analysis unpaired t-test was used routinely for values with a normal distribution. The Mann-Whitney U-test was used for samples not distributed normally; when more than two values were analyzed, we used analysis of variance with post hoc analysis by Tukey’s test (GraphPad Prism; Graphpad Software, San Diego, CA). A value of P < 0.05 was considered statistically significant.
All ODNs were manufactured by Geneworks (Thebarton, South Australia, Australia). The sequence for the immunostimulatory CpG-ODN was 5′-TCCATGACGTTCCTGACGTT-3′; nonstimulatory deoxyguanosine-deoxy- cytidyl-ODN was 5′-TCCATGAGCTTCCTGAGCTT-3′; and for the immunoregulatory (IRS869), 5′-TCCTGGAGGGGTTGT-3′.
Assessment of Renal Injury
Glomerular abnormalities were assessed on PAS-stained, Bouin’s fixed, 3-μm-thick, paraffin-embedded sections by using coded slides. Abnormalities recorded included crescent formation, defined as two or more layers of cells visible in Bowman’s space, segmental proliferation, necrosis, or hyalinosis. Severely abnormal glomeruli was defined as glomeruli with evidence of either crescent formation, accumulation of cells in Bowman’s space that did not fulfill criteria for crescent formation, >50% glomerular tuft necrosis, or severe proliferative changes as described previously.25,26 Results are expressed as the proportion of glomeruli severely affected. A minimum of 50 glomeruli was analyzed per animal to determine glomerular injury. Semiquantative analysis of tubulointerstitial damage was performed in each mouse; using a graticle (10 mm2) and assessing 10 randomly selected cortical areas (×200 magnifications), injury was defined as tubular dilatation, tubular atrophy, sloughing of tubular epithelial cells, or thickening of the basement membrane. Interstitial inflammation was defined according to the degree of leukocyte infiltration in the interstitium. Injury and inflammation were assessed separately and graded according to a scoring system, 0 to 4. Each cortical field was scored (0 to 4) according to the amount of injury and inflammation: 0, no interstitial damage or inflammation; 1, less than 25% of the tubulointerstitum damaged with minimal tubulointerstitial inflammation; 2, 25% to 50% of the tubulointerstitum damaged and mild inflammation; 3, 50% to 75% of the tubulointerstitum damaged with moderate inflammation; and 4, >75% of the tubulointerstitum damaged with diffuse inflammation.
Urine was collected, using metabolic cages, during the 24-hour period before sacrifice while serum was collected after sacrifice. Proteinuria was measured by using a modified Bradford’s assay.25 Serum creatinine and blood urea nitrogen (BUN) measurements were recorded at the end of the experiment by using an alkaline picric acid method for creatinine and an autoanalyser, whereas an enzymatic assay was used to assay BUN.
Glomerular CD4+ T Cell, CD8+ T Cells, Neutrophils, Macrophage, IgG, and C3 Deposition
Kidney sections were initially fixed in periodate lysine paraformaldehyde for 4 hours, after which they were washed with 20% sucrose solution and frozen in liquid nitrogen. Tissue sections were cut and a three-layered immunopeoxidase technique as previously described was used to stain for T cells, neutrophils, and macrophages.25 The primary antibodies used were GK1.5 for CD4+ T cells (anti-mouse CD4; American Type Culture Collection, Manassas, VA), 53–6.7 for CD8+ T cells (anti-mouse CD8; American Type Culture Collection), RB6-8C5 (anti-Gr-1; DNAX, Palo Alto, CA) for neutrophils, and FA/11 for macrophages (anti-mouse CD68, generously provided by Dr G. Koch, Medical Research Council Laboratory of Molecular Biology, Cambridge, England). The secondary antibody used was rabbit anti-rat biotin (BD Biosciences, San Jose, CA). A minimum of 20 consecutively viewed glomeruli was assessed per animal, and results are expressed as cells per glomerular cross section. Glomerular IgG and complement C3 deposition were assessed on 6-μm-thick, frozen sections by using fluorescein isothiocyanate-sheep anti-mouse Ig (1:100; Silenus, Hawthorn, Victoria, Australia) or goat anti-mouse C3 (1:100; Cappell, West Chester, PA). Scores were assigned based on the intensity of IgG/C3 deposition where 0 represents no deposition and 3 represents intense depositions.
Intrarenal Cytokine mRNA Expression
For measurement of T-bet, tumor necrosis factor (TNF), IL-1β, IFN-γ, CCL2, CCL5 (RANTES), CXCL9 (MIG), Rorγ, and GATA3 by RT-PCR, 500 ng of RNA was treated with 1 U of amplification grade DNase I (Invitrogen, Melbourne, Australia), primed with random primers (Applied Biosystems, Foster City, CA), and reverse transcribed by using a High-Capacity cDNA reverse transcription kit (Applied Biosystems). Gene-specific oligonucleotide primers designed using the Primer 3 software (Whitehead Institute for Biomedical Research, Cambridge, MA) were synthesized by Invitrogen as previously described.27 A Rotor Gene RG-3000 (Corbett Research Mortlake, Australia) using Power SYBR Green PCR master mix (Applied Biosystems) was used to perform RT-PCR. PCR products were confirmed by using melt-curve analysis, whereas mRNA expression was quantified by using serial dilutions of an exogenous standard. Primer sequences used were as previously described,27,28 expression was standardized to 18S (house-keeping gene) before being expressed as a fold increase (or decrease) relative to WT mice with glomerulonephritis.
