

Abstract
Rationale
The transcriptional code that programs maladaptive cardiac hypertrophy involves the zinc finger-containing DNA binding factor GATA-4. The highly related transcription factor GATA-6 is also expressed in the adult heart, although its role in controlling the hypertrophic program is unknown.
Objective
To determine the role of GATA-6 in cardiac hypertrophy and homeostasis.
Methods and Results
Here we performed a cardiomyocyte-specific conditional gene targeting approach for Gata6, as well as a transgenic approach to overexpress GATA-6 in the mouse heart. Deletion of Gata6-loxP with Nkx2.5-cre produced late embryonic lethality with heart defects, while deletion with β-myosin heavy chain-cre (βMHC-cre) produced viable adults with greater than 95% loss of GATA-6 protein in the heart. These later mice were subjected to pressure overload induced hypertrophy for 2 and 6 weeks, which showed a significant reduction in cardiac hypertrophy similar to that observed Gata4 heart-specific deleted mice. Gata6-deleted mice subjected to pressure overload also developed heart failure while control mice maintained proper cardiac function. Gata6-deleted mice also developed less cardiac hypertrophy following 2 weeks of angiotensin II/phenylephrine infusion. Controlled GATA-6 overexpression in the heart induced hypertrophy with aging and predisposed to greater hypertrophy with pressure overload stimulation. Combinatorial deletion of Gata4 and Gata6 from the adult heart resulted in dilated cardiomyopathy and lethality by 16 weeks of age. Mechanistically, deletion of Gata6 from the heart resulted in fundamental changes in the levels of key regulatory genes and myocyte differentiation-specific genes.
Conclusions
These results indicate that GATA-6 is both necessary and sufficient for regulating the cardiac hypertrophic response and differentiated gene expression, both alone and in coordination with GATA-4.
Keywords: Hypertrophy, transcription, differentiation, heart, genetically altered mice
Introduction
The myocardium can hypertrophy in response to injury, changes in workload, or increases in wall stress.1 In response to hypertrophic stimuli, a fundamental reprogramming occurs within the adult cardiomyocyte that results in the expression of genes encoding fetal protein isoforms. Genes such as skeletal α-actin, β-myosin heavy chain (βMHC), b-type natriuretic peptide (BNP), and atrial natriuretic factor (ANF) become highly expressed within ventricular myocytes.2, 3 Neural, humoral, and intrinsic stimuli result in the activation of membrane bound receptors that in turn activate specific intracellular signaling cascades such as mitogen-activated protein kinase (MAPK), protein kinase C (PKC), insulin-like growth factor-1 (IGF-1)/Akt, and the calcium activated protein phosphatase calcineurin.2, 3 These intracellular signaling cascades then modulate transcriptional regulatory proteins, altering gene expression to facilitate the growth of the heart. For example, transcription factors such as myocyte enhancer factor-2 (MEF-2) and nuclear factor of activated T-cells (NFAT) are directly activated by cytoplasmic signaling effectors, which in turn mediate hypertrophic gene expression in cardiomyocytes.2, 3 Similarly, the zinc finger-containing transcription factor GATA-4 can also serve as a terminal effector of the cardiac hypertrophic response following pathologic stimuli by direct phosphorylation from kinases.4, 5 GATA-4 can respond to mechanical load, vasopressin infusion, or direct stretching of the ventricles in an isolated rat heart.6, 7 However, the role of a closely related family member GATA-6 as a hypertrophic mediator is unknown, although GATA-4/6 are known to compensate for one another in the heart.8, 9
Six GATA transcription factors have been identified in vertebrates that are parsed into two subclasses based on their expression patterns. GATA-1, -2, and -3 are prominently expressed in hematopoietic cell lineages while GATA-4, -5, and -6 are expressed in various mesoderm and endoderm derived tissues such as heart, liver, lung, gonad, and gut.10, 11 GATA factors contain a highly conserved DNA binding domain consisting of two zinc fingers that interact with the nucleotide sequence element (A/T)GATA(A/G), which has been found in the promoters of most cardiac muscle-specific genes, especially those that are altered by the hypertrophic response.10 GATA-4 is expressed in the adult heart where it is thought to function as a key transcriptional regulator of numerous cardiac genes including ANF, BNP, αMHC,βMHC, and many others.10–12
Overexpression of Gata4 in culture by adenoviral gene transfer or in the hearts of transgenic mice each induced hypertrophy indicating the sufficiency of GATA-4 in this process.13 Expression of a dominant negative GATA-engrailed fusion protein or antisense Gata4 mRNA each blocked GATA-4-directed transcriptional responses and features of cardiomyocyte hypertrophy induced by phenylephrine (PE) and endothelin-1 in culture.4, 5, 13 GATA-4 is also directly phosphorylated by extracellular signal-regulated kinase (ERK1/2) and p38 MAPK as a means of enhancing hypertrophic gene expression.4, 5 In vivo, heart-specific deletion of Gata4 reduced the hypertrophic response to pressure overload stimulation and rapidly led to failure with reductions in cardiac angiogenesis.14, 15 Gata4 heterozygous mice were also more susceptible to heart failure after pressure overload stimulation, and showed more cardiac injury with doxorubicin administration.16,17 While overexpression of Gata6 in cultured neonatal myocytes was sufficient to induce a hypertrophic response,13 its role in controlling cardiac hypertrophy in vivo has not been investigated.
