

Abstract
Rationale
The mobilization of bone-marrow (BM) progenitor cells (PCs) is largely governed by interactions between stromal–cell derived factor 1 (SDF-1) and CXC-chemokine receptor 4 (CXCR4). Ischemic injury disrupts the SDF-1–CXCR4 interaction and releases BM PCs into the peripheral circulation, where the mobilized cells are recruited to the injured tissue and contribute to vessel growth. BM PCs can also be mobilized by the pharmacological CXCR4 antagonist AMD3100, but the other components of the SDF-1–CXCR4 signaling pathway are largely unknown. c-kit, a membrane bound tyrosine-kinase and the receptor for stem cell factor, has also been shown to play a critical role in BM PC mobilization and ischemic tissue repair.
Objective
To investigate the functional interaction between SDF-1–CXCR4 signaling and c-kit activity in BM PC mobilization.
Methods and Results
AMD3100 administration failed to mobilize BM PCs in mice defective in c-kit kinase activity or in mice transplanted with BM cells that expressed a constitutively active c-kit mutant. Furthermore, BM levels of phosphorylated c-kit (phospho–c-kit) declined after AMD3100 administration and after CXCR4 deletion. In cells adhering to culture plates coated with vascular cell adhesion molecule 1 (VCAM-1), SDF-1 and SCF increased phospho–c-kit levels, and AMD3100 treatment suppressed SDF-1–induced, but not SCF-induced, c-kit phosphorylation. SDF-1–induced c-kit phosphorylation also required the activation of Src non-receptor tyrosine kinase: pre-treatment of cells with a selective Src inhibitor blocked both c-kit phosphorylation and the interaction between c-kit and phosphorylated Src.
Conclusions
These findings indicate that the regulation of BM PC trafficking by SDF-1 and CXCR4 is dependent on Src-mediated c-kit phosphorylation.
Keywords: CXCR4, c-kit, Integrin, Stem cells, Bone marrow, Niche, Mobilization, Homing
Introduction
The mobilization or release of progenitor cells (PCs) from the bone marrow (BM) to the peripheral blood (PB) is highly regulated and occurs both under normal conditions and in response to stress 1-2. PB PCs have an essential role in blood-cell formation and homeostasis 3 and in the response of various tissues to noxious insults. Thus, therapies that enhance PC mobilization are being investigated as novel strategies for promoting tissue repair 4-5. Ample evidence suggests that PCs are retained in the BM by interactions between the CXC chemokine stromal-cell–derived factor 1 (SDF-1) and CXC chemokine receptor 4 (CXCR4) 6-10. CXCR4 is expressed by BM mononuclear cells (MNCs), and SDF-1 is expressed by osteoblasts, endothelial cells, and a subset of reticular cells scattered throughout the BM 7. Ischemic injury disrupts the SDF-1–CXCR4 interaction and releases BM PCs into the peripheral circulation, then the mobilized cells are recruited to the injured tissue and contribute to vessel growth 11-12. BM PCs can also be mobilized by the pharmacological CXCR4 antagonist AMD3100 13-15, but the downstream components of the SDF-1–CXCR4 signaling pathway are largely unknown.
Like CXCR4, c-kit is expressed predominantly in BM PCs, and the ligand for c-kit, stem cell factor (SCF), is constitutively produced by BM endothelial cells and fibroblasts 11. c-kit is a class III receptor tyrosine kinase, and administration of a c-kit–neutralizing antibody (ACK2) to wild-type (WT) mice released BM cells to the peripheral circulation and enhanced the engraftment of intravenously injected BM cells 12. On the other hand, PC mobilization is markedly blunted in c-kitW/W-V mutant mice, which are defective in c-kit kinase activity but have normal levels of c-kit expression and SCF binding at the cell surface 17-20. Furthermore, the kinetics of BM PC mobilization induced by the c-kit–neutralizing antibody and by the antagonism of CXCR4 with AMD3100 were similar. Collectively, these findings suggest that SCF–c-kit signaling participates in BM PC mobilization and trafficking 22-23, but whether the SDF-1–CXCR4 and SCF–c-kit pathways interact is unknown.
Here, we show that SDF-1–CXCR4 signaling is mediated, at least in part, by c-kit phosphorylation. AMD3100 administration failed to mobilize BM PCs in c-kitW/W-V mice or in mice transplanted with BM cells that expressed a constitutively active c-kit mutant, and BM levels of phosphorylated c-kit (phospho–c-kit) declined after both AMD3100 administration and induction of a CXCR4-knockout mutation. In cells adhering to plates coated with vascular cell adhesion molecule 1 (VCAM-1), SDF-1 and SCF increased phospho–c-kit levels, and AMD3100 treatment suppressed SDF-1–induced, but not SCF-induced, c-kit phosphorylation. Furthermore, pre-treatment of cells with a selective Src inhibitor blocked both c-kit phosphorylation and the interaction between c-kit and phosphorylated Src. Collectively, these results suggest that the regulation of BM PC trafficking by SDF-1 and CXCR4 is linked to Src-mediated c-kit phosphorylation.
Methods
Mice
c-kitw/w-v (WBB6F1/J-KitW/KitW-V/J) and WT mice (Male, 10-12 weeks old) were purchased from the Jackson Laboratory. CXCR4fl/fl and CXCR4BAC-GFP transgenic mice were provided by Dr. Richard J. Miller (Northwestern University) 13-14. The Mx1-cre+CXCR4fl/fl mice were bred by mating CXCR4fl/fl and Mx1-cre (The Jackson Laboratory) mice, and CXCR4 gene deletion was induced with 3 intraperitoneal injections of 250 μg poly(I)-poly(C) (GE Healthcare) administered at 2-day intervals 15. All surgical procedures were approved by Northwestern University's Institutional Animal Care and Use Committee.
Isolation of BM and PB MNCs
BM and PB MNCs were isolated as described previously 15 and as detailed in the Online Methods section.
PC colony-forming assay
PB and BM MNCs were isolated, and the PC colony-forming assay was performed in 35-mm dishes with a semi-solid methylcellulose medium containing SCF and other recombinant cytokines (MethoCult GF M3434, StemCell Technologies, Canada) as directed by the manufacturer's protocol; 2×105 PB MNCs and 2×104 BM MNCs were seeded in each dish, and colonies were counted 12 days later. To ensure that c-kitw/w-v PC levels would not be underestimated, Flt3-ligand (R&D Systems, final concentration 100 ng/mL) was added to the medium to compensate for cells that may not respond well to SCF.
BM clearance/repopulation assay
AMD3100 (5 mg/kg) was subcutaneously injected into WT and c-kitw/w-v mice to mobilize PCs from the BM. Two hours later 16, 40×106 BM MNCs that had been isolated from eGFP-transgenic mice (C57BL/6-Tg[ACTB-eGFP], The Jackson Laboratory) were injected into the tail vein and allowed to repopulate the BM for 3 hours 28, then BM MNCs were isolated from the recipients, and eGFP expression was evaluated by flow-cytometry.