Antigen Stimulated Splenocyte Cytokine Production
Spleens were removed, and a single cell suspension was obtained. Splenocytes (4 × 106 cells/ml per well) were cultured in RPMI/10% fetal calf serum with protein G-purified normal sheep IgG (10 μg/ml) at 37°C for 72 hours. Measurement of cytokine production by enzyme-linked immunosorbent assay (ELISA) was performed as previously described.27 The following antibodies were used: rat anti-mouse IFN-γ (R4-6A2; BD Pharmingen, San Diego, CA); biotinylated rat anti-mouse IFN-γ (XMG1.2; BD Pharmingen); rat anti-mouse IL-4 (11B11; American Type Culture Collection); and biotinylated rat anti-mouse IL-4 (BVD6; DNAX). For IL-17A an ELISA from DuoSet (R&D Systems, Minneapolis MN) was used. For the IL-2 ELISA, rat anti-mouse IL-2 (JES6-1A12; DNAX) and biotinylated anti-mouse IL-2 (JES6-5H4; DNAX) were used. For IL-6, rat anti-mouse IL-6 (Mab 406; R&D Systems) and biotinylated rat anti-mouse IL-6 (BAF 406; R&D Systems) were used. For IL-10, rat anti-mouse IL-10 (category number 551215; BD Pharmingen) and biotinylated rat anti-mouse IL-10 (category number 554423; BD Pharmingen) were used.
Dermal Delayed Type Hypersensitivity
Twenty-four hours before sacrifice, mice received 0.5 mg of sheep globulin in 30 μl of PBS in the left hindfoot. Thirty microliters of PBS was injected into the right hindfoot. Immediately after sacrifice, delayed type hypersensitivity (DTH) was quantified by measuring the difference in foot pad thickness (in millimeters) by using a micrometer, as previously described.29
Circulating Antigen-Specific Antibody Levels
ELISA was used to detect circulating serum antigen-specific IgG titers, as previously described.9 Horseradish peroxidase conjugated sheep anti-mouse IgG (Amersham Biosciences, Rydalmere, Australia) with serial dilutions (1:50 to 1:3200), goat anti-mouse IgG1 (1:100 dilution; Silenius, Boronia, Australia), and biotinylated rat anti-mouse IgG3 (1:50 dilution; BD Pharmingen) antibodies were used. Results are expressed as OD450 ± SEM.
Flow Cytometry Analysis
Antibodies used for fluorescence activated cell sorting (FACS) analysis were as follows: CD11c-fluorescein isothiocyanate; MHCII-PE; and B220-APC (all from BD Bioscience). FACS analysis was performed on BD FACS Canto (San Jose, CA).
Results
TLR9 Ligation Enhances Experimental Anti-GBM Glomerulonephritis and Renal Injury
To define the effect of TLR9 ligation in experimental crescentic GN, anti-GBM globulin was administered to three groups of mice. C57BL/6 (WT) mice were injected with CpG-ODN and sheep anti-mouse GBM globulin, and TLR9−/− mice (on a C57BL/6 background) were injected with CpG-ODN and sheep anti-mouse GBM globulin. As a control group, WT mice were injected with control DNA sequence, which does not ligate TLR9 (GpC) and sheep anti-mouse GBM globulin. Experiments ended after 21 days. WT mice administered CpG-ODN and sheep anti-mouse GBM globulin developed more glomerular crescentic formation, a greater proportion of severely abnormal glomeruli and more severe tubulo-interstitial injury (Figure 1, A–C) compared with TLR9−/− mice. Injury in control WT mice administered GpC and sheep anti-mouse GBM globulin (represented by the dotted line in Figure 1) did not differ significantly from injury seen in TLR9−/− mice. Renal function, assessed by urinary proteinuria (24-hour urinary collections) and serum creatinine and BUN measurements, was increased in WT mice administered CpG-ODN and sheep anti-mouse GBM globulin compared with TLR9−/− mice (Figure 1, D–F) and WT mice treated with (GpC and) sheep anti-mouse GBM globulin (dotted line). Representative histological sections demonstrating renal injury are shown at low and high power in WT and TLR9−/− mice administered CpG-ODN and sheep anti-mouse GBM globulin in Figure 2, A–D.
Figure 1.

Histological injury and renal function in WT and TLR9−/− mice administered CpG-ODN and sheep anti-mouse GBM globulin. Injury in control WT mice administered GpC and sheep anti-mouse GBM globulin is represented by the dotted line (mean value). On day 21, WT mice administered CpG-ODN and sheep anti-mouse GBM globulin (n = 17) had developed more glomerular crescents (A), proportionally more severely abnormal glomeruli (B), and more interstitial injury (C) than TLR9−/− mice (n = 16). Injury in WT mice administered GpC and sheep anti-mouse GBM globulin (n = 8) did not differ from injury seen in TLR9−/− mice administered CpG-ODN and sheep anti-mouse GBM globulin. Renal function assessed by 24-hour urinary protein excretion (D), serum creatinine (E), and BUN (F) was enhanced in WT mice administered CpG-ODN and sheep anti-mouse GBM globulin. The dotted line represents mean values obtained for WT mice administered GpC and sheep anti-mouse GBM globulin (the individual mean values and SEM are as follows: glomerular crescents, 10.1 ± 1.5%; severely abnormal glomeruli, 26.6 ± 2.3%; tubulointerstitial injury, 2.2 ± 0.1; proteinuria, 10.6 ± 0.3 g/24 hours; creatinine, 17.6 ± 0.6 μmol/L; and BUN, 18.0 ± 0.8 mmol/L). *P < 0.05; ***P < 0.001.
Figure 2.

Representative kidney sections demonstrating histological renal injury in WT and TLR9−/− mice 21 days after the administration of CpG-ODN and sheep anti-mouse glomerular basement membrane globulin. Glomerular renal injury is demonstrated at high power (original magnification, ×400) in WT mice, where severe crescentic glomerulonephritis with fibrinoid necrosis is demonstrated (A) and TLR9−/− mice where glomerular injury is less severe (B). Tubulo-interstitial injury with hypercellularity, tubular dilatation, and atrophy (original magnification, ×200) is shown in WT mice (C), and injury is attenuated in TLR9−/−mice (D).
TLR9 Ligation Increases Glomerular Cellular Effectors and Kidney mRNA Expression of Pro-Inflammatory Cytokines, Chemokines, and Key Transcription Factors
After 21 days, kidney sections from TLR9−/− mice given CpG-ODN and sheep anti-mouse GBM globulin demonstrated fewer glomerular CD4+ T cells and macrophages compared with WT mice (Figure 3, A and B). There was no difference in glomerular CD8+ T cells or neutrophil recruitment between the groups (Figure 3, C and D).