Materials and Methods
All procedures utilized in this study were described previously, although a full listing of the methods and materials is given in the online supplement. Mice used in these studies were either previously published15, 18 or generated with the tetracycline inducible cardiac specific promoter system.19 Transverse Aortic Constriction was induced in mice aged 8 to 12 weeks, after which mice were studied during the indicated time periods. Hearts were excised at the end of the experiment, used for morphometric measurements and stored in formalin or snap frozen in liquid nitrogen until further use.
Results
Heart-specific deletion of Gata6
Standard gene targeting of Gata6 in the mouse results in early embryonic lethality just prior to gastrulation,20 necessitating the use of conditional gene targeting with the cre-loxP system to investigate cardiogenesis and adult heart functions of this transcription factor. Here we crossed Gata6fl/fl mice with Nkx2.5-cre “knockin” mice and βMHC-cre transgenic mice. The Nkx2.5-cre allele, which is typically used to investigate early embryonic heart development given its early expression, resulted in efficient deletion of GATA-6 protein from the embryonic (E) day 14.5 heart as assessed by Western blotting, and complete embryonic lethality by birth (Supplemental Figure I A,B). Examination of hearts from these embryos showed prominent ventricular septal defects, irregular septal thicknesses, and some loss of trabeculae (Supplemental Figure I C).
Use of the βMHC-cre transgene results in slightly later embryonic deletion of loxP-targeted alleles from the heart,15 which bypassed embryonic lethality and produced viable adults. Western blotting of protein extracts from hearts of 1 day-old Gata6fl/flβMHC-cre mice showed a near complete absence of GATA-6 protein, as did immunohistochemistry in 8 week-old hearts of the same genotype (Figure 1A,B). Quantitation of nuclei from hearts of Gata6fl/flβMHC-cre mice showed that deletion was achieved in over 95% of all cells (P<0.05).
Figure 1.

Baseline characterization of Gata6-deleted hearts. A, Western blot for GATA-6 and GATA-4 in wildtype (Gata6fl/fl) and Gata6-deleted hearts from 1 day-old mice. Arrows indicate the long and short isoforms of GATA-6. B, Immunohistochemistry for GATA-6 (green), wheat germ agglutinin (red=membranes) and TO-PRO 3 (blue=nuclei) from hearts of 8 week-old control and Gata6-deleted mice. Original magnification x400. C, Gel shift assay for total GATA binding activity from 1 day-old heart lysates from control (Gata4fl/fl and Gata6fl/fl), Gata4-deleted (Gata4fl/flβMHC-cre) and Gata6-deleted (Gata6fl/flβMHC-cre) mice. D, Ventricular weight to body weight (VW/BW) ratio in 8 week-old mice. E, Adult cardiomyocytes isolated from 8 week-old Wt and Gata6-deleted hearts stained for α-actinin (red). Original magnification x600. F, Length measurements from isolated adult cardiomyocytes, 3 mice per group were analyzed and total number of cells is shown in the bars. G, Total surface area from isolated adult cardiomyocytes, 3 mice per group were analyzed and total number of cells analyzed is shown in the bars. H, BrdU positively stained nuclei per field from control and Gata6-deleted hearts from 3 day-old mice. I, Quantitation of phospho-histone H3 positively stained nuclei per field from control and Gata6-deleted hearts of 3 day-old mice. The number of mice or cells analyzed is shown in the bars of each panel. *P<0.05 vs control (for all panels).
