

Abstract

A highly enantio- and diastereoselective anti-aldol process (up to >99% ee, >99:1 dr) catalyzed by a proline mimetic – N-(p-dodecylphenylsulfonyl)-2-pyrrolidinecarboxamide – has been developed. Catalyst loading as low as 2 mol% can be employed. Use of industry-friendly solvents for this transformation as well as neat reaction conditions have been demonstrated. The scope of this transformation on a range of aldehydes and ketones is explored. Density Functional Theory computations reveal that the origins of enhanced diastereoselectivity is due to the presence of non-classical hydrogen bonds between the sulfonamide, the electrophile and the catalyst enamine that favor the major Anti-Re aldol TS in the Houk-List model.
Introduction
The aldol reaction has been a central focus of the chemical community since observed by Kane1 and later by Borodin,2 Kekulé3 and Wurtz.4 Subsequent decades led to numerous advances that helped to address the stereoselectivity of the reaction process.5 These accomplishments have paved the way for modern polyketide and macrolide synthesis.6 In addition to these important discoveries, a wealth of effort has been directed to the development of alternate reaction protocols, which generate a net aldol adduct but do not utilize traditional aldol starting materials.7,8,9 Zimmerman and Traxler proposed over fifty years ago that controlling syn- vs. anti-aldol adducts may be explained through the use of cyclic, chair-like transition states.10 Alternate acyclic transition states have also been proposed to address stereochemical outcome of other aldol reactions.11 Metal-based enolates have proven particularly useful in controlling both the enantio- and diastereoselectivity in aldol processes through the use of chiral directing groups.11,12 Metal-catalyzed, enantioselective protocols have also been developed which offer the ability to reduce the dependence on auxiliary-based approaches for accomplishing these transformations.13
Considerable recent excitement has been generated by the resurgence14 of organocatalysis as a practical and user-friendly method for facilitating these types of transformations.15 Many of the catalyst scaffolds are based on amino acid architecture with 2° amines proving particularly useful. Consequently, a large percentage of organocatalysts find their origins in proline. These organocatalyzed reactions often are performed at ambient or near-ambient temperatures and do not require the careful exclusion of moisture and oxygen. Organocatalyzed aldol reactions also tend to provide access to the anti-aldol adduct as the major product from the transformation. While alternate approaches to accessing enantioenriched anti-aldol adducts do exist,16,17 these protocols have often proven attractive based on stereoselectivity and practicality. The mechanistic underpinnings of the proline-catalyzed aldol reaction transformation have been previously explored;18 however, the divergent nature of the stereoselectivities based on catalyst modification and substrate scope is not fully understood. In this article, we provide a full account of our organocatalyzed process for facilitating highly enantio- and diastereoselective aldol reactions using the practical proline mimetic N-(p-dodecylphenylsulfonyl)-2-pyrrolidinecarboxamide (1)19 and a detailed analysis of the enhanced stereoselectivities of this catalyst.
Results and Discussion
Proline and proline-derived organocatalysts have proven useful in a range of transformations.20 Our interest in this field arose during our synthetic work towards the alkaloid lycopodine.21,22 We required an organocatalyst for an intramolecular Michael addition which possessed both a 2° amine and an organic acid motif. These efforts ultimately resulted in the development of a catalyst scaffold based on a proline sulfonamide. A more detailed discussion of this transformation has been published elsewhere.23
Solvent Effects
With our development of catalyst 1 for enantioselective, intramolecular keto-sulfone Michael reactions, we became intrigued by the possibility that this catalyst scaffold would have more widespread applicability. Given the considerable importance of the aldol reaction in modern synthetic organic chemistry, the application of our sulfonamide catalyst system in this setting seemed appropriate.24 While numerous examples of organocatalyzed aldol reactions have been reported based on proline, we were struck by the fact that the vast majority of these catalyst systems employed polar, aprotic solvent systems (e.g. DMSO, DMF). This solvent choice is primarily based on the poor solubility of proline and related derivatives in non-polar, aprotic solvents. We reasoned that if improved levels of stereoselectivity could be realized in non-polar media, it would allow for the more subtle differentiation between competing transition states.
While use of these polar aprotic solvents (e.g. MeCN, DMF, DMSO) are common in academic research laboratories, their polarity poses serious challenges in industrial settings with aqueous phase miscibility, product isolation issues25 and solvent reclamation.26 DMSO is a particularly challenging solvent, as solvent recycling on an industrial scale is typically impossible.27 The development of new catalysts and processes that use relatively inexpensive, non-polar solvents is crucial, due to the specific industrial advantages associated with efficient aqueous phase splits and ease of recycle at scale.28 Alternatively, the development of synthetic processes performed in the absence of solvents (or minimal solvent volumes) can effectively eliminate chemical waste in industrial processes (in some cases solvent is always required to effectively dissipate exotherms and ensure even heat transfer throughout the medium).29
The proline dodecylphenylsulfonamide 1 has greatly improved solubility properties in non-polar solvents, as compared to proline, as illustrated in Figure 2. Comparable weights (300 mg) of proline and our sulfonamide 1 were mixed with 1.0 milliliter of dichloromethane at ambient temperature. Proline is essentially insoluble in this solvent (calculated solubility < 5 mg/mL in CH2Cl2), as were proline tetrazole and proline phenylsulfonamide. In contrast, dodecylphenylsulfonamide 1 is completely soluble under these conditions.
Figure 2.

Solubility Comparison of Organocatalysts in CH2Cl2.a
a The left image illustrates that 300 mg of sulfonamide 1 is soluble in 1 mL of CH2Cl2 and right image demonstrates that 300 mg of proline is not readily soluble in 1 mL of CH2Cl2.
We first explored the optimization of the catalyst on the aldol reaction between cyclohexanone and p-nitrobenzaldehyde (Table 1). This reaction has become the standard reaction explored in most organocatalyzed aldol publications and provided us with a good metric for analysis. We were primarily interested in developing a protocol that preformed well in non-polar organic solvents. Given the high solubility of the sulfonamide catalyst in dichloromethane (DCM), we started our screening process using it as the baseline solvent (Entry 1). While the reaction performed reasonably well in that solvent, we observed dramatic improvements in yield by switching the solvent to 1,2-dichloroethane (DCE) (Entry 2). We23,30 and others31 have observed similar results by replacement of DCM with DCE. Reduction of catalyst loading led to a reduction in chemical efficiency (Entry 3). Use of ethanol as a potential proton source had minimal impact on the transformation (Entries 4–6). Fortunately, the addition of a single equivalent of water had a dramatic positive impact on the rate and selectivity of the transformation (Entry 7). Pihko and others have observed the significant importance of water in the rate, yield and stereoselectivity of the reaction.32 Excess water did not appear to have a significant additional impact on the transformation (Entry 8). Use of DCE as solvent continued to be preferred to DCM for achieving optimum levels of diastereoselectivity (Entries 7 and 9). The reaction could alternatively be performed in water as solvent (Entry 10) at reduced catalyst loading or in 2-methyltetrahydrofun (2-Me-THF, Entry 11) with excellent levels of selectivity. Reduction in the reaction temperature to 4°C led to the outstanding levels of chemical yield and stereoselectivities with 99% ee, >99:1 dr and 95% chemical yield (Entry 12). It is important to note that one equivalent of water is not readily soluble under the DCE, 4°C reaction conditions. Immiscible phases can be visually observed – indicating that far less than 1 equivalent of water is actually present in the organic phase under these reaction conditions. It is possible that the organocatalyst is acting in part as a phase transfer catalyst between the aqueous and organic phases. We have also developed an alternative procedure requiring just 2 mol % catalyst loading and reduced equivalents of the cyclohexanone (Entry 14). This protocol was performed in the absence of solvent at room temperature with a single of equivalent of water and again provides excellent levels of chemical yield and stereoselectivity.
Table 1.
Optimization of Reaction Conditions.a
| Entry | Conditionsa | mol % (1) | Temp | Time (h) | Yield | eeb | drc |
|---|---|---|---|---|---|---|---|
| 1 | CH2Cl2 | 20 | rt | 36 | 51 | 97 | 15:1 |
| 2 | DCE | 20 | rt | 46 | 92 | 95 | 12:1 |
| 3 | DCE | 10 | rt | 36 | 55 | 95 | 11:1 |
| 4 | DCE/EtOH (99:1) | 20 | rt | 36 | 96 | 97 | 14:1 |
| 5 | DCE/EtOH (99:1) | 10 | rt | 36 | 60 | 95 | 18:1 |
| 6 | EtOH | 20 | rt | 36 | 82 | 80 | 6:1 |
| 7 | DCE, H2O (1 equiv.) | 20 | rt | 14 | 96 | 97 | 36:1 |
| 8 | DCE, H2O (10 equiv.) | 20 | rt | 16 | 96 | 98 | 31:1 |
| 9 | CH2Cl2, H2O (1 equiv.) | 20 | rt | 16 | 95 | 97 | 18:1 |
| 10 | H2O | 10 | rt | 36 | 95 | 95 | 20:1 |
| 11 | 2-Me-THF, H2O (1 equiv.) | 20 | rt | 16 | 85 | 94 | 30:1 |
| 12 | DCE/EtOH (99:1) | 20 | 4°C | 36 | 97 | 97 | 53:1 |
| 13 | DCE, H2O (1 equiv.) | 20 | 4°C | 30 | 95 | 99 | >99:1e |
| 14d | neat, H2O (1 equiv.) | 2 | rt | 36 | 96 | 96 | >99:1 |
All reactions were performed at 2 M concentration of 6 in solution and with five equivalents of 5.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.
Performed using two equivalents of 5.
1H NMR expansions are provided in the Supporting Information to demonstrate the diastereomeric selectivity for product 7.