Figure 3.

Effectors of cell-mediated renal injury in WT and TLR9−/− mice, 21 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin. More glomerular CD4+ T cells (A) and glomerular macrophages (B) were seen in WT mice compared with TLR9−/− mice. There was no difference in glomerular CD8+ T cell (C) or neutrophil (D) recruitment. **P < 0.01; ***P < 0.001.
To better characterize the observed differences in renal injury between WT and TLR9−/− mice, we analyzed renal mRNA expression of the renal injurious pro-inflammatory cytokines, chemokines, and the key Th1, Th2, and Th17 transcription factors. IFN-γ, TNF, and IL-1β kidney mRNA expression were decreased in TLR9−/− mice (Figure 4, A–C). Although there was no change in MCP1/CCL2 kidney mRNA expression (P = 0.17), RANTES/CCL5 and MIG/CXCL9 were also decreased in TLR9−/− mice (Figure 4, D–F). T-bet, the master Th1 transcription factor, kidney mRNA expression showed a trend to decrease (P = 0.18) in TLR9−/− mice, whereas Rorγ, the master Th17 transcription factor, and GATA3, key Th2 transcription factor, expression were decreased in TLR9−/− mice (Figure 4, G–I).
Figure 4.

Renal mRNA expression of cytokines, chemokines, and T cell transcription factors, 21 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin. mRNA expression of the signature Th1 produced cytokine, IFN-γ (A), and pro-inflammatory cytokines, TNF (B) and IL-1β (C), were decreased in TLR9−/− mice. mRNA expression of CCL2 (D) was unchanged (P = 0.17), whereas CCL5 (E) and CXCL9 (F) expression were decreased in TLR9−/− mice. Analyzing mRNA expression of the key transcription factors showed a trend to decrease in T-bet (G; P = 0.18), whereas Rorγ (H) and GATA3 expression (I) were decreased in TLR9−/− mice. *P < 0.05; **P < 0.01; ***P < 0.001.
TLR9 Ligation Enhances Cellular Nephritogenic Systemic Immune Responses on Day 21 and Day 7
Twenty-four hours before the end of 21-day experiments, sheep globulin was injected, and differences in footpad swelling were measured. Measurable DTH was only seen in WT mice treated with CpG-ODN and sheep anti-mouse GBM globulin (Δ footpad thickness: WT mice, 0.16 ± 0.02 mm; TLR9−/− mice, 0.02 ± 0.02 mm; P < 0.001). WT mice treated with GpC and sheep anti-mouse GBM globulin did not develop DTH responses (Δ footpad thickness: 0.01 ± 0.01 mm; P = NS).
More splenocytes were obtained from WT mice (4.1 ± 0.4 × 107 cells) than TLR9−/− mice (2.4 ± 0.1 × 107 cells; P < 0.001), or WT mice treated with GpC and sheep anti-mouse GBM globulin (2.5 ± 0.2 × 107 cells). Splenic cytokine production showed enhanced systemic immunity in WT mice treated with CpG-ODN and sheep anti-mouse GBM globulin compared with TLR9−/− mice administered CpG-ODN and sheep anti-mouse GBM globulin. IFN-γ production was increased in WT administered CpG-ODN and sheep anti-mouse GBM globulin (Figure 5A); the dotted line represents the mean value of WT mice given GpC and sheep anti-mouse GBM globulin. IL-17A was increased in WT mice administered CpG-ODN and sheep anti-mouse GBM globulin (Figure 5C) as was IL-6 (Figure 5D). Levels of IL-2, IL-4, and IL-10 cytokine production were unchanged (Figure 5, B, E, and F). We also assessed immune responses in WT and TLR9−/− mice 7 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin (Figure 5, G and H). Production of IFN-γ and IL-17A was increased in WT mice. Flow cytometric analysis demonstrated increased splenic dendritic cell activation (WT 4.7 ± 0.3% versus TLR9−/− mice 3.7 ± 0.1% CD11c+MHCII cells; P < 0.01), with increased numbers of plasmacytoid dendritic cells in WT mice (WT 1.2 ± 0.2% versus TLR9−/− mice 0.8 ± 0.1% CD11c+B220+ cells; P < 0.05). These results demonstrated that nephritogenic Th1 and Th17 immune responses are enhanced after the administration of CpG-ODN and sheep anti-mouse GBM globulin to WT mice.
Figure 5.

Systemic immune responses to the nephritogenic antigen measured in WT and TLR9−/− mice, 21 and 7 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin and control WT mice administered GpC and sheep anti-mouse GBM globulin (represented by the dotted line). On day 21, antigen-stimulated production of IFN-γ (A), IL-17A (C), and IL-6 (D) was increased in WT mice administered CpG-ODN and sheep anti-mouse GBM globulin compared with TLR9−/− mice. There was no change in IL-2 (B), IL-4 (E), or IL-10 (F) production between the groups and control WT mice administered GpC and sheep anti-mouse GBM globulin. Mean values obtained for controls and WT mice administered GpC and sheep anti-mouse GBM globulin are represented by the dotted line (mean and SEM values for WT mice treated with GPC and sheep anti-mouse GBM globulin were as follows: IFN-γ, 236.8 ± 78.9 pg/4 × 106cells; IL-2, 252.9 ± 23.2 pg/4 × 106cells; IL-17A, 24.2 ± 6.2 pg/4 × 106cells; IL-6, 22.8 ± 5.6 pg/4 × 106cells; IL-4, 59.3 ± 12.4 pg/4 × 106cells; and IL-10, 66.0 ± 13.6 pg/4 × 106cells). IFN-γ (G) and IL-17A (H) cytokine production were increased in WT compared with TLR9−/− mice 7 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin. **P < 0.01; ***P < 0.001.