Flow cytometry
Flow cytometry analyses of gene expression in isolated BM and PB MNCs were performed as previously described 15.
Transplantation of retrovirally transduced c-kitWT and c-kitD816V BM MNCs
MIG-HyKITWT and MIG- HyKITD816V retroviral vectors were generated by co-transfecting 293FT cells with a packaging plasmid, pIK6.1 MCV.ecopac.UTd, and one of two backbone plasmids: pMSCV-HyKITWT-IRES-eGFP (pMIG-HyKITWT), or pMSCV-HyKITD816V-IRES-eGFP (pMIG- HyKITD816V) 17. Methods for virus generation, BM MNC isolation and ex vivo expansion, viral infection and BM transplantation are described in Online Methods section.
Tissue sectioning and immunofluorescent staining
Mice were subcutaneously injected with AMD3100 (5 mg/kg) or PBS and sacrificed 15 minutes later. Femurs were harvested, fixed, and immunofluorescently stained as described in Online Methods section.
Cell adhesion assay
The BM-MNC adhesion assay was performed as described previously 15.
Western blotting and co-immunoprecipitation
The Western blotting and co-immunoprecipitation was performed as previously described 18-19, and further details can be found in Online Methods section.
Quantitative real-time RT-PCR
Real time RT-PCR was performed via standard techniques20 as detailed in the Online Methods section.
Statistics
Data are presented as mean ± SEM. Comparisons between 2 means were assessed for significance with the unpaired Student's t test; comparisons between 3 or more means were assessed for significance via ANOVA and Bonferroni MCP. Statistical significance was assigned if P<0.05.
Results
AMD3100-induced PC mobilization is impaired in c-kitw/w-v mice
We initiated our investigation into the potential involvement of c-kit in CXCR4-mediated BM PC trafficking by quantifying the levels of colony forming PCs in the peripheral blood (PB) and BM of WT mice and mice defective in the kinase activity of c-kit (c-kitw/w-v mice). PB PC levels significantly increased, and BM PC levels significantly declined, 2 hours after AMD3100 injection in WT mice but not in c-kitw/w-v mice (Figures 1A-B). To determine whether the impaired mobilization observed in c-kitw/w-v mice was limited to specific subpopulation(s) of PCs, we developed a short-term, in vivo BM clearance/repopulation assay. WT and c-kitw/w-v mice were treated with or without AMD3100, then 40×106 BM MNCs from eGFP-transgenic mice were intravenously injected 2 hours later 16, 21. Three hours after cell injection 22, MNCs were isolated from the BM, and flow cytometry analysis was performed for eGFP expression, to identify injected cells that had repopulated the BM, and for the expression of CXCR4, c-kit, Lin, and Sca-1. In the absence of AMD3100 treatment, eGFP expression was negligible (<0.01% of cells) in BM MNCs from both c-kitw/w-v and WT mice. In AMD3100-treated mice, the proportion of BM MNCs that expressed eGFP did not differ significantly between strains (Figure 1C) and neither did the proportion of eGFP+ MNCs that coexpressed CXCR4 (Figure 1D), Lin (Figure 1E), or the proportion of eGFP+Lin− cells. However, the proportion of eGFP+Lin− cells that co-expressed CXCR4 (c-kitw/w-v: 16.3%, WT: 30.6%; P<0.01), Sca-1 and CXCR4 (c-kitw/w-v: 16.6%, WT: 29.7%; P<0.01), or c-kit and CXCR4 (c-kitw/w-v: 5.7%, WT: 17.8%; P<0.001) was significantly lower in c-kitw/w-v mice than in WT mice (Figures 1F). These significant differences could occur if the c-kitW/W-V mutation impaired the recruitment of PCs to the BM, but expression of the PC chemoattractant SDF-1 was similar in both strains (Online Figure I). Thus, the c-kitW/W-V mutation appeared to block the mobilization of Lin−CXCR4+ cells that co-expressed Sca1 or c-kit, and because the Lin−Sca1+ and Lin−c-kit+ phenotypes are typically associated with PC identity, these observations suggest that a loss of c-kit kinase activity impairs the AMD3100-induced mobilization of CXCR4+ PCs.
Figure 1. AMD3100-induced PC mobilization is impaired in c-kitw/w-v mice.

(A-B) WT and c-kitW/W-V mice received subcutaneous injections of AMD3100 (5 mg/kg) or PBS; 2 hours later, the numbers of colony-forming PCs in the PB-MNC (A) and BM-MNC (B) populations were evaluated via the colony-forming assay. (C-F) In the BM clearance/repopulation assay, WT and c-kitw/w-v mice were treated with AMD3100 for 2 hours to mobilize endogenous PCs from the BM, then 40×106 eGFP-transgenic BM MNCs were injected intravenously to compete with the endogenous PCs for repopulation of the BM. Three hours later, BM MNCs were isolated from the recipients and analyzed via flow cytometry to determine the proportion of injected (eGFP+) cells in the total BM MNC population (C), the proportion of eGFP+ BM MNCs that were CXCR4+ (D), Lin+, or Lin− (E), and the proportion of eGFP+Lin− BM MNCs that were CXCR4+, Sca-1+CXCR4+ and c-kit+CXCR4+ (F). Values are mean ± SEM; n=10 per group.
AMD3100 fails to mobilize PCs that express a constitutively-active c-kit mutant (c-kitD816V)
Because AMD3100-induced BM PC mobilization is impaired in c-kit–kinase defective mice, we investigated whether increasing the level of activated c-kit would enhance AMD3100-induced mobilization. WT BM MNCs were transfected with retroviral vectors coding for eGFP and WT c-kit (c-kitWT) or eGFP and a constitutively active c-kit mutant (c-kitD816V) 17, 23 (Figures 2 A-B), then transplanted into WT mice after lethally irradiating the endogenous BM. Three weeks later, the numbers of BM MNCs in the two recipient strains were similar. Furthermore, the expression of eGFP and of the transgene were driven by the same promoter (CMV), so the high proportion of eGFP+ BM and PB MNCs (Figure 2C) confirmed that transgene expression persisted for at least 3 weeks after transplantation. The constitutive activation of c-kit in BM MNCs from mice transplanted with c-kitD816V–transduced cells was verified via Western blot (Online Figure II).
Figure 2. AMD3100 fails to mobilize PCs that express a constitutively-active c-kit mutant (c-kitD816V).