Catalyst Structure Effects on Stereoselectivities
Next, we screened a range of known organocatalysts under our optimized reaction conditions to provide a comparative analysis. We recognize that each catalyst structure may have reaction parameters which are favorable for optimum performance, but a standardized set of conditions were important to gauge how much of the high diastereoselectivity is a function of the unique structural characteristics of the new sulfonamide catalyst 1. We chose to look at reaction conditions containing 1% ethanol or one equivalent of water to provide two separate data sets for evaluation. With each catalyst we screened, conditions B (which employed one equivalent of water) provided higher levels of diastereoselectivity than the ethanol conditions (conditions A). The increased chemical yield with proline under conditions A is likely due to the improved solubility of proline with 1% ethanol present in the reaction mixture. As mentioned previously, immiscible phases are readily formed under conditions B. From the data collected, it appears that the long alkyl chain on the sulfonamide is acting principally to improve solubility in the transformations – leading to improved chemical yields. That said, proline sulfonamides as a group provided superior levels of diastereoselectivity as compared to both proline and proline tetrazole.
We have developed a model to explain the considerable improvements in diastereoselectivity that are uniquely observed with proline sulfonamide-catalyzed aldol reactions (Table 2). The intermolecular aldol reaction between the proline-sulfonamide enamine of cyclohexanone and benzaldehyde was explored computationally using DFT (B3LYP/6-31G*) geometries and thermochemistries,33 augmented with SCS-MP2 energies extrapolated to infinite basis (extrapolated from cc-pVTZ and cc-pVQZ results).34 The use of the two-point extrapolation to infinite basis addresses the problem of basis set truncation, a major source of error in computations. Solvation single points were computed for DCM at the B3LYP/6–31+G** level, using the PCM solvation model and UAKS radii.
Table 2.
Comparison of Organocatalysts.a
| Entry | Conditionsa | Catalyst | Yield | eeb | drc |
|---|---|---|---|---|---|
| 1 | A | 8 | 84 | 96 | 6:1 |
| 2 | B | 8 | 22 | 98 | 13:1 |
| 3 | A | 9 | 81 | 96 | 2:1 |
| 4 | B | 9 | 91 | 98 | 7:1 |
| 5 | A | 10 | 34 | 97 | 24:1 |
| 6 | B | 10 | 42 | 99 | 65:1 |
| 7 | A | 11 | 55 | 99 | 40:1 |
| 8 | B | 11 | 49 | 98 | 83:1 |
| 9 | A | 12 | 54 | 98 | 35:1 |
| 10 | B | 12 | 52 | 99 | 92:1 |
| 11 | A | 1 | 97 | 97 | 53:1 |
| 12 | B | 1 | 95 | 99 | >99:1 |
Conditions A: DCE/EtOH (99:1), 4°C, 36 h. Conditions B: DCE, H2O (1 equiv.), 4°C, 30 h. All reactions were performed at 2 M concentration of 6 in solution and with five equivalents of 5.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.
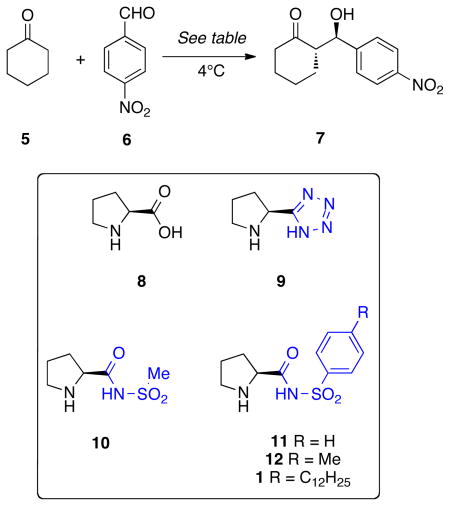
The mechanism of the aldol reaction catalyzed by proline has been reported previously by Houk and coworkers.18 A strong hydrogen-bonding network between the carboxylic acid and the aldehyde oxygen in the stereodetermining C-C bond forming transition state (TS) is key for stereocontrol and catalysis. In addition, staggering of the substituents around the forming C-C bond minimizes steric repulsions in the TS. There are four possible diastereomeric enamine aldol transition states that meet these two requirements: TS-Anti-Re, TS-Anti-Si, TS-Syn-Re, and TS-Syn-Si. Anti/Syn refers to the arrangement of the enamine with respect to the organic acid moiety while re/si denotes the facial attack of the aldehyde electrophile. The most stable transition structures are shown in Figure 3.
Figure 3.

35 The lowest energy transition structures for the aldol reaction between cyclohexanone enamine of proline-sulfonamide and benzaldehyde leading to the formation of each of the diastereomeric products. Anti/syn refers to the arrangement of the enamine with respect to the organic acid moiety while re/si denotes the facial attack of the aldehyde electrophile. Distances are in Ångstroms, energies in kcal/mol. Green lines designate possible stabilizing electrostatic interactions. Structures and thermodynamic corrections computed using B3LYP/6–31G* in the gas phase. Numbers in parenthesis include solvation corrections for DCE. ∞ designates infinite basis set extrapolation from the Dunning cc-pVTZ and cc-pVQZ basis sets.
The major diastereomer arises from the TS-Anti-Re, and the minor isomer from the TS-Anti-Si. All theoretical methods employed (B3LYP/6–31G*, B3LYP/6–31G*//SCS-MP2/∞, and B3LYP/6–31G*//MP2/∞) agree in the substantial energetic preference for TS-Anti-Re. Our calculations indicate that the energetic difference between these two TS is >4.5 kcal/mol (or >3.7 with solvation corrections). It should be noted that the computed difference in this series is 3 kcal/mol more selective than observed for proline.18
The greatly enhanced diastereoselectivity of proline sulfonamide catalysts over proline is due to two non-classical hydrogen bonds: between the sulfonamide oxygens and (1) the hydrogens of the aldehyde electrophile (distance O-HCOPh = 2.9 Å) and (2) the cyclohexyl enamine (distance O-EnamineH = 3.9 and 2.6 Å) that stabilize the TS-Anti-Re C-C bond-forming TS. In the disfavored TS-Anti-Si, these favorable interactions are replaced by steric repulsions from the intercalating phenyl ring (analogous distance O-EnamineH = 4.9 and 4.0 Å). This loss of stabilizing electrostatic interactions and gain of repulsive steric interactions in the TS-Anti-Si cause the proline-sulfonamide catalysts to be ~3 kcal/mol more selective than other simpler proline-type catalysts.
An extensive conformational analysis was performed in order to determine the most stable non-classical hydrogen-bonding pattern by the sulfonamide. Three such arrangements for the TS-Anti-Re are shown in Figure 4. The sulfonamide conformation found in TS-Anti-Re, in which the sulfonamide oxygens are in close proximity to the aldehyde hydrogen and the cyclohexyl protons, were found to be much more stable than TS-Anti-Re′ or TS-Anti-Re″, in which one of these stabilizing non-classical hydrogen bonding interactions are missing. This preference is somewhat diminished with solvation corrections (DCE results shown in Figure 4), as expected for any electrostatic interactions.
Figure 4.
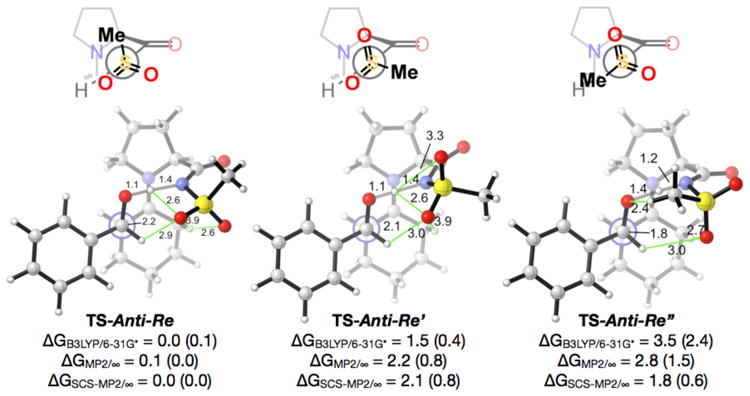
35 Three Anti-Re transition structures for the aldol reaction between cyclohexanone enamine of proline-sulfonamide and benzaldehyde, showing three different conformations of the sulfonamide. Distances are in Ångstroms, energies in kcal/mol. Green lines designate possible stabilizing electrostatic interactions. Structures and thermodynamic corrections computed using B3LYP/6–31G* in the gas phase. Numbers in parenthesis include solvation corrections for DCE. ∞ designates infinite basis set extrapolation from the Dunning cc-pVTZ and cc-pVQZ basis sets.
Non-classical hydrogen bonds, or electrostatic interactions, are prevalent in literature. Corey was one of the pioneers to propose formyl δ+CH—Oδ− non-classical hydrogen bonds as stabilizing interactions.36 Non-classical hydrogen bonds have been subsequently invoked by other groups to explain stabilizing relationships.37 With respect to organocatalysis, Houk and co-workers pointed out that stabilizing electrostatic interactions between the CH vicinal to the forming iminum and the forming alkoxide (referred to as the δ+NCH—Oδ− interactions) contributes to the stereoselectivity of proline-catalyzed aldol reactions.18a These non-classical hydrogen bonding electrostatic interactions were also demonstrated as stabilizing interactions in stereoselective additions of chiral alcohols to ketenes.38 The magnitude of these stabilizing interactions have also been quantified and have been demonstrated to be significant even in the solution.39
We did consider the possibility that the enhanced diastereoselectivity of the proline sulfonamide catalysts over proline may be due to increased torsional strain around the forming bond that stabilize the TS-Anti-Re over other transition states. Interestingly, juxtaposing the proline sulfonamide transition structures with those published for proline,18b we find no evidence that proline-sulfonamide transition structures exhibit drastically different torsional strain than proline transition structures. The torsional differences that do exist cannot explain the >3 kcal/mol greater selectivity exhibited by proline sulfonamides compared to proline.
Variation of Sulfonamide Scaffold
We also explored additional modification of the sulfonamide scaffold (Table 3). For this series, the aldol reaction was conducted at room temperature. The baseline experiment with the sulfonamide 1 provides high chemical yield, diastereoselectivity and enantioselectivity (Entry 1). Recently, we reported the development of a second generation (compound 13) of our catalyst system in which a dodecyl ester is placed on the aromatic ring.40 This catalyst proved more effective than catalyst 1 in the formation of cyclohexenones containing a stereogenic all-carbon quaternary center. Interestingly, this catalyst 13 proved significantly less reactive than its parent 1 in the aldol reaction (Entry 2). While the enantioselectivity and diastereoselectivity were comparable to catalyst 1, a nearly four-fold decrease in reactivity was observed. This difference is likely due to the presumed increased acidity of the sulfonamide N-H in the modified catalyst 13. In addition to this electronic impact, catalyst 13 is also less soluble than catalyst 1. This solubility difference may be explained by the difference in the dodecyl chain – linear, homogenous C12H25 chain in catalyst 13 and a mixture of branched and linear isomers (e.g. 1-dodecyl, 2-doceyl etc) in catalyst 1. As a complement to this study, we also explored the removal of the carbonyl of the amide carbon (catalyst 14). This previously unknown catalyst 14 led to dramatically lower levels of diastereoselectivity (Entry 3). Removal of the amide carbonyl should decrease the sulfonamide N-H acidity. Finally, we also probed the placement of an additional Lewis base on the sulfonamide scaffold through pyridyl sulfonamide 15. While catalyst 15 had not been previously prepared, a closely related desmethyl version was utilized by Nakamura and co-workers in an aldol reaction between acetaldehyde and 4,6-dibromoisatin with modest success (77% yield, 80% ee).41 In our system, pyridyl catalyst 15 gave greatly reduced diastereoselectivity in the aldol reaction (Entry 4). These experiments point to the delicate balance between the nature of the sulfonamide moiety and reaction rate and diastereoselectivity.
Table 3.
Comparison of Proline Sulfonamides.a
| Entry | Catalyst | Time (h) | Yield | eeb | drc |
|---|---|---|---|---|---|
| 1 | 1 | 16 | 96 | 97 | 36:1 |
| 2 | 13 | 60 | 97 | 97 | 26:1 |
| 3 | 14 | 18 | 96 | -d | 1.5:1 |
| 4 | 15 | 24 | 88 | -d | 2.4:1 |
All reactions were performed at 2 M concentration of 6 in solution and with five equivalents of 5.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.
Not determined due to poor diastereoselectivity.