TLR9 Ligation Enhances Systemic Humoral Immunity, But Not Glomerular IgG or Complement Deposition
WT mice given CpG-ODN and sheep anti-mouse GBM globulin demonstrated enhanced levels of serum antigen-specific mouse IgG levels (Figure 6A) compared with TLR9−/− mice given CpG-ODN and sheep anti-mouse GBM globulin. Antigen-specific mouse IgG subclass analyses demonstrated no difference in IgG1 subclass production; WT mice produced more of the Th1 produced IgG subclasses, IgG2c and IgG3, than TLR9−/− mice (Figure 6, B–D). It is unlikely that the enhanced humoral responses resulted in increased renal injury. Using a semiquantative scoring system, we analyzed kidney sections by direct immunofluorescence for glomerular IgG and complement, C3 deposition. Decreased glomerular IgG deposition was seen in WT mice compared with TLR9−/− mice (Figure 7, A–C), whereas no difference in glomerular C3 deposition was seen between the groups (Figure 7, D–F).
Figure 6.

Antigen-specific serum antibody titers measured in WT and TLR9−/− mice 21 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin. Serum antigen-specific IgG (A) was increased in WT mice compared with TLR9−/− mice. Analysis of subclass titers showed no change in IgG1 (B) between the groups, whereas IgG2c (C) and IgG3 (D) levels were decreased in TLR9−/− mice. *P < 0.05; **P < 0.01.
Figure 7.
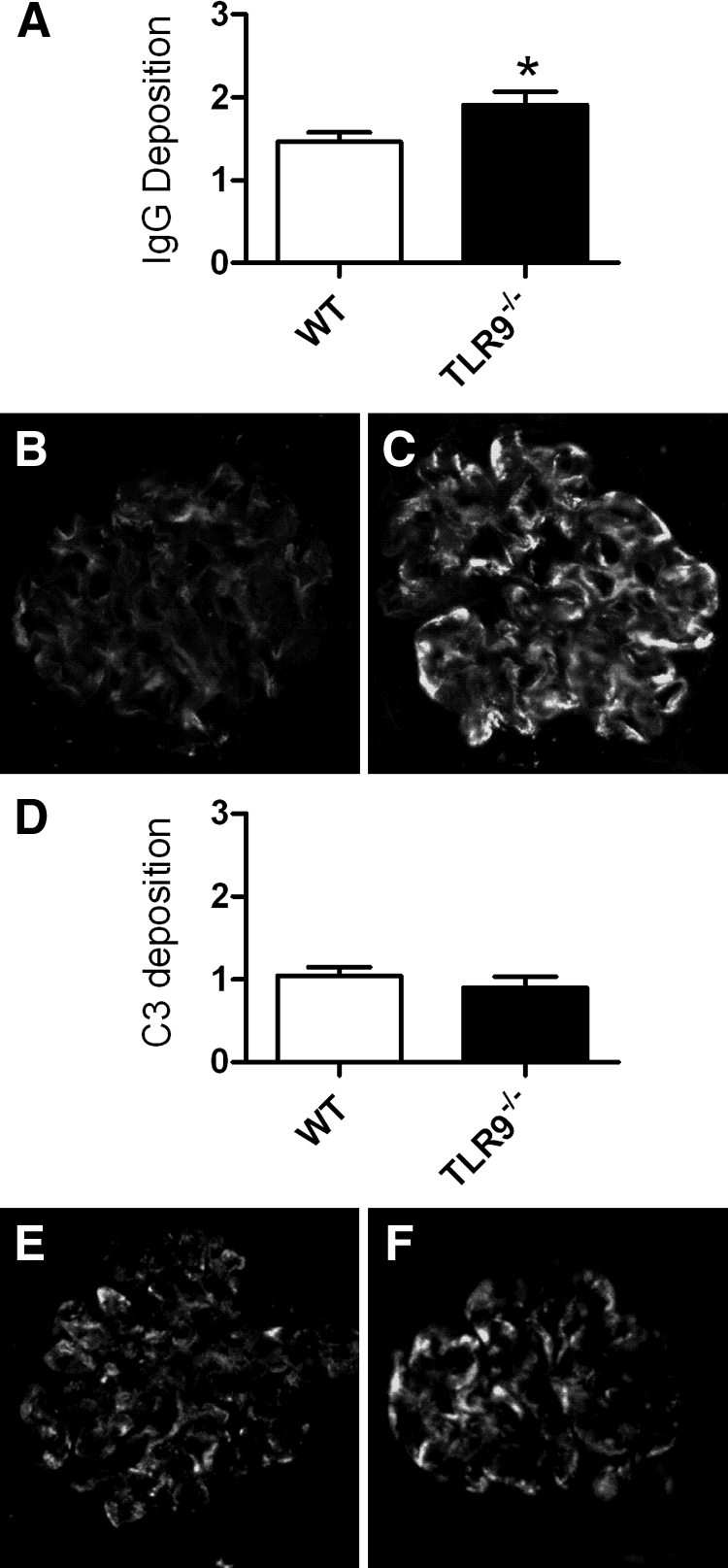
Glomerular IgG and complement, C3, deposition analyzed by direct immunofluorescence 21 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin. Semiquantative analysis of glomerular IgG deposition demonstrated less intense IgG staining in WT mice (A), representative glomeruli from WT (B), and TLR9−/− (C) mice are shown. There was no difference in glomerular C3 staining intensity (D); representative glomeruli from WT (E) and TLR9−/− (F) mice are shown. *P < 0.05.
Pre-Emptive Administration of a Specific TLR9 Inhibitor Decreases Nephritogenic Immunity and Protects Mice from Experimental Crescentic GN
To assess if an inhibitory nucleotide (TLR9 inhibitor) could limit renal injury attributable to CpG-ODN and anti-GBM globulin, we injected mice with immunoregulatory sequence (IRS869) or PBS (control mice) before administration of CpG-ODN and sheep anti-mouse GBM globulin. Subsequently, we administered the IRS869 (or control) twice weekly; experiments ended after 21 days. Glomerular crescentic injury, the proportion of severely abnormal glomeruli and tubulointerstitial injury, was attenuated in mice treated with IRS869 compared with control mice (Figure 8, A–C). Renal function assessed by 24-hour urine proteinuria and serum creatinine decreased with administration of TLR9 inhibitor (Figure 8, D and E), with a trend to decrease in serum BUN (Figure 8F). Representative sections demonstrating kidney injury in mice treated with IRS869 or control are shown at high and low power (Figure 8, G–J).