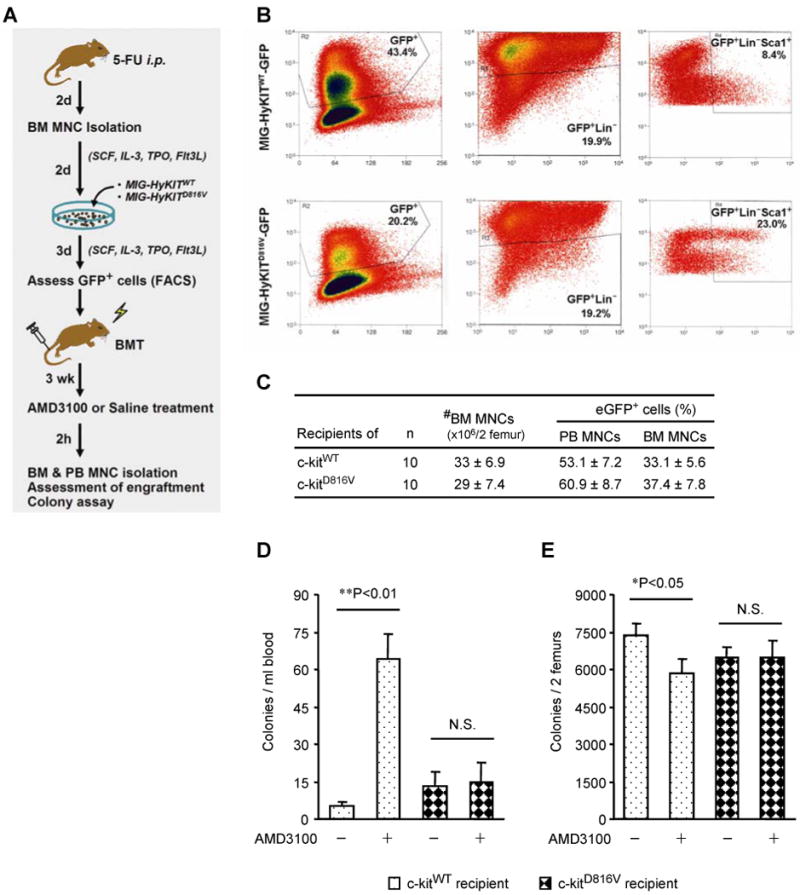
(A) WT BM MNCs were isolated, transduced ex vivo with retroviral vectors coding for eGFP-linked WT c-kit (MSCV-HyKITWT-IRES-eGFP [MIG-HyKITWT]) or eGFP-linked constitutively active c-kit (MSCV-HyKITD816V-IRES-eGFP [MIG-HyKITD816V]), and transplanted (independently) into WT mice after radiative ablation of the endogenous BM. Three weeks after BM transplantation, the recipient mice were subcutaneously injected with AMD3100 (5 mg/kg) (+) or PBS (−); 2 hours later, PB and BM MNCs were isolated, and the numbers of PCs transduced with WT c-kit or constitutively active c-kit (i.e., the number of GFP+ PCs) in the PB and BM were quantified via the colony-forming assay. (B) Flow cytometry analysis for eGFP expression in HyKITWT- or HyKITD816V-transduced BM MNCs before transplantation. (C) The number of BM MNCs and the proportions of BM and PB MNCs in recipient mice that expressed GFP three weeks after BM transplantation. (D-E) The number of HyKITWT- or HyKITD816V-transduced colony-forming PCs (i.e., GFP+ colonies) in the PB (D) and BM (E) of mice treated with AMD3100 or PBS. Values are mean ± SEM; n=10 per group.
On the third week after BM transplantation, the recipient mice were treated with AMD3100 or PBS, and the numbers of eGFP+ PCs in the BM and PB were determined 2 hours later. AMD3100 treatment significantly increased the number of eGFP+ PB PCs (Figure 2D), and significantly decreased the number of eGFP+ BM PCs (Figure 2E), in mice transplanted with MNCs that overexpressed c-kitWT but not in mice transplanted with c-kitD816V–expressing MNCs. Thus, either a deficiency (Figures 1A-B) or the constitutive activation of c-kit kinase activity (Figures 2D-E) impaired AMD3100-induced BM PC mobilization. Collectively, these observations suggest that CXCR4-mediated BM PC mobilization requires a change in the phosphorylation state of c-kit.
SDF-1/CXCR4 signaling upregulates c-kit phosphorylation in BM MNCs in vitro
Previous reports suggest that c-kit participates in PC mobilization through an α4-integrin–mediated mechanism 24, and that interactions between α4-integrin and VCAM-1 support the adhesion and retention of MNCs in the BM 15. Thus, we performed a series of in vitro experiments to determine whether the phosphorylation state of c-kit is altered by α4-integrin–mediated adhesion and CXCR4 signaling. Freshly isolated WT and c-kitw/w-v BM MNCs were applied to VCAM-1–coated or uncoated plates, allowed to adhere for 15 minutes, incubated with or without AMD3100 for another 15 minutes, and then c-kit phosphorylation at tyrosine719 was evaluated via Western blotting. The VCAM-1 coating did not induce c-kit phosphorylation in c-kitw/w-v cells, but phospho–c-kit levels were notably higher in adherent WT cells (i.e., cells from VCAM-1–coated plates) than in nonadherent WT cells (i.e., cells from uncoated plates) (Figure 3A). In adherent WT cells, treatment with an α4-integrin–blocking antibody (Figure 3B) reduced phospho–c-kit levels, whereas treatment with the CXCR4 ligand SDF-1 markedly increased phospho–c-kit levels (Figure 3C). Furthermore, both SDF-1 and SCF (i.e., c-kit ligand) induced c-kit phosphorylation, but c-kit levels were highest when the cells were incubated with both factors (Figure 3C), and AMD3100 treatment suppressed SDF-1–induced, but not SCF-induced, c-kit phosphorylation (Figure 3D). Notably, similar results were obtained after treating HEK293 cells that transiently expressed CXCR4 and c-kit with SDF-1, SCF, or AMD3100 alone and in combination (Figures 3E-F), and AMD3100 treatment did not downregulate c-kit phosphorylation in BM MNCs transduced with the constitutively-active c-kitD816V mutant (Figure 3G). Collectively, these observations suggest that MNC adhesion is associated with an increase in phospho–c-kit levels, and that in adherent MNCs, SDF-1 upregulates, and AMD3100 downregulates (in the presence of exogenous SDF-1), c-kit phosphorylation. Thus, AMD3100 appears to induce BM MNC mobilization through a decline in phospho–c-kit levels. Furthermore, SDF-1– and SCF-induced c-kit activation may occur independently and regulate different cellular activities.
Figure 3. SDF-1/CXCR4 signaling upregulates c-kit phosphorylation in BM MNCs and in HEK293 cells in vitro.