We hypothesize that the sulfonamide conformation is key for stereocontrol. The electronics of the sulfonamide moiety is critical in determining this conformation of the sulfonamide (Figure 5). In catalysts 1 and 13, the attenuation of the developing negative charge on the nitrogen via conjugation to the carbonyl, allows the sulfonamide conformational freedom to electrostatically complement the incoming electrophile in the critical C-C bond forming TS (Bottom, Figure 5). The exposure of the aldehyde hydrogen to the catalyst organic base moiety in the anti-re TS is ideally suited to benefit energetically from this conformational freedom. This is in contrast to the re-face approach, in which the sterically bulky phenyl group is universally poorly accommodated by the any conformation of the sulfonamide (Figure 3). Thus, sulfonamides in catalysts 1 and 13 are conformationally more flexible and can change conformations to better electrostatically suit the anti-re TS, leading to high selectivity. The sulfonamide in catalyst 14 is more conformationally rigid (the double bond between N=S makes the sulfonamide more rigid – Top, Figure 5) and poorly stabilizes the anti-re TS – leading to the observed poor selectivity.
Figure 5.

Effect of Carbonyl Motif on Catalyst Performance
The lower diastereoselectivities observed with catalysts 14 and 15 can be rationalized through analysis of their structural features. In catalyst 14, the sulfonamide is the only functional group stabilizing the nitrogen anion. We hypothesize that the remarkable degradation in diastereoselectivity of catalyst 14 is due to the sulfonamide conformation being locked to achieve this conjugation. This in effect results in the loss of electrostatic distinction between the anti-re and anti-si TS, and leads to loss of selectivity. The change from sp2 to sp3 hybridization at the starred carbon in catalyst 14 likely introduces flexibility in the sulfonamide group that may decrease the differentiation between the Anti-Re and Anti-Si TS, leading to lower selectivities. Wang and co-workers have shown that further increase in the electron withdrawing capacity of the sulfonamide scaffold by incorporation of a trifluoromethyl moiety can counterbalance this effect.24n In catalyst 15, we believe the additional internal base may participate in the key proton transfer event of the critical C-C bond forming transition state, as previously suggested by Nakamura.41 Such participation will impart conformational rigidity to the sulfonamide moiety, leading to poor selectivity.
Alternate explanations for the differences in selectivities between the various sulfonamide catalysts are possible. One alternative is based on the pKa differences between the various catalysts. The pKa of sulfonamides are generally viewed as similar to carboxylic acids24c and can conceivably be modulated through either the addition of electron-withdrawing or electron-donating moiety on the sulfonamide aromatic ring40 or modification of the carbonyl moiety. Wu and co-workers have previously postulated that the repulsive interaction between the proton being transferred and the electrophile substituent destabilize the Anti-Si TS and could contribute to the enhanced selectivity of proline-amide catalyzed aldol reactions.42 The modulation of the sulfonamide pKa could effect this interaction and may be a factor in the resulting differential selectivities.
Substrate Scope
We next set out to screen a range of aromatic aldehydes to probe the scope of this transformation (Table 4). In the vast majority of cases, the transformation performed remarkable well. High levels of enantioselectivity, diastereoselectivity and chemical yield were observed in most cases. Aldol reactions on even sterically congested aldehydes 16b and 16c proved reasonable effective. Single ortho substitution did not appear adversely impact reactivity (Entries b–c and e–f). Current limitations to the protocol appear to include electron rich aldehydes, which showed sluggish reactivity (Entry g).
Table 4.
Scope of Aldehyde Moiety at 4°C.a
| Entry | R | Time (h) | Yield (Product) | eeb | drc |
|---|---|---|---|---|---|
| a | pentafluorophenyl- | 36 | 91 (17a) | >99 | >99:1 |
| b | 2,4-dinitrophenyl- | 72 | 51 (17b) | 97 | >99:1 |
| c | 2,6-dichlorophenyl- | 72 | 94 (17c) | 99 | >99:1 |
| d | 4-chlorophenyl- | 48 | 68 (17d) | 99 | >99:1 |
| e | 2-nitrophenyl- | 72 | 91 (17e) | 99 | >99:1 |
| f | 2-chlorophenyl- | 72 | 79 (17f) | 98 | >99:1 |
| g | 4-methoxyphenyl- | 72 | 16 (17g) | 98 | 80:1 |
| h | phenyl- | 60 | 60 (17h) | 99 | >99:1 |
| i | 4-pyridyl- | 24 | 98 (17i) | 99 | 29:1 |
| j | 1-naphthyl- | 72 | 19 (14j) | 99 | >99:1 |
| k | 2-naphthyl- | 72 | 51 (14k) | >99 | >99:1 |
All reactions were performed at 2 M concentration of 16 in solution and with five equivalents of 5.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.

We also wanted to screen the scope of these reactions performed at room temperature (Table 5). While it was expected that increased reaction temperature would increase reactivity while possibly reducing stereoselectivity, the added practicality of avoiding externally cooling a reaction was attractive. Fortunately, the overall stereoselectivities in most cases at room temperature were still high. In fact, the dinitrophenyl case gave significantly higher yields at room temperature than at 4°C (Entry 2). One substrate class that was ineffective at lower temperatures was aliphatic aldehydes. Fortunately, it appears that improved reactivity provided by conducting the experiment at room temperature did facilitate a modestly effective transformation with 59% ee and 32% chemical yield (Entry 6). These types of aldehydes are particularly challenging as self-aldol condensation is often a major competing transformation.43 Currently, 4-hydroxyproline derivatives have proven most effective at facilitating related aldol reactions with aliphatic aldehydes.32c,d
Table 5.
Scope of Aldehyde Moiety at room temperature.a
| Entry | R | Time (h) | Yield (Product) | eeb | drc |
|---|---|---|---|---|---|
| 1 | pentafluorophenyl- | 13 | 97 (17) | >99 | >99:1 |
| 2 | 2,4-dinitrophenyl- | 40 | 99 (17) | 96 | 57:1 |
| 3 | 2,6-dichlorophenyl- | 48 | 99 (17) | 99 | >99:1 |
| 4 | 4-pyridyl- | 12 | 92 (17) | 99 | 50:1 |
| 5 | 2-naphthyl- | 48 | 63 (17k) | >99 | >99:1 |
| 6 | phenylethyl- | 72 | 32 (17l) | 59 | 29:1 |
All reactions were performed at 2 M concentration of 16 in solution and with five equivalents of 5.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.