Figure 8.

Histological injury and renal function in WT mice pre-emptively administered control or TLR9 inhibitor followed by administration of CpG-ODN and sheep anti-mouse glomerular basement membrane globulin. Pre-emptive administration of TLR9 inhibitor to WT mice (n = 8) attenuated histological renal injury with fewer glomerular crescents (A), proportionally less severely abnormal glomeruli (B), and less interstitial injury (C) than control mice (n = 8). Renal function as assessed by 24-hour protein excretion (D), and serum creatinine (E) was also decreased after pre-emptive administration of a TLR9 inhibitor, whereas serum BUN (F) demonstrated a trend to decrease. *P < 0.05; **P < 0.01. Representative kidney sections from WT mice treated with PBS or TLR9 inhibitor before administration of CpG-ODN and sheep anti-mouse glomerular basement membrane globulin. Representative high power (original magnification, ×400) sections show severe crescentic injury with fibrinoid necrosis with prior administration of control (G), whereas attenuated glomerular injury was seen with pre-emptive administration of TLR 9 Inhibitor (H). Representative photomicrographs of the interstitial injury (original magnification, ×200) demonstrate that the marked injury seen after treatment with PBS (I) decreased with pre-emptive TLR9 inhibition (J).
Assessment of antigen stimulated splenocyte cytokine production demonstrated a decrease in IFN-γ and IL-17A nephritogenic systemic immune responses after the administration of IRS869 (Figure 9, A and B) There was no change in humoral immune responses between the IRS869 and control treated groups (Figure 9, C–F). After direct immunofluorescent staining of glomeruli for complement and antibody deposition, no difference was detectable in glomerular IgG or C3 deposition (data not shown).
Figure 9.

Systemic immune responses and antigen-specific serum antibody titers in WT mice treated with control or TLR9 inhibitor before CpG-ODN and sheep anti-mouse glomerular basement membrane globulin. On day 21, antigen-stimulated production of IFN-γ (A) and IL-17A (B) were decreased with pre-emptive administration of a TLR9 inhibitor. There was no change in serum antigen specific IgG titers (C) or in the IgG subclasses: IgG1 (D), IgG2c (E), or IgG3 (F). *P < 0.05; **P < 0.01.
Administration of a TLR9 Inhibitor from Day 7, but Not from Day 14, After Disease Initiation Partially Protects Mice from Experimental Crescentic GN
To determine whether TLR9 inhibition could ameliorate renal injury after disease initiation, we administered IRS869 (or control) 7 days after the administration of CpG-ODN and sheep anti-mouse GBM globulin; experiments ended after 21 days. Although this protocol resulted in a nonsignificant decrease in glomerular crescent formation, the number of severely abnormal glomeruli and interstitial injury was decreased in mice treated with IRS869 (Figure 10, A–C). There was a nonsignificant change in 24-hour urinary protein, but serum creatinine and BUN were decreased in mice given IRS869 (Figure 10, D–F). Splenocyte production of IFN-γ (control, 601 ± 201 pg/ml; TLR9 inhibitor, 298 ± 87 pg/ml; P = 0.2) and IL17A (control, 177 ± 75 pg/ml; TLR9 inhibitor, 85 ± 35 pg/ml; P = 0.3) was not different between the two groups. Glomerular leukocyte recruitment was decreased in mice treated with the TLR9 inhibitor. In addition to decreased glomerular macrophages (1.5 ± 0.1 vs. 1.0 ± 0.1 cells/glomerular cross section; P < 0.01), glomerular CD4+ T cells (1.2 ± 0.2 vs. 0.6 ± 0.1 cells/glomerular cross section; P < 0.05) were also decreased. Administration of a TLR9 inhibitor 14 days after initiation of disease did not alter the progression of histological renal injury or renal function (Figure 10, G–L).
Figure 10.

Histological injury and renal function in WT mice treated with control or TLR9 inhibitor 7 and 14 days after the administration of CpG-ODN and sheep anti-mouse glomerular basement membrane globulin. Analysis of kidney sections on day 21 after administering the TLR9 inhibitor on day 7 demonstrated a trend to decrease in glomerular crescent formation (A) with significantly fewer severely abnormal glomeruli (B), and significantly less interstitial injury (C). Assessment of renal function showed no significant change in 24-hour protein excretion (D); however, serum creatinine and BUN was decreased (E and F); n = 8 for both experimental groups. Administration of the TLR9 inhibitor 14 days after sheep anti-mouse glomerular basement membrane globulin did not decrease histological renal injury (G–I) or renal function (J–L); n = 10 for both experimental groups. *P < 0.05; **P < 0.01.
Bone Marrow Cells are the Major Contributors to TLR9 Induced Crescentic GN
We defined the relative contributions of BM and TC TLR9 expression in renal injury induced by injecting CpG-ODN and sheep anti-mouse GBM globulin into TLR9 bone marrow chimeric mice. Chimeric mice were generated by injecting intact or deficient BM into irradiated mice. WT BM transplanted into WT mice (BM+TC+; “sham” chimeras) were a positive control. TLR9 tissue cell (TC) intact, BM TLR9 deficient (BM-TC+) and TC TLR9 deficient, BM intact (BM+TC−) chimeras were studied. Compared with BM+TC+ mice, crescentic and interstitial injury were decreased in BM+TC− mice, whereas all histological parameters were decreased in BM-TC+ mice (Figure 11, A–C). Functional injury assessed by proteinuria was decreased in BM-TC+ mice compared with BM+TC+ mice (Figure 11D). Although there was no change in serum creatinine (BM+TC+, 24.0 ± 0.8 μmol/L; BM-TC+, 23.8 ± 1.3 μmol/L; BM+TC− 24.6 ± 1.2 μmol/L), there was a trend to decrease in BUN in BM-TC+ mice (BM+TC+, 14.1 ± 1.9 mmol/L; BM-TC+, 9.6 ± 0.6 mmol/L; BM+TC−, 13.3 ± 1.8 mmol/L).