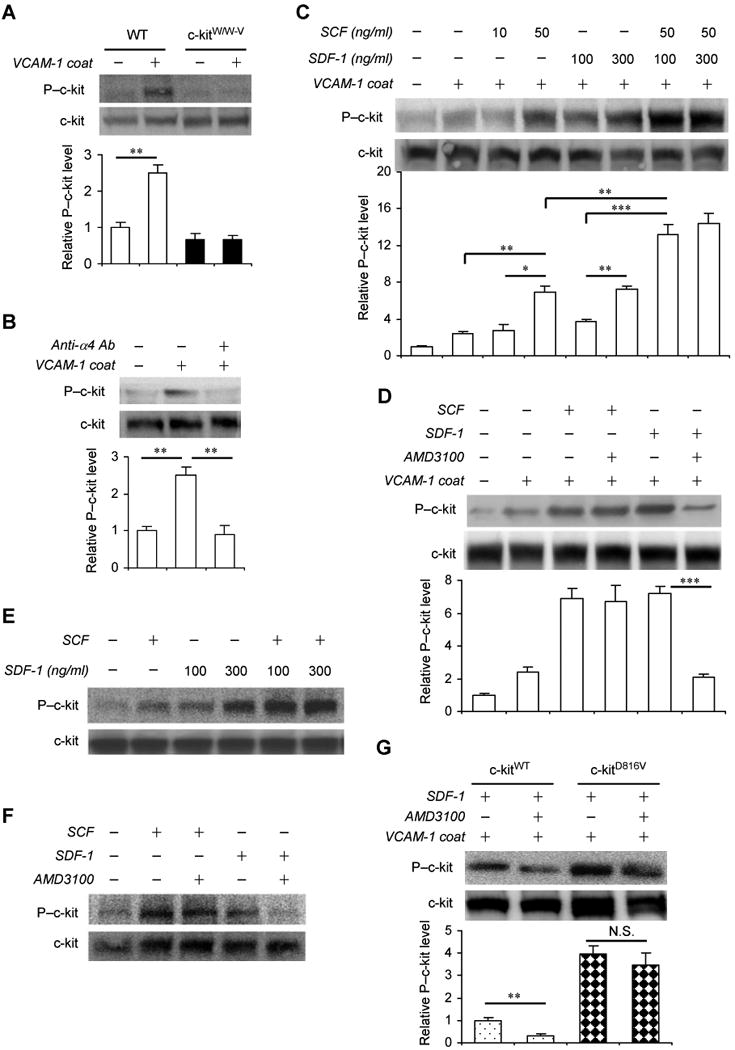
(A-D) BM MNCs were isolated from WT and c-kitW/W-V mice and applied to uncoated plates or to plates coated with the stromal protein VCAM-1 to mimic adhesion in the BM. Fifteen minutes later, the levels of phospho-c-kit (P-c-kit) and total c-kit were analyzed by Western blot in WT and c-kitW/W-V cells (A), in WT cells incubated with or without an anti-α4-integrin antibody (Anti-α4 Ab) (B) and in WT cells incubated with the indicated combinations of SCF, SDF-1, or AMD3100 (C-D). P–c-kit levels were quantified densitometrically, normalized to the total c-kit level, and expressed as the fold-difference from the levels measured in WT cells applied to uncoated plates and incubated without treatment. Values are mean ± SEM (*P<0.05, **P<0.01, ***P<0.001; n=6-8 per treatment). (E-F) HEK293 cells were co-transfected with pMyc-CXCR4 and pMSCV-HyKITWT-IRES-eGFP plasmids via standard techniques; 24 hours later, the transfected cells were treated with the indicated combinations of SCF, SDF-1, and AMD3100 for 15 min, lysed, and then P–c-kit and total c-kit levels were evaluated via Western blot. Representative results from 4 experiments are shown. (G) WT BM MNCs were transduced with retroviral vectors coding for WT (c-kitWT) or constitutively active (c-kitD816V) c-kit as described in Figure 2, then the cells were applied to VCAM-1–coated plates, stimulated with SDF-1 for 15 min in the presence or absence of AMD3100, and the levels of P–c-kit and total c-kit were analyzed via Western blot. Values are mean ± SEM. (**P<0.01, N.S., not significant; n=6 per treatment).
Antagonism or genetic deletion of CXCR4 downregulates BM c-kit phosphorylation in vivo
To determine whether CXCR4 also regulates BM MNC c-kit phosphorylation in vivo, WT mice were treated with AMD3100 or PBS and sacrificed 30 minutes later, then BM phospho–c-kit levels were evaluated by Western blot; phospho–c-kit levels were distinctly lower in mice administered AMD3100 than in PBS-treated mice (Figure 4A). These findings were corroborated by breeding Mx1-cre+ mice with CXCR4fl/fl mice 13 to generate Mx1-cre+CXCR4fl/fl offspring, which carry an inducible CXCR4 knockout mutation that is triggered by poly(I)-poly(C) injection 15. Two weeks after the CXCR4 knockout mutation was induced, real-time RT-PCR analyses confirmed that CXCR4 expression had fallen to just 5% of the levels observed in CXCR4-expressing mice (Figure 4B), and BM phospho–c-kit levels had also declined (Figure 4C-D). These observations were corroborated by administering AMD3100 or PBS to CXCR4BACGFP mice, which carry a bacterial artificial chromosome coding for GFP expression driven by the human CXCR4 genomic regulatory sequence 14. The mice were sacrificed 30 minutes after treatment, then sections of the femoral BM (Figure 4E) were immunofluorescently stained for total c-kit (Figure 4F) and phospho–c-kit (Figure 4G). The proportion of CXCR4-expressing (i.e., GFP+) cells that co-expressed phospho–c-kit was significantly lower in sections harvested from AMD3100-treated mice than from mice administered PBS (Figure 4H). Notably, phospho–c-kit and CXCR4 were co-expressed by the majority of cells located near the endosteum and surrounding the sinusoidal vessels, where high numbers of PCs are typically found. These observations confirm that CXCR4 regulates c-kit phosphorylation in the BM.
Figure 4. Antagonism or genetic deletion of CXCR4 downregulates c-kit phosphorylation in the BM in vivo.