While DCE proved an excellent solvent for facilitating highly stereoselective aldol reactions, it has been designated a Class 1 genotoxin.44,45 Consequently, we sought to explore alternative solvent choices, which might be viewed as more industrial friendly (Scheme 2). 2-Methyltetrahydrofuran (2-Me-THF) is rapidly becoming an industrially friendly alternative to chlorinated solvents.46 The cost of production of this solvent has come down significantly in recent years. Unlike THF, 2-Me-THF provides good phase splits with water, which can facilitate easier solvent recycling. We were pleased to see that 2-Me-THF can be substituted for DCE with only minimal disruption in the levels of enantioselectivity. The experiments were conducted at room temperature and provided high levels of chemical yield, enantioselectivity and diastereoselectivity. These examples coupled with the entry detailed in Table 1 indicate that there is significant promise in utilizing the catalyst 1 in non-chlorinated solvents such as 2-Me-THF.
Scheme 2.

Scope of Aldehyde Moiety at 2-Methyltetrahydrofuran as solvent.a,b,c
a All reactions were performed at 2 M concentration of 16 in solution and with five equivalents of 5. b Enantiomeric excess was determined by chiral HPLC analysis. c Diastereomeric ratios (dr) were determined by 1H NMR analysis.
We also explored the utility of neat reaction conditions using the low catalyst loading conditions (2 mol % 1, neat, rt) on a range of aldehyde substituents (Table 6). Please note that these 2 mol % catalyst experiments were performed with a reduced loading of 5 (2 equiv.) and only a single equivalent of both the aldehyde and water. These transformations generally performed well - with enantioselectivities greater than 90% ee in each case. The diastereoselectivity in neat reactions was somewhat reduced as compared to the experiments conducted in an organic solvent. We attribute that result to the impact of the solvent’s polarity on the diastereomeric transition states as compared to the neat component reactions. It should be noted that in most cases, both 5 and the aldehyde 16 were not readily miscible with the one equivalent of water. We hypothesize that the polar pyrrolidine portion and the aliphatic sidearm of the sulfonamide catalyst may function as a phase transfer agent for this transformation.
Table 6.
Scope of Aldehyde Moiety under Low Catalyst Loading.a
| Entry | R | Time (h) | Yield (Product) | eeb | drc |
|---|---|---|---|---|---|
| 1 | 2,4-dinitrophenyl- | 48 | 50 (17b) | 93 | >99:1 |
| 2 | 4-chlorophenyl- | 48 | 49 (17d) | 92 | >99:1 |
| 3 | 2-chlorophenyl- | 72 | 67 (17f) | 99 | >99:1 |
| 4 | 4-hydroxyphenyl- | 72 | 22 (17m) | 90 | 26:1 |
| 5 | phenyl- | 48 | 75 (17h) | 99 | 55:1 |
| 6 | 4-pyridyl- | 36 | 91 (17i) | 91 | 7:1 |
| 7 | 1-naphthyl- | 72 | 36 (17j) | 88 | 96:1 |
All reactions were performed with two equivalents of 5.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.

Given the procedural ease for accomplishing these aldol reactions, we thought it would be valuable to demonstrate the utility of this protocol for conducting a mole scale reaction (Scheme 3). The experiment was conducted in a single 500 milliliter round bottom capped flask using 235 grams of cyclohexanone and 181 grams of 4-nitrobenzaldehyde with just 2 mol % (10.1 grams) of the sulfonamide catalyst 1 and a single equivalent of water (21.6 milliliters). Shown in the scheme below is the photograph of the experiment. The solid stirring around in the reaction is the product 7 which was filtered and washed with hexanes to isolate. Over 60% of the catalyst 1 could be recovered via simple purification of the mother liquor through a plug of silica gel followed by a single recrystallization in methanol.
Scheme 3.

Large Scale Example of Aldol Reaction.a,b
a Enantiomeric excess was determined by chiral HPLC analysis. b Diastereomeric ratios (dr) were determined by 1H NMR analysis.
We investigated the impact of substitution of the cyclohexanone ring in the aldol process (Scheme 4). This reaction worked similarly well with either the methyl or tert-butyl substituted cyclohexanones. In each case, high levels of stereoselectivity and chemical yield were observed. In each experiment, a total of three stereogenic centers are established.
Scheme 4.

Use of Substituted Cyclohexanones in Aldol Reactions.a,b,c
a All reactions were performed at 2 M concentration of 6 in solution and five equivalents of ketone 18. b Enantiomeric excess was determined by chiral HPLC analysis. c Diastereomeric ratios (dr) were determined by 1H NMR analysis.
The use of acetone as a potential nucleophile in the aldol process was also screened (Table 7). This reaction appeared to work most effectively using our optimized conditions [DCE, H2O (1 equiv.), 4°C] to provide 66% yield of the desired product in a reasonable (84% ee) enantioselectivity. Performing the transformation at room temperature led to reduced chemical yields and enantioselectivity. Interestingly, the same reaction performed in the absence of added water gave comparable levels of enantioselectivity to the optimized conditions. We are unsure as to the rational of this result, but this outcome may be in part due to the hydroscopic nature of acetone, which was not rigorously dried prior to use.
Table 7.
Use of Acetone in Organocatalyzed Reaction.
| Entry | Aldehyde | Mol % of 1 | Conditions | Time (h) | Yield | eea |
|---|---|---|---|---|---|---|
| 1 | 6 | 20 | DCE, H2O (1 equiv.), 4°C | 62 | 66 | 84 |
| 2 | 6 | 20 | DCE/EtOH (99:1), 4°C | 72 | 64 | 82 |
| 3 | 6 | 20 | DCE, rt | 30 | 80 | 80 |
| 4d | 6 | 2 | neat, H2O (1 equiv.), rt | 48 | 52 | 71 |
| 5 | 16h | 20 | DCE/EtOH (99:1), 4°C | 72 | 42 | 87 |
All reactions were performed at 2 M concentration of aldehyde in solution and was five equivalents of 20 unless otherwise noted.
Determined by chiral HPLC analysis.
This reaction was performed with two equivalents of 20.

We explored the impact of utilizing cyclopentanone instead of cyclohexanone in the aldol process (Table 8). Interestingly, the level of diastereoselectivty (anti/syn) was greatly eroded as compared to the cyclohexanone examples. This diastereoselectivity difference between cyclopentanone and cyclohexanone was first observed by Gong47 and Zhao48 independently; however, we are unaware of a mechanistic explanation for the difference outside of the increased reactivity of cyclopentanone-derived enamines.49 We screened both the low catalyst loading conditions [2 mol % 1, H2O (1 equiv.), neat] and the optimized 20 mol % catalyst loading conditions [DCE, H2O (1 equiv.) 4°C]. Under both sets of conditions, similar results were obtained.
Table 8.
Use of Cyclopentanone in Aldol Reaction.a
| Entry | Aldehyde | Mol % (1) | Conditions | Yield | eeb | drc |
|---|---|---|---|---|---|---|
| 1d | 6 | 2 | neat, H2O (1 equiv), 15 h, rt | 87 | 77 (syn), 91 (anti) | 1.05:1 |
| 3 | 6 | 2 | neat, 24 h, rt | 63 | 73 (syn), 74 (anti) | 3.2:1 |
| 4 | 6 | 20 | DCE, H2O (1 equiv), 16 h, 4°C | 94 | 87 (syn), 95 (anti) | 1.35:1 |
| 5 | 6 | 20 | DCE/EtOH (99:1), 36 h, 4°C | 87 | 85 (syn), 92 (anti) | 2:1 |
| 6 | 16f | 20 | DCE, H2O (1 equiv), 48 h, 4°C | 51 | 81 (syn), 93 (anti) | 1.5:1 |
All reactions were performed at 2 M concentration of aldehyde in solution and was five equivalents of 22 unless otherwise noted.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.
This reaction was performed with two equivalents of 22.

We explored the potential use of 4-pyranone in the aldol reaction process with a series of aromatic aldehydes (Table 9). 4-Pyranones are present in a wide range of natural product scaffolds.50 Xiao and co-workers have focused specifically on exploiting the utility of organocatalysis for the heteroatom-containing cyclic ketone substrates.51 In each case, our optimized reaction conditions [DCE, H2O (1 equiv.) 4°C] provided superior results to alternative procedures performed at room temperature or using alternative additives. Aldehyde 16d was particularly effective as excellent levels of enantioselectivity and diastereoselectivity were achieved (Entry 4).
Table 9.
Use of 4-Pyranone in Aldol Reactions.a
| Entry | Aldehyde | Conditions | Time (h) | Yield | eeb | drc |
|---|---|---|---|---|---|---|
| 1 | 6 | DCE/EtOH (99:1), rt | 16 | 16 | 88 | 4:1 |
| 2 | 6 | DCE, H2O (1 equiv.), 4°C | 52 | 92 | 96 | 5:1 |
| 3 | 16d | DCE, H2O (1 equiv.), rt | 16 | 52 | 94 | 13:1 |
| 4d | 16d | DCE, H2O (1 equiv.), 4°C | 72 | 60 | 97 | >99:1 |
| 5 | 16h | DCE, H2O (1 equiv.), rt | 16 | 27 | 90 | 41:1 |
| 6 | 13h | DCE, H2O (1 equiv.), 4°C | 72 | 47 | 94 | 41:1 |
All reactions were performed at 2 M concentration of aldehyde in solution and was five equivalents of 24.
Determined by chiral HPLC analysis.
Determined by 1H NMR analysis.