Figure 11.

Assessment of histological renal injury and renal function in BM chimeric mice, WT → WT mice (BM+, TC+, n = 10), TLR9−/− → WT (BM-TC+, n = 10), and WT→ TLR9−/− mice (BM+TC−, n = 8). Compared with BM+TC+ “sham” chimeras, histological renal injury decreased in both sets of chimeras, with a more pronounced decrease in injury observed in BM-TC+ mice (A–C). Proteinuria was decreased in BM-TC+ mice (D) compared with BM+TC+ mice. *P < 0.05; **P < 0.01; ***P < 0.001.
These results suggest that both BM and TC TLR9 are required for maximal renal injury, but BM TLR9 plays the predominant role.
Discussion
TLRs link innate and adaptive immune systems making them attractive therapeutic targets in inflammatory diseases. In the current set of experiments, we have shown how TLR9 ligation enhances glomerular inflammation through the recruitment of key cellular effectors resulting in severe renal injury in autologous phase anti-GBM GN. TLR9 specificity of the ligand was confirmed by demonstrating that administration of the ligand (and sheep anti-mouse GBM globulin) to TLR9−/− mice had no effect on immune responses or disease outcome. Pre-emptive treatment with a TLR9 inhibitor decreased cellular nephritogenic immunity, attenuating renal injury, whereas late treatment decreased glomerular leukocyte recruitment and resultant injury. These results highlight the therapeutic potential of TLR9 inhibition in severe crescentic GN.
TLRs are involved in several experimental models of immune mediated kidney injury. These include TLR2 and TLR4 in experimental crescentic GN,14,15,16 and TLR9 in experimental immune complex GN,30 IgA nephropathy31 where injurious roles were demonstrated and lupus nephritis, where injurious19 and protective roles are described.20 In the current studies, TLR9 ligation (in WT mice) was required for full expression of Th1 and Th17 nephritogenic systemic immunity resulting in glomerular inflammation with severe renal injury.
In experimental planted antigen models of GN, CD4+ T cells are involved in both the initiation of the nephritogenic immune response32 and the effector phase of disease.33 Alterations in the balance of Th1/Th2 CD4+ T cell phenotype are responsible for different patterns and effects in glomerular inflammation and injury.7 The absence of either the key Th1 defining cytokine chain, IL-12p40,25 or Th1 transcription factor, T-bet,27 attenuates renal injury, whereas administering IL-12, the key Th1 cytokine, enhances renal injury,34 confirming the role of Th1 cell polarization in severe glomerular and renal injury. Recently, Th17 cells have been implicated in experimental immune-mediated renal disease. In response to a foreign glomerular antigen, Th17 cells induced proliferative GN with impaired renal function.35 Furthermore, IL-23 and IL-17 are required for initiation and progression of disease in a model of experimental crescentic GN.10 A protective role for IL-4 and IL-10, the key Th2 cytokines, has been demonstrated in this model.9 We found that after administration of CpG-ODN and sheep anti-mouse GBM globulin, WT mice developed the hallmarks of classical cellular (Th1 and Th17) mediated nephritogenic immune responses with enhanced glomerular crescent formation, increased CD4+ T cell recruitment, increased expression of the pro-inflammatory renal injurious cytokines (TNF24 and IL-1β36), and decreased renal function. Th1 specific mediated responses included the following: increased DTH, increased IFN-γ production with increased glomerular macrophages, and enhanced expression of CCL5 and CXCL9 in kidneys of WT mice. Evidence of Th17-mediated responses included increased systemic IL-17A (and IL-6) with increased Rorγ expression in the kidneys of WT mice. Therefore, we concluded that CpG-ODN and sheep anti-mouse GBM globulin induced nephritogenic Th1 and Th17 responses resulting in severe kidney injury.
TLR9 is expressed on B cells, monocytes, and dendritic cells, and recently TLR9 expression has been identified on T cells.37 CpG-ODN stimulation of TLR9 receptors on B lymphocytes can induce humoral immunity,6 and in experimental lupus nephritis TLR9 is required for complete autoantibody production.38 The role of antibodies in experimental crescentic GN is less clear; experiments in antibody deficient mice (mμ chain deficient) demonstrated full expression of disease can progress without antibody.33 We found that CpG-ODN stimulation enhanced serum sheep anti-mouse globulin titers, an increase most pronounced in Th1-driven IgG2c and IgG3 subclasses, which fix complement and facilitate glomerular injury.39 The increased serum IgG did not result in increased glomerular IgG deposition, which was surprisingly decreased in WT mice. C3 deposition was not different between WT and TLR9−/− mice suggesting that antibody production and deposition did not influence severity of renal injury, highlighting the predominant role of cellular effector mechanisms and glomerular leukocytes in the induction of crescentic nephritis in experimental anti-GBM GN.
Therapies to treat human crescentic GN have improved renal and patient outcomes but are still associated with considerable morbidity and mortality. Selective targeting of key molecules involved in disease pathogenesis offers the potential reward of attenuating organ injury with fewer adverse effects. The newer specific TLR9 inhibitor, IRS869, successfully limits inflammatory cytokine production in both human and mouse cells23 and holds promise for treatment of human autoinflammatory diseases. We hypothesized that the TLR9 inhibitor would limit renal injury attributable to TLR9 ligation. After pre-emptive administration of IRS869, we found Th1 and Th17 cytokine production was decreased, and renal injury was attenuated, with no change in humoral responses. These results suggested a partial inhibition of TLR9 activity, predominantly inhibiting cell mediated immunity. In addition to enhancing adaptive immune responses, TLR9 ligation is known to induce leukocyte recruitment and activate macrophages,5 the key glomerular effector cell in this model. Administering IRS869 after disease initiation, on day 7, decreased glomerular leukocyte recruitment, which resulted in attenuated renal injury. When the inhibitor was administered on day 14, injury was not attenuated, presumably because immune responses were established at this stage. These results highlight the potential of TLR9 inhibition, if used early in the disease process to decrease the progression of established renal injury.