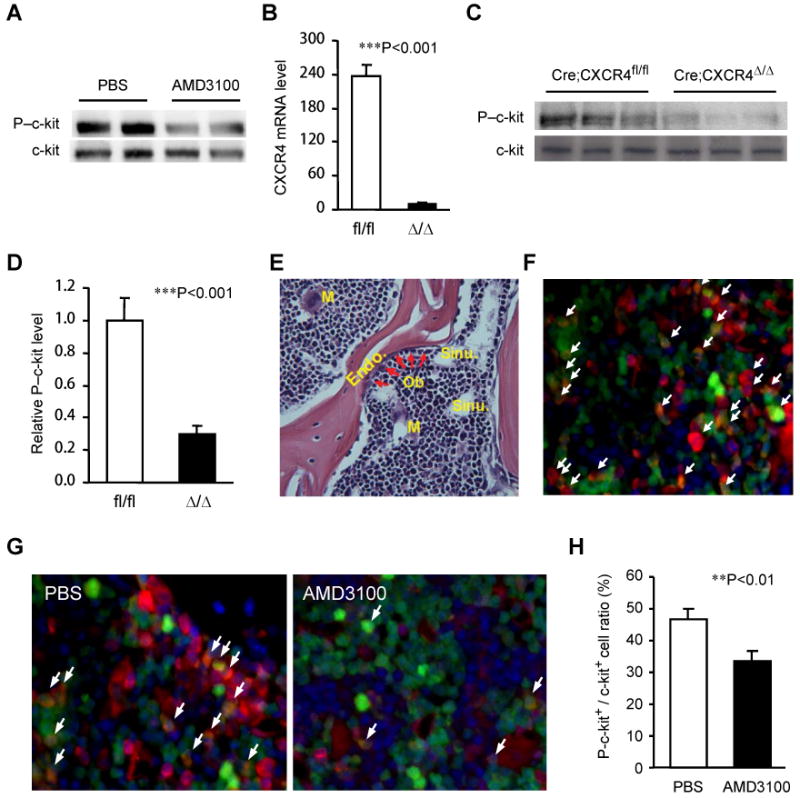
(A) WT mice were subcutaneously injected with AMD3100 (5 mg/kg) or PBS; 30 minutes later, phospho–c-kit (P-c-kit) and total c-kit levels were evaluated via Western blot. (B-D) Mx1-creCXCR4fl/fl mice were treated with or without 3 intraperitoneal injections of 250 μg poly(I)-poly(C) at 2 day intervals. Two weeks after the last injection, CXCR4 expression in BM MNCs from the untreated (i.e., CXCR4fl/fl) mice (fl/fl) and from the treated (i.e., CXCR4-knockout) mice (Δ/Δ) was assessed via real-time RT-PCR (B), and the levels of P–c-kit in BM lysates were evaluated by Western blot (C), quantified densitometrically, and normalized to the levels of total c-kit (D) (n=4). (E-F) Bone tissue harvested from CXCR4BAC-eGFP transgenic mice was evaluated histologically. Sections were H & E stained to show the trabecular region of the femurs (M, megakaryocyte; BV, blood vessel; OB, osteoblast; Endo, endosteum; Sinu, Sinusoidal vessel) (E) or immunofluorescently stained to reveal partial co-localization of c-kit (red) and CXCR4 (green) (F). (G-H) CXCR4BAC-eGFP transgenic mice were treated with AMD3100 (5 mg/kg) or PBS, sacrificed 30 minutes later, and bone tissues were harvested and immunofluorescently stained for the presence of P–c-kit (red) (G); then, the proportion of P-c-kit+ cells in the total c-kit+-cell population was quantified (H). Values are mean ± SEM; n=5 mice per group.
We also investigated whether the expression of CXCR4 and c-kit changed in response to BM MNC mobilization. Flow cytometry analysis of BM and PB MNCs isolated from CXCR4BACGFP-transgenic mice indicated that both CXCR4 (i.e., GFP fluorescence) and c-kit were expressed by a much greater proportion of BM MNCs (CXCR4: 93%; c-kit: ∼26%) than PB MNCs (CXCR4: ∼55%; c-kit: ∼0.60%) (Online Figure IIIA). CXCR4 expression was similarly distributed when evaluated by staining cells with a fluorescent anti-CXCR4 antibody (data not shown). Because c-kit expression is ∼40-fold more common among BM MNCs than PB MNCs, MNC mobilization should substantially increase the proportion of PB MNCs that express c-kit, unless c-kit expression is altered during mobilization. However, the proportion of PB MNCs that expressed c-kit was unchanged 2 hours after AMD3100 treatment (Online Figure IIIB), while the proportion of PB MNCs that expressed GFP (i.e., the number of circulating, BM-derived cells) increased from ∼60% to ∼89%. Thus, AMD3100-induced MNC mobilization is associated with a decline in c-kit expression, as well as c-kit phosphorylation.
SDF-1/CXCR4–induced c-kit phosphorylation is mediated by Src kinase
CXCR4 is a G-protein coupled receptor, and previous reports suggest that signaling through G protein-coupled receptors activates the Src family of protein tyrosine kinases (SFKs) 25; furthermore, the SDF-1/CXCR4 axis has been shown to activate Lyn, an SFK that is expressed in hematopoietic cells 26. Thus, we investigated whether Src kinase has a role in SDF-1/CXCR4-induced c-kit phosphorylation.
SDF-1 treatment markedly increased Src phosphorylation at tyrosine 416 (which signals Src activation) in HEK293 cells that transiently expressed CXCR4 and c-kit, and Src phosphorylation was followed by c-kit phosphorylation (Src: ∼2 min; c-kit: ∼10 min) (Figure 5A). SDF-1 also induced Src and c-kit phosphorylation in freshly isolated mouse BM MNCs adhered to VCAM-1–coated plates, whereas co-treatment with AMD3100 attenuated the SDF-1–induced phosphorylation of both proteins (Figure 5B), and co-treatment with the selective Src kinase inhibitor SU6656 dose-dependently blocked c-kit phosphorylation (Figure 5C). Furthermore, c-kit co-immunoprecipitated with phosphorylated Src (Figure 5D) in adherent BM MNCs, which suggests that phospho-Src binds to c-kit in vivo, and the levels of c-kit–bound phospho-Src increased after SDF-1 treatment (Figure 5D-E); once again, the effects of SDF-1 treatment declined in BM MNCs co-treated with ADM3100 (Figure 5D) or SU6656 (Figure 5E). Collectively, these observations suggest that SDF-1-induced c-kit phosphorylation requires the activation of Src and, consequently, that SDF-1 and CXCR4 regulate BM PC trafficking through a Src- and c-kit–dependent pathway.
Figure 5. SDF-1/CXCR4-induced c-kit phosphorylation is mediated by Src kinase activation.