The potential use of 4-thiopyranone in the aldol reaction process was also explored (Scheme 5). Thiopyranones have been exploited as polyproprioate surrogates through Raney-Ni removal of the carbon-sulfur bonds after coupling.52 The aldol reaction proved sufficiently effective that they could be performed at room temperature with high levels of stereoselectivity. We attribute the difference between pyranone and thiopyranone to the modulated reactivity imparted by the presence of the sulfur moiety.
Scheme 5.

Use of 4-Thiopyranone in Aldol Reactions.a,b,c
a All reactions were performed at 2 M concentration of aldehyde in solution and was five equivalents of 26. b Enantiomeric excess was determined by chiral HPLC analysis. c Diastereomeric ratios were determined by 1H NMR analysis.
Glycolate aldol reactions have proven particular significant for construction of 1,2-anti and 1,2-syn diol functionality.53 We were pleased to see that our organocatalyzed process could again provide reasonable levels of stereoselectivity in this type of transformation (Scheme 6). While the chemical yield and enantioselectivity appear for this transformation to be insensitive to reaction temperature, the diastereoselectivity was improved when the reaction was carried out at lower temperatures.
Scheme 6.

Use of Glycolates in Aldol Reactions.a,b,c
a All reactions were performed at 2 M concentration of 6 in solution and was five equivalents of 24. b Enantiomeric excess was determined by chiral HPLC analysis. c Diastereomeric ratios (dr) were determined by 1H NMR analysis.
Conclusion
A practical asymmetric aldol process has been developed based on the proline aryl sulfonamide scaffold. These reactions have been shown to be effective on a wide range of substrates in generally high levels of enantioselectivity. The presence of a single equivalent of water was shown to be beneficial to the reaction yield, enantioselectivity and diastereoselectivity. Particular utility has been showcased in non-polar organic solvents such as DCE and 2-Me-THF. Non-polar solvents increase the practicality of the chemistry for applications in industrial settings as the solvents is more readily recyclable. Alternatively, the sulfonamide catalyzed aldol reaction was also shown to be effective in aqueous media with generally good levels of stereoselectivity. We hypothesized that the proline sulfonamide organocatalyst may be acting as a phase transfer catalyst between the aqueous and non-polar media. Catalyst loading as low as 2 mol % was demonstrated using neat reaction conditions with generally high levels of stereoselectivity. The practicality of this protocol has been demonstrated on a one mole scale reaction conducted in a single 500 milliliter round-bottom flask. Detailed computational analysis established that non-classical hydrogen bonds to the sulfonamide lead to improve stereoselectivity in this system. We anticipate that this catalyst system will find application in natural product synthesis and industrial processes.
Experimental Section
Sulfonamide 14
To a solution of 1 (0.422 g, 1.00 mmol) in THF (10 mL) was added lithium aluminum hydrid (0.190 g, 5.0 mmol) slowly at 0°C. After stirring at 0°C for 2 h, the solution was quenched with H2O (10 mL) and extracted with CH2Cl2 (3 × 25 mL). The dried (Na2SO4) extract was concentrated in vacuo and purified chromatography over silica gel, eluting with 10% MeOH/dichloromethane, to give 14 (0.346 g, 0.848 mmol, 85%) as a colorless oil: [α]D23 = +21.4 (c=1.9, CHCl3); IR (neat) 3276, 2954, 2916, 2856, 1451, 1408, 1331, 1146, 1097, 825, 645 cm−1; 1H NMR 400 MHz, CDCl3) δ 7.77 (d, J = 7.6 Hz, 2H), 7.27–7.33 (m, 2H), 3.87 (br s, 2H), 3.31–3.33 (m, 1H), 2.57–3.04 (m, 4H), 0.75–1.86 (m, 28H); 13C NMR (100 MHz, CDCl3) δ 137.3, 128.3, 127.7, 127.0, 57.2, 47.2, 46.3, 40.0, 38.9, 38.2, 36.7, 36.4, 31.9, 29.7, 29.3, 28.9, 27.5, 27.2, 25.8, 22.7, 22.0, 20.6, 14.1, 12.1; HRMS (EI+) calcd. for C23H40N2O2S (M+) 408.2811, found 408.2787.
Z-L-sulfonamide 32
To a solution of Z-L-proline 30 (1.25 g, 5.0 mmol) in CH2Cl2 (50 mL) was added sulfonamide 31 (1.04 g, 6.0 mmol), EDCI (0.96 g, 5.0 mmol) and DMAP (0.122 g, 1.0 mmol) respectively. The reaction mixture was stirred at room temperature for 48 h before being partitioned between EtOAc (100 mL) and water (50 mL). The organic layer was washed with half-saturated brine (2 × 50 mL). The dried (Na2SO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 20–70% EtOAc/CH2Cl2, to give 32 (1.59 g, 3.95 mmol, 79%) as a white solid. Mp: 136–137°C; [α]D23 = −129.7 (c=3.1, CHCl3); IR (neat) 3069, 2953, 2871, 1704, 1684, 1455, 1420, 1357, 1190, 1132, 1108, 739, 700, 665 cm−1; 1H NMR (400 MHz, DMSO-d6, two rotamers) δ 8.37 (s, 1H), 8.30 (s, 1H), 7.59–7.76 (m, 2H), 7.21–7.35 (m, 5H), 4.78–5.03 (m, 2H), 4.02–4.06 (m, 1H), 2.33 (s, 3H), 2.27 (s, 3H), 1.71–2.10 (m, 3H); 13C NMR (100 MHz, MeOD, two rotamers) δ 181.3, 155.9, 155.6, 154.9, 148.9, 148.7, 139.5, 139.3, 137.9, 137.8, 136.6, 128.2, 128.0, 127.7, 127.3, 127.2, 126.2, 127.7, 66.7, 66.2, 62.5, 62.1, 47.1, 46.7, 31.1, 30.3, 23.8, 23.1, 17.2; HRMS (EI+) calcd. for C19H21N3O5S (M+) 403.1202, found 403.1219.
Sulfonamide 15
To a solution of Z-L-sulfamide 32 (0.12 g, 0.298 mmol) in MeOH (3 mL) was added Pd/C (12 mg, 10%). The mixture was stirred at rt under an atmosphere of hydrogen. After 13 h, the reaction was filtered through Celite and silica gel pad, and the filtrate was concentrated in vacuo to give white solid. The crude product was purified by chromatography over silica gel, eluting with 10% MeOH/CH2Cl2, to give the product 15 (71.2 mg, 0.265 mmol, 89%) as a white solid. Mp: 192–194°C; [α]D23 = −44.5 (c=0.2, EtOH); IR (neat) 3432, 3070, 3031, 2992, 1610, 1564, 1455, 1381, 1315, 1264, 1248, 1151, 1089, 789, 696, 649, 560 cm−1; 1H NMR (400 MHz, MeOD) δ 8.41 (s, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 7.2 Hz, 1H), 4.02–4.06 (m, 1H), 3.21–3.39 (m, 2H), 2.45 (s, 3H), 1.93–2.42 (m, 4H); 13C NMR (100 MHz, MeOD) δ 173.3, 157.3, 148.9, 137.8, 136.5, 121.9, 62.3, 45.8, 29.2, 23.4, 16.9; HRMS (ES+) calcd. for C11H16N3O3S (M+1) 270.0895, found 270.0912.
General Procedure for Aldol Reaction – Procedure A (DCE, 20 mol% catalyst, 4°C)
To a solution of aldehyde (0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in DCE (0.24 mL) was added sulfonamide 123 (42.2 mg, 0.1 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at 4°C. After stirring at same temperature, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the corresponding aldol product.
General Procedure for Aldol Reaction – Procedure B (DCE, 20 mol% catalyst, room temperature)
To a solution of aldehyde (0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in DCE (0.24 mL) was added sulfonamide 123 (42.2 mg, 0.1 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at room temperature. After stirring at same temperature, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the corresponding aldol product.
General Procedure for Aldol Reaction – Procedure C (neat, 2 mol% catalyst, room temperature)
To a solution of aldehyde (0.5 mmol) and cyclohexanone (0.098 g, 0.1 mL, 1.0 mmol, 2 equiv.) was added sulfonamide 123 (4.2 mg, 0.01 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at room temperature. After stirring at same temperature, reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the corresponding aldol product.
General Procedure for Aldol Reaction – Procedure D (DCE/EtOH (99:1), 20 mol% catalyst, room temperature)
To a solution of aldehyde (0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in DCE/EtOH (99:1, 0.24 mL) was added sulfonamide 123 (42.2 mg, 0.1 mmol) at room temperature. After stirring at same temperature, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the corresponding aldol product.
General Procedure for Aldol Reaction – Procedure E (2-Methyltetrahydrofuran, 20 mol% catalyst, room temperature)
To a solution of aldehyde (0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in 2-methyltetrahydrofuran (0.24 mL) was added sulfonamide 1 (42.2 mg, 0.1 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at room temperature. After stirring at same temperature, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the corresponding aldol product.
2-[Hydroxy-(4-nitro-phenyl)-methyl]-cyclohexan-1-one (7):1
Procedure A: To a solution of p-nitrobenzaldehyde (75.5 mg, 0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in DCE (0.24 mL) was added sulfonamide 123 (42.2 mg, 0.1 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at 4°C. After stirring at same temperature for 30 h, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the known aldol product 754 (118 mg, 0.474 mmol, 95%, 99% ee, >99:1 dr). Procedure B: To a solution of p-nitrobenzaldehyde (75.5 mg, 0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in DCE (0.24 mL) was added sulfonamide 123 (42.2 mg, 0.1 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at room temperature. After 14 h, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the aldol product 7 (119 mg, 0.478 mmol, 96%, 97% ee, 36:1 dr). Procedure C: To a solution of p-nitrobenzaldehyde (75.5 mg, 0.5 mmol) and cyclohexanone (98.0 mg, 0.1 mL, 1.0 mmol, 2 equiv.) was added sulfonamide 123 (4.2 mg, 0.01 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at room temperature. After 36 h, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the aldol product 7 (119 mg, 0.478 mmol, 96%, 96% ee, >99:1 dr). Procedure D: To a solution of p-nitrobenzaldehyde (75.5 mg, 0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in DCE/EtOH (99:1, 0.24 mL) was added sulfonamide 123 (42.2 mg, 0.1 mmol) at room temperature. After 36 h, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the aldol product 7 (119 mg, 0.478 mmol, 96%, 97% ee, 14:1 dr). Procedure E: To a solution of p-nitrobenzaldehyde (75.5 mg, 0.5 mmol) and cyclohexanone (0.245 g, 0.26 mL, 2.5 mmol, 5 equiv.) in 2-Me-THF (0.24 mL) was added sulfonamide 123 (42.2 mg, 0.1 mmol) and water (0.5 mmol, 9 mg, 1 equiv.) at room temperature. After stirring at same temperature for 16 h, the reaction was loaded directly onto silica gel and was purified by chromatography, eluting with 10–30% EtOAc/hexanes, to give the known aldol product 7 (106 mg, 0.425 mmol, 85%, 94% ee, 30:1 dr). [α]D23 = +8.0 (c=1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 8.7 Hz, 2H), 7.53 (d, J = 8.7 Hz, 2H), 4.92 (dd, J = 8.4, 3.2 Hz, 1H), 4.10 (d, J = 3.2 Hz, 1H), 2.38–2.62 (m, 3H), 2.11–2.16 (m, 1H), 1.38–1.87 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 214.8, 148.4, 147.6, 127.9, 123.6, 74.0, 57.2, 42.7, 30.8, 27.7, 24.7; HPLC: Daicel Chiralpak AD. Hexanes-i-PrOH, 90:10, 1.0 mL min−1, 254 nm: tR (major) = 41.1 min; tR (minor) = 32.7 min.
Supplementary Material
Figure 1.