Using bone marrow chimeric mice, we demonstrated that both bone marrow and tissue cell TLR9 were required for maximal histological renal injury, although bone marrow TLR9 was more important. This is consistent with previous studies demonstrating the importance of immune cells in the progression of TLR- mediated experimental lupus injury.40
In conclusion, we have shown that TLR9 ligation enhances cellular and humoral responses increasing glomerular leukocyte recruitment and exacerbating renal injury in autologous phase anti-GBM GN. Injury is predominantly mediated by bone marrow TLR9. The administration of a TLR9 inhibitor led to attenuation in renal injury mediated through decreased production of Th1 and Th17 nephritogenic immune responses and decreased recruitment of glomerular leukocytes. These studies confirm and significantly extend results seen in other experimental studies highlighting a potential therapeutic role for TLR9 inhibition in treatment of autoimmune diseases.
Acknowledgments
We thank Ms. Alice Wright and Sophia Ling for technical assistance.
Footnotes
Address reprint requests to Stephen R. Holdsworth, M.B.B.S., F.R.A.C.P., Ph.D., M.D., Centre for Inflammatory Diseases, Monash University Department of Medicine, Monash Medical Centre, 246 Clayton Rd, Clayton, VIC 3168, Australia. E-mail: stephen.holdsworth@med.monash.edu.au.
Supported by a program grant (S.R.H. and A.R.K.; 334067) and a postgraduate research scholarship (S.A.S. 519426) from the National Health and Medical Research Council of Australia.
Parts of this work were published in abstract form (program and abstracts of the American Society of Nephrology, 2009 Annual Meeting, October 27 to November 1, 2009; San Diego, CA: abstract TH-PO824).
None of the authors disclosed any relevant financial relationships.
References
- Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007;220:251–269. doi: 10.1111/j.1600-065X.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- Krieg AM. A role for Toll in autoimmunity. Nat Immunol. 2002;3:423–424. doi: 10.1038/ni0502-423. [DOI] [PubMed] [Google Scholar]
- Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. 2000;1:305–310. doi: 10.1038/79747. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Holdsworth SR, Kitching AR, Tipping PG. Th1 and Th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int. 1999;55:1198–1216. doi: 10.1046/j.1523-1755.1999.00369.x. [DOI] [PubMed] [Google Scholar]
- Tipping PG, Holdsworth SR. T cells in crescentic glomerulonephritis. J Am Soc Nephrol. 2006;17:1253–1263. doi: 10.1681/ASN.2005091013. [DOI] [PubMed] [Google Scholar]
- Kitching AR, Tipping PG, Huang XR, Mutch DA, Holdsworth SR. Interleukin-4 and interleukin-10 attenuate established crescentic glomerulonephritis in mice. Kidney Int. 1997;52:52–59. doi: 10.1038/ki.1997.303. [DOI] [PubMed] [Google Scholar]
- Paust HJ, Turner JE, Steinmetz OM, Peters A, Heymann F, Holscher C, Wolf G, Kurts C, Mittrucker HW, Stahl RA, Panzer U. The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J Am Soc Nephrol. 2009;20:969–979. doi: 10.1681/ASN.2008050556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook HT, Singh SJ, Wembridge DE, Smith J, Tam FW, Pusey CD. Interleukin-4 ameliorates crescentic glomerulonephritis in Wistar Kyoto rats. Kidney Int. 1999;55:1319–1326. doi: 10.1046/j.1523-1755.1999.00354.x. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, Bonventre JV, Hughes J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson MG. Toll-like receptors and renal disease. Nephron Exp Nephrol. 2009;113:e1–e7. doi: 10.1159/000228077. [DOI] [PubMed] [Google Scholar]
- Brown HJ, Lock HR, Sacks SH, Robson MG. TLR2 stimulation of intrinsic renal cells in the induction of immune-mediated glomerulonephritis. J Immunol. 2006;177:1925–1931. doi: 10.4049/jimmunol.177.3.1925. [DOI] [PubMed] [Google Scholar]
- Brown HJ, Lock HR, Wolfs TG, Buurman WA, Sacks SH, Robson MG. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J Am Soc Nephrol. 2007;18:1732–1739. doi: 10.1681/ASN.2006060634. [DOI] [PubMed] [Google Scholar]
- Brown HJ, Sacks SH, Robson MG. Toll-like receptor 2 agonists exacerbate accelerated nephrotoxic nephritis. J Am Soc Nephrol. 2006;17:1931–1939. doi: 10.1681/ASN.2005111167. [DOI] [PubMed] [Google Scholar]
- Fu Y, Xie C, Chen J, Zhu J, Zhou H, Thomas J, Zhou XJ, Mohan C. Innate stimuli accentuate end-organ damage by nephrotoxic antibodies via Fc receptor and TLR stimulation and IL-1/TNF-alpha production. J Immunol. 2006;176:632–639. doi: 10.4049/jimmunol.176.1.632. [DOI] [PubMed] [Google Scholar]
- Pawar RD, Patole PS, Ellwart A, Lech M, Segerer S, Schlondorff D, Anders HJ. Ligands to nucleic acid-specific toll-like receptors and the onset of lupus nephritis. J Am Soc Nephrol. 2006;17:3365–3373. doi: 10.1681/ASN.2006030263. [DOI] [PubMed] [Google Scholar]
- Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, Strutz F, Bauer S, Rutz M, Wagner H, Grone HJ, Schlondorff D. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J. 2004;18:534–536. doi: 10.1096/fj.03-0646fje. [DOI] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Zeuner RA, Ishii KJ, Lizak MJ, Gursel I, Yamada H, Klinman DM, Verthelyi D. Reduction of CpG-induced arthritis by suppressive oligodeoxynucleotides. Arthritis Rheum. 2002;46:2219–2224. doi: 10.1002/art.10423. [DOI] [PubMed] [Google Scholar]