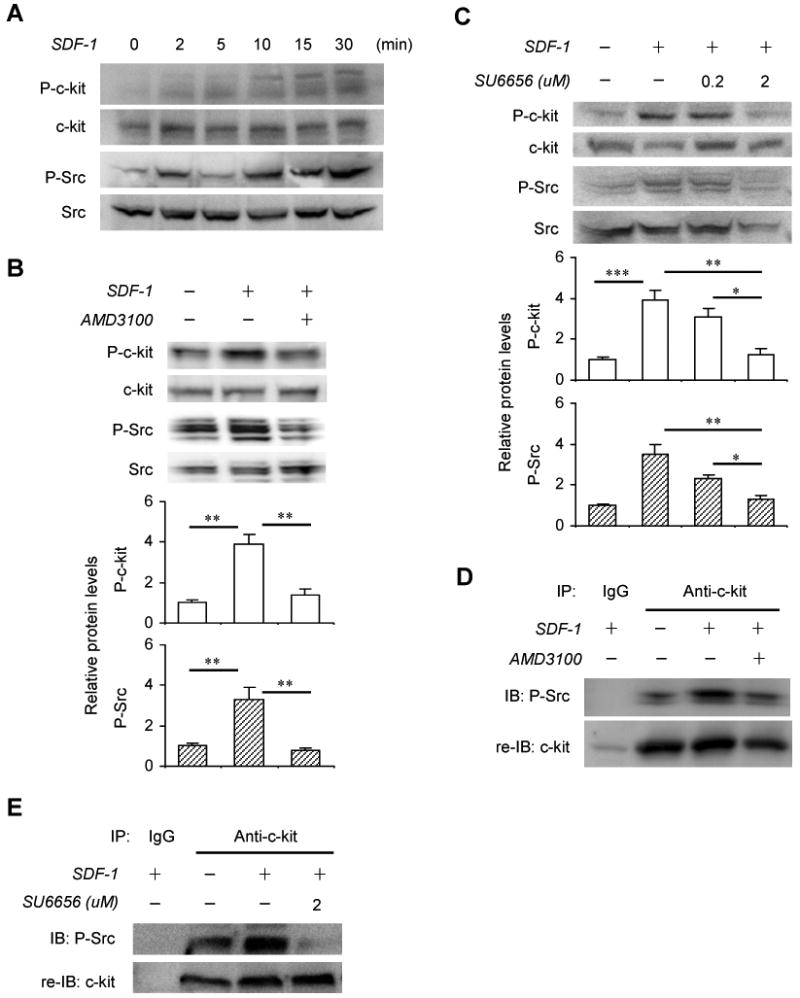
(A) HEK293 cells were co-transfected with pMyc-CXCR4 and pMSCV-HyKITWT-IRES-eGFP plasmids; 24 hours later, the transfected cells were treated with SDF-1 (200 ng/mL) for 0-30 minutes, lysed, then the levels of phospho-c-kit (P-ckit), total c-kit, phospho-Src (P-Src), and total Src were evaluated via Western blot. Representative results from 3 experiments are shown. (B-E) BM MNCs were isolated from WT mice and applied to plates coated with VCAM-1. (B, D) Fifteen minutes later, the cells were treated with the indicated combinations of SDF-1 (200 ng/mL) and AMD3100 (100 ng/mL) for 15 min. (C, E) The cells were treated with the indicated concentrations of SU6656 for 45 minutes, and then with or without SDF-1 (200ng/mL) for 15 minutes. (B-C) The cells were lysed, and the levels of P-c-kit, total c-kit, P-Src, and total Src were evaluated via Western blot. P–c-kit and P-Src levels were quantified densitometrically, normalized to total c-kit and total Src levels, respectively, and expressed as the fold-difference from the levels measured in untreated cells. Values are mean ± SEM. (*P<0.05, **P<0.01, ***P<0.001; n=6 per treatment). (D-E) Cell lysates were immunoprecipitated (IP) with anti–c-kit antibodies or control IgG antibodies, immunoblotted (IB) with anti-P-Src antibodies, and re-immunoblotted (re-IB) with anti–c-kit antibodies. Representative results from 3 experiments are shown.
Discussion
The mobilization of PCs from the BM to the peripheral circulation is a crucial step in the response to ischemic injury. Researchers in other laboratories have shown that BM PC retention is largely governed by interactions between SDF-1 and CXCR4, and that the selective CXCR4 antagonist AMD3100 disrupts these interactions, thereby releasing PCs into the peripheral circulation. For these reasons, AMD3100 and other treatments that enhance PC mobilization are currently being investigated for use in cell-based therapeutic approaches designed to promote tissue repair 27, 15, 28 and to treat a number of diseases 29-30. Our findings provide the first evidence that SDF-1–CXCR4 binding retains PCs in the BM by transactivating c-kit, and that CXCR4 antagonism mobilizes PCs by downregulating c-kit phosphorylation. Furthermore, phospho–c-kit has been shown to activate phosphatidylinositol 3 kinase (PI3K) (through the p85 regulatory subunit), Ras-mitogen-activated protein kinase (through growth factor receptor-bound protein 2), and integrins (e.g., VLA-4/VCAM-1), which are linked to the regulation of cell adhesion and mobility31, and results from other labs suggest that growth factors such as granulocyte colony stimulating factor (G-CSF) mobilize BM PCs by triggering the enzymatic cleavage of membrane-bound SCF–c-kit and SDF-1–CXCR4 complexes 32-33. Thus, SDF-1–CXCR4–c-kit signaling could function as a “final common pathway” of fundamental importance to BM PC retention and mobilization.
Because G-CSF–induced PC mobilization requires an increase in c-kit activation 9, the decline in c-kit phosphorylation reported here may seem counterintuitive. However, G-CSF-induced mobilization occurs 3 to 5 days after administration and is accompanied by an increase in the number of PCs present in the perivascular niche 34, whereas AMD3100-induced mobilization occurs within a few hours and, consequently, is unlikely to be preceded by the perivascular accumulation of PCs. Furthermore, the two agents appear to mobilize different PC subpopulations, and more PCs are mobilized when G-CSF and AMD3100 administration are combined than when G-CSF is administered alone. 35-37 Thus, the available evidence suggests that the mechanisms of G-CSF– and AMD3100-induced PC mobilization differ substantially. G-CSF and other slow-acting agents may increase c-kit phosphorylation by upregulating SCF, which (by itself) has only a modest effect on mobilization but potently promotes PC proliferation 38; if so, G-CSF–induced mobilization may be delayed until an adequate surplus of PCs is available for release to the PB 34. Conversely, fast-acting agents, such as AMD3100 or the α4-integrin–blocking antibody, may mobilize PCs directly by reducing c-kit phosphorylation in the perivascular niche 39. This hypothesis is also supported by recent evidence that PCs can be rapidly mobilized by the administration of a c-kit neutralizing antibody 12.
Both the loss and the constitutive activation of c-kit kinase activity impaired AMD3100-induced BM PC mobilization. In c-kit kinase-defective mice, BM PC levels were lower than in WT mice, and systemically administered CXCR4+ PCs could not repopulate the BM. These observations suggest that c-kit–kinase inactivation blocks the retention of CXCR4+ PCs in the BM and, consequently, that the cells susceptible to AMD3100-induced mobilization are (in effect) already mobilized. In mice transplanted with BM cells that expressed a constitutively active c-kit mutant, AMD3100 treatment did not alter PC levels in the BM or the PB, which suggests that CXCR4-mediated BM PC mobilization requires c-kit kinase deactivation. Collectively, these observations indicate that PCs are retained in the BM by the kinase activity of c-kit, and that AMD3100 mobilizes CXCR4+ BM PCs by downregulating c-kit kinase activity.
Our observations also indicate that SDF-1/CXCR4–induced c-kit transactivation is mediated by SFKs. CXCR4 is a G-protein coupled receptor, and two families of G-protein α subunits (Gαs and GαI) activate SFKs (c-Src and Hck) by binding directly to the catalytic domain and increasing substrate access to the active site 25. CXCR4-dependent stimulation of SFKs (Lyn) has also been associated with the activation of PI3K 26. However, SFKs display considerable redundancy both in their activation pathways and in the downstream effectors that mediate their biological function 40, so whether the G-proteins, PI3K, or other upstream components link the SDF-1/CXCR4 axis to SFK activation and, subsequently, to c-kit phosphorylation and BM PC retention, has yet to be determined.