N-(p-Dodecylphenylsulfonyl)-2-pyrrolidinecarboxamide.
Scheme 1.

Development of an Organocatalyzed, Intramolecular Michael Addition and Application to Lycopodine.
Acknowledgments
Financial support was provided by the National Institutes of Health (NIH) (GM63723) and Oregon State University. National Science Foundation (CHE-0722319) and the Murdock Charitable Trust (2005265) are acknowledged for their support of the NMR facility. The authors would like to thank Professor Max Deinzer and Dr. Jeff Morré (OSU) for mass spectra data and Synthetech, Inc. for the generous gift of Cbz-proline. Finally, the authors are grateful to Dr. Roger Hanselmann (Rib-X Pharmaceuticals) for their helpful discussions. We acknowledge Christopher M. Sullivan and Matthew Peterson of the Center for Genome Research and Biocomputing (CGRB), Oregon State University, for computing cluster management.
Footnotes
SUPPORTING INFORMATION PARAGRAPH. Complete experimental procedures are provided, including 1H and 13C spectra, of all new compounds. Cartesian coordinates and energies for structures present in Figure 3 and 4 are also provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kane R. Ann Phys Chem. 1838;44:475. Ser. 2. [Google Scholar]
- 2.(a) Borodin A. J Prakt Chem. 1864;93:413–425. [Google Scholar]; (b) Podlech J Angew Chem Int Ed . 2010:49. doi: 10.1002/anie.201002023. Early View Release August 16, 2010. [DOI] [Google Scholar]
- 3.Kekulé A. Ber Dtsch Chem Ges. 1869;2:365–368. [Google Scholar]
- 4.Wurtz A. Bull Soc Chim Fr. 1872;17:436–442. [Google Scholar]
- 5.(a) Geary LM, Hultin PG. Tetrahedron: Asymm. 2009;20:131–173. [Google Scholar]; (b) Mahrwald R, editor. Modern Aldol Reactions. Wiley-VCH; 2004. [Google Scholar]
- 6.(a) Paterson I. In: Asymmetric Synthesis. 2. Ojima I, editor. Wiley-VCH; 2008. pp. 293–298. [Google Scholar]; (b) Brodmann T, Lorenz M, Schaeckel R, Simsek S, Kalesse M. Synlett. 2009:174–192. [Google Scholar]
- 7.(a) Jung ME, Salehi-Rad R. Angew Chem Int Ed. 2009;48:8766–8769. doi: 10.1002/anie.200904607. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jung ME, D’Amico DC. J Am Chem Soc. 1993;115:12208–12209. [Google Scholar]
- 8.Lohse-Frafel N, Carreira EM. Eur J Chem. 2009;15:12065–12081. doi: 10.1002/chem.200900529. [DOI] [PubMed] [Google Scholar]
- 9.Bahadoor AB, Flyer A, Micalizio GC. J Am Chem Soc. 2005;127:3694–3695. doi: 10.1021/ja050039+. [DOI] [PubMed] [Google Scholar]
- 10.Zimmerman HE, Traxler MD. J Am Chem Soc. 1957;79:1920–1923. [Google Scholar]
- 11.(a) Mahrwald R, editor. Modern Aldol Reactions. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (b) Mahrwald R. Aldol Reactions. Springer Dordrecht Heidelberg; New York: 2009. [Google Scholar]
- 12.Evans DA, Nelson JV, Taber TR. Top Stereochem. 1982;13:1–115. [Google Scholar]
- 13.(a) Johnson JS, Evans DA. Acc Chem Res. 2000;33:325–355. doi: 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]; (b) Carreira EM, Fettes A, Marti C. Org React. 2006;67:1–216. [Google Scholar]; (c) Miynarski J, Paradowska J. Chem Soc Rev. 2008;37:1502–1511. doi: 10.1039/b710577k. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Brindle CS. Chem Soc Rev. 2010;39:1600–1632. doi: 10.1039/b923537j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Prelog V, Wihelm M. Helv Chim Acta. 1954;37:1634–1660. [Google Scholar]; (b) Yamada SI, Otani G. Tetrahedron Lett. 1969:4237–3240. [Google Scholar]; (c) Eder U, Sauer G, Wiechert R. Angew Chem, Int Ed Engl. 1971;10:496–497. [Google Scholar]; (d) Hiroi K, Achiwa K, Yamada SI. Chem Pharm Bull. 1972;20:246–257. [Google Scholar]; (e) Hiroi K, Yamada SI. Chem Pharm Bull. 1973;21:47–53. [Google Scholar]; (f) Otani G, Yamada SI. Chem Pharm Bull. 1973;21:2112–2118. [Google Scholar]; (g) Hajos ZG, Parrish DR. J Org Chem. 1974;39:1615–1621. [Google Scholar]; (h) Hajos ZG, Parrish DR. Org Synth. 1990;7:363–368. Coll. [Google Scholar]
- 15.For recent reviews, see: Zlotin SG, Kucherenko AS, Belskaya IP. Russ Chem Rev. 2009;78:737–784.Melchiorre P, Marigo M, Carlone A, Bartoli G. Angew Chem, Int Ed. 2008;47:6138–6171. doi: 10.1002/anie.200705523.Dondoni A, Massi A. Angew Chem, Int Ed. 2008;47:4338–4360.Longbottom DA, Franckevicius V, Kumarn S, Oelke AJ, Wascholowski V, Ley SV. Aldrichim Acta. 2008;41:3–11.Verkade JMM, van Hemert LJC, Quaedflieg PJLM, Rutjes FPJT. Chem Soc Rev. 2008;37:29–41. doi: 10.1039/b713885g.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016.Ting A, Schaus SE. Eur J Org Chem. 2007;2007:5797–5815.
- 16.Select Auxiliary-Based Examples: Oppolzer W, Marco-Contelles J. Helv Chim Acta. 1986;69:1699–1703.Walker MA, Heathcock CH. J Org Chem. 1991;56:5747–5750.Gennari C, Hewkin CT, Molinari F, Bernardi A, Comotti A, Goodman JM, Paterson I. J Org Chem. 1992;57:5173–5177.Abiko A, Liu JF, Masamune S. J Am Chem Soc. 1997;119:2586–2587.Paterson I, Wallace DJ, Cowden CJ. Synthesis. 1998:639–652.Evans DA, Tedrow JS, Shaw JT, Downey CW. J Am Chem Soc. 2002;124:392–393. doi: 10.1021/ja0119548.Andrus MB, Meredith EL, Hicken EJ, Simmons BL, Glancey RR, Ma W. J Org Chem. 2003;68:8162–8169. doi: 10.1021/jo034870l.Ghosh AK, Kim JH. Org Lett. 2003;5:1063–1066. doi: 10.1021/ol034086n.Crimmins MT, McDougall PJ. Org Lett. 2003;5:591–594. doi: 10.1021/ol034001i.Zhang W, Carter RG, Yokochi AFT. J Org Chem. 2004;69:2569–2572. doi: 10.1021/jo035829l.Fanjul S, Humle AN. J Org Chem. 2008;73:9788–9791. doi: 10.1021/jo801720v.
- 17.Select Transition-Metal Mediated, Catalytic Asymmetric-Based Examples: Evans DA, MacMillan DWC, Campos KR. J Am Chem Soc. 1997;119:10859–10860.Yamashita Y, Ishitani H, Shimizu H, Kobayashi S. J Am Chem Soc. 2002;124:3292–3302. doi: 10.1021/ja016293t.Denmark SE, Pham SM, Stavenger RA, Su X, Wong KT, Nishigaichi Y. J Org Chem. 2006;71:3904–3922. doi: 10.1021/jo060243v.