- Dong L, Ito S, Ishii KJ, Klinman DM. Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus-prone NZB × NZW mice. Arthritis Rheum. 2005;52:651–658. doi: 10.1002/art.20810. [DOI] [PubMed] [Google Scholar]
- Duramad O, Fearon KL, Chang B, Chan JH, Gregorio J, Coffman RL, Barrat FJ. Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflammation. J Immunol. 2005;174:5193–5200. doi: 10.4049/jimmunol.174.9.5193. [DOI] [PubMed] [Google Scholar]
- Timoshanko JR, Sedgwick JD, Holdsworth SR, Tipping PG. Intrinsic renal cells are the major source of tumor necrosis factor contributing to renal injury in murine crescentic glomerulonephritis. J Am Soc Nephrol. 2003;14:1785–1793. doi: 10.1097/01.asn.0000073902.38428.33. [DOI] [PubMed] [Google Scholar]
- Kitching AR, Turner AL, Wilson GR, Semple T, Odobasic D, Timoshanko JR, O'Sullivan KM, Tipping PG, Takeda K, Akira S, Holdsworth SR. IL-12p40 and IL-18 in crescentic glomerulonephritis: IL-12p40 is the key Th1-defining cytokine chain, whereas IL-18 promotes local inflammation and leukocyte recruitment. J Am Soc Nephrol. 2005;16:2023–2033. doi: 10.1681/ASN.2004121075. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Pusey CD. Oral administration of glomerular basement membrane prevents the development of experimental autoimmune glomerulonephritis in the WKY rat. J Am Soc Nephrol. 2001;12:61–70. doi: 10.1681/ASN.V12161. [DOI] [PubMed] [Google Scholar]
- Phoon RK, Kitching AR, Odobasic D, Jones LK, Semple TJ, Holdsworth SR. T-bet deficiency attenuates renal injury in experimental crescentic glomerulonephritis. J Am Soc Nephrol. 2008;19:477–485. doi: 10.1681/ASN.2007030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, O'Sullivan KM, Jones LK, Lo C, Semple T, Kumanogoh A, Kikutani H, Holdsworth SR, Kitching AR. Endogenous CD100 promotes glomerular injury and macrophage recruitment in experimental crescentic glomerulonephritis. Immunology. 2009;128:114–122. doi: 10.1111/j.1365-2567.2009.03098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odobasic D, Kitching AR, Semple TJ, Timoshanko JR, Tipping PG, Holdsworth SR. Glomerular expression of CD80 and CD86 is required for leukocyte accumulation and injury in crescentic glomerulonephritis. J Am Soc Nephrol. 2005;16:2012–2022. doi: 10.1681/ASN.2004060437. [DOI] [PubMed] [Google Scholar]
- Anders HJ, Banas B, Linde Y, Weller L, Cohen CD, Kretzler M, Martin S, Vielhauer V, Schlondorff D, Grone HJ. Bacterial CpG-DNA aggravates immune complex glomerulonephritis: role of TLR9-mediated expression of chemokines and chemokine receptors. J Am Soc Nephrol. 2003;14:317–326. doi: 10.1097/01.asn.0000042169.23931.73. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Suzuki Y, Narita I, Aizawa M, Kihara M, Yamanaka T, Kanou T, Tsukaguchi H, Novak J, Horikoshi S, Tomino Y. Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol. 2008;19:2384–2395. doi: 10.1681/ASN.2007121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping PG, Huang XR, Qi M, Van GY, Tang WW. Crescentic glomerulonephritis in CD4- and CD8-deficient mice: requirement for CD4 but not CD8 cells. Am J Pathol. 1998;152:1541–1548. [PMC free article] [PubMed] [Google Scholar]
- Li S, Holdsworth SR, Tipping PG. Antibody independent crescentic glomerulonephritis in mu chain deficient mice. Kidney Int. 1997;51:672–678. doi: 10.1038/ki.1997.97. [DOI] [PubMed] [Google Scholar]
- Kitching AR, Tipping PG, Holdsworth SR. IL-12 directs severe renal injury, crescent formation, and Th1 responses in murine glomerulonephritis. Eur J Immunol. 1999;29:1–10. doi: 10.1002/(SICI)1521-4141(199901)29:01<1::AID-IMMU1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Summers SA, Steinmetz OM, Li M, Kausman JY, Semple T, Edgtton KL, Borza DB, Braley H, Holdsworth SR, Kitching AR. Th1 and Th17 cells induce proliferative glomerulonephritis. J Am Soc Nephrol. 2009;20:2518–2524. doi: 10.1681/ASN.2009030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoshanko JR, Kitching AR, Iwakura Y, Holdsworth SR, Tipping PG. Contributions of IL-1beta and IL-1alpha to crescentic glomerulonephritis in mice. J Am Soc Nephrol. 2004;15:910–918. doi: 10.1097/01.asn.0000115704.86897.f4. [DOI] [PubMed] [Google Scholar]
- Chiffoleau E, Heslan JM, Heslan M, Louvet C, Condamine T, Cuturi MC. TLR9 ligand enhances proliferation of rat CD4+ T cell and modulates suppressive activity mediated by CD4+ CD25+ T cell. Int Immunol. 2007;19:193–201. doi: 10.1093/intimm/dxl136. [DOI] [PubMed] [Google Scholar]
- Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudino L, Azeredo da Silveira S, Nakata M, Izui S. Molecular and cellular basis for pathogenicity of autoantibodies: lessons from murine monoclonal autoantibodies. Springer Semin Immunopathol. 2006;28:175–184. doi: 10.1007/s00281-006-0037-0. [DOI] [PubMed] [Google Scholar]
- Patole PS, Pawar RD, Lech M, Zecher D, Schmidt H, Segerer S, Ellwart A, Henger A, Kretzler M, Anders HJ. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol Dial Transplant. 2006;21:3062–3073. doi: 10.1093/ndt/gfl336. [DOI] [PubMed] [Google Scholar]