There are nine known SFKs, several of which, as well as multiple isoforms of the same SFK, are expressed in any given tissue. SU6656 inhibits all SFKs (without directly altering c-kit activity) 41, and the available antibodies are not specific for the phosphorylated forms of individual family members, so we used antibodies against activated SFKs that are found predominantly in the hematopoietic system (i.e., Lyn, Hck, and Fgr). Thus, the results reported here cannot identify which specific SFK mediates c-kit activation. We have begun a series of experiments with mice deficient in one or more SFK to characterize the roles of the individual family members.
In conclusion, the results presented here demonstrate that SDF-1/CXCR4 signaling retains PCs in the BM through the Src-mediated activation of c-kit, and that CXCR4 antagonism induces PC mobilization by downregulating c-kit phosphorylation. Because BM PCs have an essential role in tissue repair, the mechanism identified by our studies could be an important component of the physiological response to ischemic injury. Furthermore, SDF-1, CXCR4, and c-kit also have a role in normal development and in diseases such as arthritis and cancer 42-43, so the implications of our findings may extend to a wide variety of pathological and developmental processes.
Novelty and Significance.
What Is Known?
Progenitor cells (PCs) are mobilized from the bone marrow (BM) to the peripheral circulation in response to ischemic injury, and ample evidence suggests that this process is governed by interactions between stromal-cell–derived factor 1 (SDF-1) and CXC chemokine receptor 4 (CXCR4).
Within the BM, CXCR4 is expressed by mononuclear cells (including PCs), and SDF-1 is expressed by osteoblasts, endothelial cells, and a subset of reticular cells.
Like CXCR4, c-kit is expressed predominantly in BM PCs, and the signaling pathways regulated by c-kit activation contribute to BM PC mobilization and trafficking.
What New Information Does This Article Contribute?
The binding of SDF-1 to CXCR4 upregulates c-kit phosphorylation (i.e., c-kit activity) in BM PCs.
Disruption of the SDF-1–CXCR4 interaction (i.e., CXCR4 antagonism) mobilizes PCs by downregulating c-kit phosphorylation.
SDF-1/CXCR4-induced c-kit phosphorylation is mediated by the Src family of non-receptor tyrosine kinases (SFKs).
Summary
In response to ischemic injury, PCs are mobilized from the BM to the peripheral circulation and recruited to the injured tissue, where they contribute to vessel growth. The retention and release of BM PCs is governed by both SDF-1/CXCR4 signaling and c-kit signaling, but whether these two pathways interact has not been investigated previously. The results presented here are the first to show that SDF-1/CXCR4 signaling retains PCs in the BM by transactivating c-kit, that CXCR4 antagonism mobilizes PCs by downregulating c-kit phosphorylation, and that SDF-1/CXCR4–induced c-kit transactivation is mediated by SFKs. Because BM PCs have an essential role in tissue repair, the mechanism identified by our studies could be an important component of the physiological response to ischemic injury and, consequently, may be a viable target for therapies designed to promote BM PC mobilization and the recovery of injured cardiovascular tissue. Furthermore, SDF-1, CXCR4, c-kit, and SFKs also have a role in normal development and in diseases such as arthritis and cancer, so the implications of our findings may extend to a wide variety of pathological and developmental processes.
Supplementary Material
Acknowledgments
We thank Dr. Gang Pei (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, China) for providing the pMyc-CXCR4 plasmid, and Drs. Zhizhou Kuang, Wuqiang Zhu, Ms. Shengying Yang, Sol Misener, Christine Kamide, Lenyr Munoz, and Mr. Andy Scarpelli, Divakar Mithal (Northwestern University) for technical help. We thank Dr. W. Kevin Meisner for editorial assistance.
Sources of Funding: This work was supported by the NIH Grant 1R01 HL093439-01A2, American Heart Association Grant 0430135N (to G.Q.), and the State Scholarship Fund of China (to M.C.).
Nonstandard Abbreviations and Acronyms
- BM
bone marrow
- CXCR4
CXC-chemokine receptor 4
- MNC
mononuclear cells
- PB
peripheral blood
- PC
progenitor cell
- SCF
stem cell factor
- SDF-1
stromal–cell derived factor 1
- SFK
Src family of protein tyrosine kinase
- VCAM-1
vascular cell adhesion molecule 1
Footnotes
Disclosures
None.
References
- 1.Laird DJ, von Andrian UH, Wagers AJ. Stem cell trafficking in tissue development, growth, and disease. Cell. 2008;132:612–630. doi: 10.1016/j.cell.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 2.Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- 3.Fliedner TM. The role of blood stem cells in hematopoietic cell renewal. Stem Cells. 1998;16:361–374. doi: 10.1002/stem.160361. [DOI] [PubMed] [Google Scholar]
- 4.Dimmeler S, Zeiher AM, Schneider MD. Unchain my heart: The scientific foundations of cardiac repair. J Clin Invest. 2005;115:572–583. doi: 10.1172/JCI24283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orlic D, Kajstura J, Chimenti S, Limana F, Jakoniuk I, Quaini F, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci U S A. 2001;98:10344–10349. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, Cote D, Rowe DW, Lin CP, Scadden DT. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by cxcl12-cxcr4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 8.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195:1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S. Recruitment of stem and progenitor cells from the bone marrow niche requires mmp-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, Nagler A, Ben-Hur H, Many A, Shultz L, Lider O, Alon R, Zipori D, Lapidot T. Dependence of human stem cell engraftment and repopulation of nod/scid mice on cxcr4. Science. 1999;283:845–848. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- 11.Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90:1345–1364. [PubMed] [Google Scholar]