- 18.(a) Bahmanyar S, Houk KN. J Am Chem Soc. 2001;123:12911–12912. doi: 10.1021/ja011714s. [DOI] [PubMed] [Google Scholar]; (b) Bahmanyar S, Houk KN, Martin HJ, List B. J Am Chem Soc. 2003;125:2475–2479. doi: 10.1021/ja028812d. [DOI] [PubMed] [Google Scholar]; (c) Allemann C, Gordillo R, Clemente FR, Cheong PHY, Houk KN. Acc Chem Res. 2004;37:558–569. doi: 10.1021/ar0300524. [DOI] [PubMed] [Google Scholar]; (d) Calderon F, Doyagez EG, Cheong PHY, Fernandez-Mayoralas A, Houk KN. J Org Chem. 2008;73:7916–7920. doi: 10.1021/jo800934b. [DOI] [PubMed] [Google Scholar]; (e) Clemente FR, Houk KN. Angew Chem Int Ed. 2004;43:5766–5768. doi: 10.1002/anie.200460916. [DOI] [PubMed] [Google Scholar]; (f) Cheong PHY, Houk KN. Synthesis. 2005:1533–1537. [Google Scholar]
- 19.For a preliminary account of this work, see: Yang H, Carter RG. Org Lett. 2008;10:4649–4652. doi: 10.1021/ol801941j.
- 20.(a) Chem Rev. 2007;(12):5413–5883. [Google Scholar]; (b) Dondoni A, Massi A. Angew Chem Int Ed. 2008;47:4638–4660. doi: 10.1002/anie.200704684. [DOI] [PubMed] [Google Scholar]; (c) MacMillan DWC. Nature. 2008;455:304–308. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]; (d) Merino P, Marques-Lopez E, Tejero T, Herrera RP. Tetrahedron. 2009;65:1219–1234. [Google Scholar]; (e) Xu L-W, Luo J, Lu Y. Chem Commun. 2009:1807–1821. doi: 10.1039/b821070e. [DOI] [PubMed] [Google Scholar]; (f) Palomo C, Oiarbide M, Lopez R. Chem Soc Rev. 2009;38:632–653. doi: 10.1039/b708453f. [DOI] [PubMed] [Google Scholar]; (g) Bella M, Gasperi T. Synthesis. 2009:1583–1614. [Google Scholar]; (h) Bertelsen S, Jørgensen KA. Chem Soc Rev. 2009;38:2178–2189. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]; (i) Alba AN, Companyo X, Viciano M, Rios R. Curr Org Chem. 2009;13:1431–1474. [Google Scholar]; (j) Liu X, Lin L, Feng X. Chem Commun. 2009:6145–6158. doi: 10.1039/b913411e. [DOI] [PubMed] [Google Scholar]; (k) Raj M, Singh VK. Chem Commun. 2009:6687–6703. doi: 10.1039/b910861k. [DOI] [PubMed] [Google Scholar]; (l) Merino P, Marques-Lopez E, Tejero T, Herrera RP. Synthesis. 2010:1–26. [Google Scholar]; (m) Grondal C, Jeanty M, Enders D. Nature Chem. 2010;2:167–178. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]; (n) Xu LW, Li L, Shi ZH. Adv Synth Catal. 2010;252:243–279. [Google Scholar]
- 21.(a) Kobayashi J, Morita H. Alkaloids. 2005;61:1–57. doi: 10.1016/s1099-4831(05)61001-2. [DOI] [PubMed] [Google Scholar]; (b) Ma X, Gang DR. Nat Prod Rep. 2004;21:752–772. doi: 10.1039/b409720n. [DOI] [PubMed] [Google Scholar]
- 22.(a) Bödeker K. Justus Liebigs Ann Chem. 1881;208:363. [Google Scholar]; (b) Achmatowicsz O, Uzieblo W. Rocz Chem. 1938;18:88–95. [Google Scholar]; (c) Ayer WA, Iverach GG. Tetrahedron Lett. 1962:87–92. doi: 10.1016/s0040-4039(00)72888-7. [DOI] [PubMed] [Google Scholar]; (d) Rogers D, Quick A, Hague M. Acta Cryst. 1974;B30:552–553. [Google Scholar]; (e) Hague M, Rogers D. J Chem Soc, Perkin Trans 2. 1975:93–98. [Google Scholar]
- 23.Yang H, Carter RG. J Org Chem. 2010;75:4929–4938. doi: 10.1021/jo100916x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berkessel A, Koch B, Lex J. Adv Synth Catal. 2004;346:1141–1146.Dahlin N, Bøegevig A, Adolfsson H. Adv Synth Catal. 2004;346:1101–1105.Cobb AJA, Shaw DM, Longbottom DA, Gold JB, Ley SV. Org Biomol Chem. 2005;3:84–96. doi: 10.1039/b414742a.Sundén H, Dahlin N, Ibrahem I, Adolfsson H, Córdova A. Tetrahedron Lett. 2005;46:3385–3389.Bellis E, Vasiatou K, Kokotos G. Synthesis. 2005:2407–2413.Wu Y, Zhang Y, Yu M, Zhao G, Wang S. Org Lett. 2006;8:4417–4420. doi: 10.1021/ol061418q.Silva F, Sawicki M, Gouverneur V. Org Lett. 2006;8:5417–5419. doi: 10.1021/ol0624225.Vogt H, Baumann T, Nieger M, Braese S. Eur J Org Chem. 2006;2006:5315–5338.Hayashi Y, Sumiya T, Takahashi J, Gotoh H, Urushima T, Shoji M. Angew Chem Int Ed. 2006;45:958–961. doi: 10.1002/anie.200502488.Aratake S, Itoh T, Okano T, Nagae N, Sumiya T, Shoji M, Hayashi Y. Chem Eur J. 2007;13:10246–10256. doi: 10.1002/chem.200700363.Huang J, Zhang X, Armstrong DW. Angew Chem Int Ed. 2007;46:9073–9077. doi: 10.1002/anie.200703606.For related series, see: Wang X-J, Zhao Y, Liu J-T. Org Lett. 2007;9:1343–1345. doi: 10.1021/ol070217z.Zu L, Xie H, Wang J, Wang W. Org Lett. 2008;10:1211–1214. doi: 10.1021/ol800074z.
- 25.Reichardt C. Solvents and Solvent Effects in Organic Chemistry. 3. WILEY-VCH; Weinheim: 2003. p. 653. [Google Scholar]
- 26.Dunn PJ, PG 10th Annual Green Chemistry and Engineering conference; Abstract 184. [See also Chem Eng News. 2006;84(30):36.].
- 27.Vinci D, Donaldson M, Hallett JP, John EA, Pollet P, Thomas CA, Grilly JD, Jessop PG, Liotta CL, Eckert CA. Chem Commun. 2007:1427–1429. doi: 10.1039/b616806j. [DOI] [PubMed] [Google Scholar]
- 28.(a) Aycock DF. Org Proc Res Dev. 2007;11:156–159. [Google Scholar]; (b) Butters M, Catterick D, Craig A, Curzons A, Dale D, Gillmore A, Green SP, Marziano I, Sherlock J-P, White W. Chem Rev. 2006;106:3002–27. doi: 10.1021/cr050982w. [DOI] [PubMed] [Google Scholar]; (c) Constable DJC, Jimenez-Gonzalez C, Henderson RK. Org Proc Res Dev. 2007;11:133–137. [Google Scholar]; (d) Delhaye L, Ceccato A, Jacobs P, Kottgen C, Merschaert A. Org Proc Res Dev. 2007;11:160–164. [Google Scholar]; (e) Fuenfschilling PC, Hoehn P, Mutz JP. Org Proc Res Dev. 2007;11:13–18. [Google Scholar]; (f) Anderson NG. Practical Process Research & Development. Academic Press; 2000. [Google Scholar]
- 29.(a) Metzger JO. Org Synth Highlights V. 2003:82–92. [Google Scholar]; (b) Varma RS, Ju Y. Green Separation Processes. 2005:53–87. [Google Scholar]; (c) Walsh PJ, Li H, de Parrodi CA. Chem Rev. 2007;107:2503–2545. doi: 10.1021/cr0509556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlson EK, Rathbone LK, Yang H, Collett ND, Carter RG. J Org Chem. 2008;73:5155–5158. doi: 10.1021/jo800749t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Wang J, E-Nunu T, Zu L, Jiang W, Wei S, Wang W. Chem Commun. 2007;50:7–509. doi: 10.1039/b611502k. [DOI] [PubMed] [Google Scholar]
- 32.Water effects in organocatalyzed aldol reactions: Nyberg AI, Usano A, Pihko PM. Synlett. 2004:1891–1896.Pihko PM, Laurikainen KM, Usano A, Nyberg AI, Kaavi JA. Tetrahedron. 2006;62:317–328.Hayashi Y, Sumiya T, Takahashi J, Gotoh H, Urushima T, Shoji M. Angew Chem, Int Ed. 2006;45:958–961. doi: 10.1002/anie.200502488.Aratake S, Itoh T, Okano T, Nagae N, Sumiya T, Shoji M, Hayashi Y. Chem Eur J. 2007;13:10246–10256. doi: 10.1002/chem.200700363.Li X-J, Zhang G-W, Wang L, Hu M-Q, Ma J-A. Synlett. 2008:1255–1259.Effects of water on organocatalyzed Mannich reactions: Itoh T, Yokoya M, Miyauchi K, Nagata K, Ohsawa A. Org Lett. 2003;5:4301–4304. doi: 10.1021/ol030103x.Notz W, Watanabe S, Chowdari NS, Zhong G, Betancort JM, Tanaka F, Barbas CF., III Adv Synth Cat. 2004;346:1131–1140.
- 33.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 34.(a) Weigend F, Köhn A, Hättig C. J Chem Phys. 2002;116:3175. [Google Scholar]; (b) Helgaker T, Klopper W, Koch H, Noga J. J Chem Phys. 1997;106:9639–9646. [Google Scholar]
- 35.Legault CY. CYLview BETA 1.0. Copyright © 2006–2010. [Google Scholar]
- 36.(a) Corey EJ, Rohde JJ, Fischer A, Azimioara MD. Tetrahedron Lett. 1997;38:33–36. [Google Scholar]; (b) Corey EJ, Rohde JJ. Tetrahedron Lett. 1997;38:37–40. [Google Scholar]; (c) Corey EJ, Barnes-Seeman D, Lee TW. Tetrahedron Lett. 1997;38:1699–1702. [Google Scholar]; (d) Corey EJ, Barnes-Seeman D, Lee TW. Tetrahedron Lett. 1997;38:4351–4354. [Google Scholar]
- 37.For a selected subset of examples, see: Gung BW, Xue W, Roush WR. J Am Chem Soc. 2002;124:10692–10697. doi: 10.1021/ja026373c.Fujiyama R, Goh K, Kiyooka SI. Tetrahedron Lett. 2005;46:1211–1215.Paton RS, Goodman JM. Org Lett. 2006;7:4299–4302. doi: 10.1021/ol061671q.Paton RS, Goodman JM. J Org Chem. 2008;73:1253–1263. doi: 10.1021/jo701849x.
- 38.Cannizzaro CE, Houk KN. J Am Chem Soc. 2004;126:10992–11008. doi: 10.1021/ja035899+. [DOI] [PubMed] [Google Scholar]
- 39.Cannizzaro CE, Houk KN. J Am Chem Soc. 2002;124:7163–7169. doi: 10.1021/ja012417q. [DOI] [PubMed] [Google Scholar]
- 40.Yang H, Carter RG. Org Lett. 2010;12:3108–3111. doi: 10.1021/ol1011955. [DOI] [PubMed] [Google Scholar]
- 41.Hara N, Nakamura S, Shibata N, Toru T. Chem Eur J. 2009;15:6790–6793. doi: 10.1002/chem.200900944. [DOI] [PubMed] [Google Scholar]; (b) Nakamura S, Hara N, Nakashima H, Kubo K, Shibata N, Toru T. Chem Eur J. 2008;14:8079–8081. doi: 10.1002/chem.200800981. [DOI] [PubMed] [Google Scholar]
- 42.Tang Z, Jiang F, Yu L-T, Cui X, Gong L-Z, Mi AQ, Jiang Y-Z, Wu Y-D. J Am Chem Soc. 2003;125:5262–5263. doi: 10.1021/ja034528q. [DOI] [PubMed] [Google Scholar]
- 43.List B, Pojarliev P, Castello C. Org Lett. 2001;3:573–575. doi: 10.1021/ol006976y. [DOI] [PubMed] [Google Scholar]
- 44.Müller L, Mauthe RJ, Riley CM, Andino MM, De Antonis D, Beels C, DeGeorge J, De Knaep AGM, Ellison D, Fagerland JA, Frank R, Fritschel B, Galloway S, Harpur E, Humfrey CDN, Jacks AS, Jagota N, Mackinnon J, Mohan G, Ness DK, O’Donovan MR, Smith MD, Vudathala G, Yotti L. Regul Toxicol Pharmacol. 2006;44:198–211. doi: 10.1016/j.yrtph.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 45.(a) Robinson DI. Org Process Res Dev. 2010;14:946–959. [Google Scholar]; (b) McGovern T, Jacobson-Kram D. TrAC Trends Anal Chem. 2006;25:790–795. [Google Scholar]
- 46.Aycock DF. Org Process Res Dev. 2007;11:157–159. [Google Scholar]
- 47.Tang Z, Yang ZH, Chen XH, Cun LF, Mi AQ, Jiang YZ, Gong LZ. J Am Chem Soc. 2005;127:9285–9289. doi: 10.1021/ja0510156. [DOI] [PubMed] [Google Scholar]
- 48.Samantha S, Liu J, Dodda R, Zhao CG. Org Lett. 2005;7:5321–5323. doi: 10.1021/ol052277f. [DOI] [PubMed] [Google Scholar]
- 49.Shechter H, Collis MJ, Dessy R, Okuzumi Y, Chen A. J Am Chem Soc. 1962;84:2905–2910. [Google Scholar]
- 50.(a) Turner WB. J Chem Soc, Perkin Trans. 1978;1:1621. [Google Scholar]; (b) Goehrt A, Grabley S, Thiericke R, Zeeck A. Lieb Ann. 1996:627–634. [Google Scholar]; (c) Pittayakhajonwut P, Suvannakad R, Thienhirun S, Prabpai S, Kongsaeree P, Tanticharoen M. Tetrahedron Lett. 2005;46:1341–1344. [Google Scholar]; (d) Lopez SN, Sierra Manuel G, Gattuso SJ, Furlan RL, Zacchino SA. Phytochem. 2006;67:2152–2158. doi: 10.1016/j.phytochem.2006.06.018. [DOI] [PubMed] [Google Scholar]; (e) Krick A, Kehraus S, Gerhaeuser C, Klimo K, Nieger M, Maier A, Fiebig HH, Atodiresei I, Raabe G, Fleischhauer J, Koenig GM. J Nat Prod. 2007;70:353–360. doi: 10.1021/np060505o. [DOI] [PubMed] [Google Scholar]; (f) Ahmed SA, Ross SA, Slade D, Radwan MM, Khan IA, ElSohly MA. Tetrahedron Lett. 2008;49:6050–6053. doi: 10.1016/j.tetlet.2008.07.178. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Tantray MA, Shawl AS, Ali N, Khuroo MA. Chem Nat Prod. 2008;44:424–426. [Google Scholar]
- 51.Chen JR, Li XY, Xing XN, Xiao WJ. J Org Chem. 2006;71:8198–8202. doi: 10.1021/jo0615089. [DOI] [PubMed] [Google Scholar]
- 52.(a) Jheengut V, Ward DE. J Org Chem. 2007;72:7805–7808. doi: 10.1021/jo701546f. [DOI] [PubMed] [Google Scholar]; (b) Beye GE, Goodman JM, Ward DE. Org Lett. 2009;11:1373–1376. doi: 10.1021/ol900192q. [DOI] [PubMed] [Google Scholar]
- 53.(a) Crimmins MT, She J. J Am Chem Soc. 2004;126:12790–12791. doi: 10.1021/ja0455852. [DOI] [PubMed] [Google Scholar]; (b) Vaughn JF, Hitchcock SR. Tetrahedron: Asymm. 2005;15:3449–3455. [Google Scholar]; (c) Davies SG, Nicholson RL, Smith AD. Org Biomol Chem. 2005;3:348–359. doi: 10.1039/b415943h. [DOI] [PubMed] [Google Scholar]; (d) Andrus MB, Liu J, Ye Z, Cannon JF. Org Lett. 2005;7:3861–3864. doi: 10.1021/ol051096a. [DOI] [PubMed] [Google Scholar]; (e) Denmark SE, Chung WJ. J Org Chem. 2008;73:4582–4595. doi: 10.1021/jo8006539. [DOI] [PubMed] [Google Scholar]; (f) Fanjul S, Hulme AN. J Org Chem. 2008;73:9788–9791. doi: 10.1021/jo801720v. [DOI] [PubMed] [Google Scholar]; (g) Gawas D, Kazmaier U. J Org Chem. 2009;74:1788–1790. doi: 10.1021/jo8025466. [DOI] [PubMed] [Google Scholar]
- 54.Wang C, Jiang Y, Zhang X, Huang Y, Li B, Zhang G. Tetrahedron Lett. 2007;48:4281–4285. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.