- 12.Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science. 2007;318:1296–1299. doi: 10.1126/science.1149726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nie Y, Han YC, Zou YR. Cxcr4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205:777–783. doi: 10.1084/jem.20072513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran PB, Banisadr G, Ren D, Chenn A, Miller RJ. Chemokine receptor expression by neural progenitor cells in neurogenic regions of mouse brain. The Journal of comparative neurology. 2007;500:1007–1033. doi: 10.1002/cne.21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin G, Ii M, Silver M, Wecker A, Bord E, Ma H, Gavin M, Goukassian DA, Yoon YS, Papayannopoulou T, Asahara T, Kearney M, Thorne T, Curry C, Eaton L, Heyd L, Dinesh D, Kishore R, Zhu Y, Losordo DW. Functional disruption of alpha4 integrin mobilizes bone marrow-derived endothelial progenitors and augments ischemic neovascularization. J Exp Med. 2006;203:153–163. doi: 10.1084/jem.20050459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Larochelle A, Fricker S, Bridger G, Dunbar CE, Abkowitz JL. Mobilization as a preparative regimen for hematopoietic stem cell transplantation. Blood. 2006;107:3764–3771. doi: 10.1182/blood-2005-09-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang Z, Kreisel F, Cain J, Colson A, Tomasson MH. Neoplasia driven by mutant c-kit is mediated by intracellular, not plasma membrane, receptor signaling. Mol Cell Biol. 2007;27:267–282. doi: 10.1128/MCB.01153-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou J, Zhu Y, Cheng M, Dinesh D, Thorne T, Poh KK, Liu D, Botros C, Tang YL, Reisdorph N, Kishore R, Losordo DW, Qin G. Regulation of vascular contractility and blood pressure by the e2f2 transcription factor. Circulation. 2009;120:1213–1221. doi: 10.1161/CIRCULATIONAHA.109.859207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin G, Kishore R, Dolan CM, Silver M, Wecker A, Luedemann CN, Thorne T, Hanley A, Curry C, Heyd L, Dinesh D, Kearney M, Martelli F, Murayama T, Goukassian DA, Zhu Y, Losordo DW. Cell cycle regulator e2f1 modulates angiogenesis via p53-dependent transcriptional control of vegf. Proc Natl Acad Sci U S A. 2006;103:11015–11020. doi: 10.1073/pnas.0509533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang YL, Zhu W, Cheng M, Chen L, Zhang J, Sun T, Kishore R, Phillips MI, Losordo DW, Qin G. Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing cxcr4 expression. Circ Res. 2009;104:1209–1216. doi: 10.1161/CIRCRESAHA.109.197723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, Calandra G, Bridger G, Dale DC, Srour EF. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with amd3100, a cxcr4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colvin GA, Lambert JF, Dooner MS, Cerny J, Quesenberry PJ. Murine allogeneic in vivo stem cell homing. J Cell Physiol. 2007;211:386–391. doi: 10.1002/jcp.20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitayama H, Tsujimura T, Matsumura I, Oritani K, Ikeda H, Ishikawa J, Okabe M, Suzuki M, Yamamura K, Matsuzawa Y, Kitamura Y, Kanakura Y. Neoplastic transformation of normal hematopoietic cells by constitutively activating mutations of c-kit receptor tyrosine kinase. Blood. 1996;88:995–1004. [PubMed] [Google Scholar]
- 24.Papayannopoulou T, Priestley GV, Nakamoto B. Anti-vla4/vcam-1-induced mobilization requires cooperative signaling through the kit/mkit ligand pathway. Blood. 1998;91:2231–2239. [PubMed] [Google Scholar]
- 25.Ma YC, Huang J, Ali S, Lowry W, Huang XY. Src tyrosine kinase is a novel direct effector of g proteins. Cell. 2000;102:635–646. doi: 10.1016/s0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 26.Ptasznik A, Urbanowska E, Chinta S, Costa MA, Katz BA, Stanislaus MA, Demir G, Linnekin D, Pan ZK, Gewirtz AM. Crosstalk between bcr/abl oncoprotein and cxcr4 signaling through a src family kinase in human leukemia cells. J Exp Med. 2002;196:667–678. doi: 10.1084/jem.20020519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aicher A, Kollet O, Heeschen C, Liebner S, Urbich C, Ihling C, Orlandi A, Lapidot T, Zeiher AM, Dimmeler S. The wnt antagonist dickkopf-1 mobilizes vasculogenic progenitor cells via activation of the bone marrow endosteal stem cell niche. Circ Res. 2008;103:796–803. doi: 10.1161/CIRCRESAHA.107.172718. [DOI] [PubMed] [Google Scholar]
- 28.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, Topol EJ, Penn MS. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003;362:697–703. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 29.De Clercq E. The amd3100 story: The path to the discovery of a stem cell mobilizer (mozobil) Biochem Pharmacol. 2009;77:1655–1664. doi: 10.1016/j.bcp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 30.Shepherd RM, Capoccia BJ, Devine SM, Dipersio J, Trinkaus KM, Ingram D, Link DC. Angiogenic cells can be rapidly mobilized and efficiently harvested from the blood following treatment with amd3100. Blood. 2006;108:3662–3667. doi: 10.1182/blood-2006-06-030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ronnstrand L. Signal transduction via the stem cell factor receptor/c-kit. Cell Mol Life Sci. 2004;61:2535–2548. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- 32.Winkler IG, Levesque JP. Mechanisms of hematopoietic stem cell mobilization: When innate immunity assails the cells that make blood and bone. Exp Hematol. 2006;34:996–1009. doi: 10.1016/j.exphem.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 33.To LB, Haylock DN, Dowse T, Simmons PJ, Trimboli S, Ashman LK, Juttner CA. A comparative study of the phenotype and proliferative capacity of peripheral blood (pb) cd34+ cells mobilized by four different protocols and those of steady-phase pb and bone marrow cd34+ cells. Blood. 1994;84:2930–2939. [PubMed] [Google Scholar]
- 34.Li Z, Li L. Understanding hematopoietic stem-cell microenvironments. Trends Biochem Sci. 2006;31:589–595. doi: 10.1016/j.tibs.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 35.DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, Maziarz RT, Jacobsen E, Nademanee A, McCarty J, Bridger G, Calandra G. Phase iii prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-hodgkin's lymphoma. J Clin Oncol. 2009;27:4767–4773. doi: 10.1200/JCO.2008.20.7209. [DOI] [PubMed] [Google Scholar]
- 36.Donahue RE, Jin P, Bonifacino AC, Metzger ME, Ren J, Wang E, Stroncek DF. Plerixafor (amd3100) and granulocyte colony-stimulating factor (g-csf) mobilize different cd34+ cell populations based on global gene and microrna expression signatures. Blood. 2009;114:2530–2541. doi: 10.1182/blood-2009-04-214403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fowler CJ, Dunn A, Hayes-Lattin B, Hansen K, Hansen L, Lanier K, Nelson V, Kovacsovics T, Leis J, Calandra G, Maziarz RT. Rescue from failed growth factor and/or chemotherapy hsc mobilization with g-csf and plerixafor (amd3100): An institutional experience. Bone Marrow Transplant. 2009;43:909–917. doi: 10.1038/bmt.2008.409. [DOI] [PubMed] [Google Scholar]
- 38.Glaspy JA, Shpall EJ, LeMaistre CF, Briddell RA, Menchaca DM, Turner SA, Lill M, Chap L, Jones R, Wiers MD, Sheridan WP, McNiece IK. Peripheral blood progenitor cell mobilization using stem cell factor in combination with filgrastim in breast cancer patients. Blood. 1997;90:2939–2951. [PubMed] [Google Scholar]
- 39.Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008;78:413–421. doi: 10.1093/cvr/cvn081. [DOI] [PubMed] [Google Scholar]
- 40.Thomas SM, Brugge JS. Cellular functions regulated by src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 41.Tatton L, Morley GM, Chopra R, Khwaja A. The src-selective kinase inhibitor pp1 also inhibits kit and bcr-abl tyrosine kinases. J Biol Chem. 2003;278:4847–4853. doi: 10.1074/jbc.M209321200. [DOI] [PubMed] [Google Scholar]
- 42.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated sdf-1/cxcl12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 43.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor cxcr4 downregulated by von hippel-lindau tumour suppressor pvhl. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.