Western blotting for GATA-6 usually detects 2 isoforms of GATA-6 arising from the same gene through alternative translational start sites.21, 22 However, their relative importance or potency in vivo is not known, although the shorter isoform is homologous in size with the single isoform present for GATA-4 and GATA-5, and hence is used in most studies.
We also generated Gata4fl/flβMHC-cre mice in parallel for comparison throughout our study, and remarkably, only the loss of Gata6 from the newborn heart (3 days-old) produced a reduction in total GATA DNA binding activity as measured by gel shift analysis (Figure 1C, Supplemental Figure II, see discussion). However, this reduction in total GATA binding activity in Gata6 heart-deleted mice may also reflect an observed significant downregulation of GATA-4 protein (Figure 1A). Finally, we also confirmed that the Gata6-loxP allele was not hypomorphic on its own, as protein expression of GATA-6 in the heart was identical to βMHC-cre control hearts (supplemental Figure III A).
While Gata6fl/flβMHC-cre mice were overtly normal well into adulthood, we did identify differences in their hearts compared with βMHC-cre and/or Gata6fl/fl mice (referred to as “control” because no differences were indentified in these 2 groups versus wildtype mice). For example, at 8 weeks of age Gata6fl/flβMHC-cre mice showed a small, albeit significant reduction in heart size compared with control mice (Figure 1D). Isolation and measurement of adult cardiomyocyte dimensions from Gata6fl/flβMHC-cre mice actually showed a significant increase in length and total area, despite the fact that these hearts are smaller (Figure 1E,F,G). This result suggested that loss of Gata6 impacted cardiomyocyte number in the heart, and indeed, measurement of myocyte DNA synthesis during the early neonatal period by BrdU incorporation and phospho-histone H3 analysis showed a significant reduction (Figure 1H,I). These results suggest that Gata6fl/flβMHC-cre mice have slightly smaller hearts with fewer myocytes compared with control mice. Despite these subtle differences, cardiac function was not different at baseline up to one year of age as assessed by echocardiography (Supplemental Table I).
Reduced pathologic hypertrophy in Gata6 heart-deleted mice
We previously showed that deletion of Gata4 from the heart resulted in less pathologic cardiac hypertrophy after 4 weeks of pressure overload stimulation.15 The adult heart also expresses GATA-6, which is nearly identical to GATA-4 in the DNA binding Zn-finger domains, suggesting that if it indeed recognizes and regulates the same downstream genes, it might similarly affect the hypertrophic response. Indeed, we determined that both GATA-4 and GATA-6 protein levels are increased in pressure overload-induced hypertrophic hearts, consistent with a very prominent increase in total GATA DNA binding activity by this stimulus (Figure 2A,B). To begin to elucidate the functional role that GATA-6 might play in regulating the cardiac hypertrophic response we subjected 8 week-old Gata6fl/flβMHC-cre mice to 2 weeks of transverse aortic constriction (TAC) stimulation. We first confirmed that the pressure overload stimulus was similar between groups (Supplemental Figure III B). Survival up to 6 weeks after surgery was similar between different groups (data not shown). Remarkably, Gata6fl/flβMHC-cre mice showed a significant reduction in the hypertrophic response compared to either Gata6fl/fl or βMHC-cre control groups at both the whole organ and cellular level (Figure 2C,D and Supplemental Figure III C). This defect in cardiac hypertrophy in the absence of GATA-6 also correlated with a greater sensitivity to cardiac dysfunction and heart failure, as ventricular fractional shortening was significantly reduced only in Gata6fl/flβMHC-cre mice after 2 weeks of TAC (Figure 2E). Similarly, hearts from Gata6fl/flβMHC-cre mice generally showed a greater induction of mRNA for fetal genes associated with heart failure and stress stimulation (Figure 2F,G,H). Despite the onset of heart failure and greater stress reactivity, no difference in cardiomyocyte death rates were observed between any of the surgical groups (data not shown).
Figure 2.

GATA-6 is required for pressure overload-induced cardiac hypertrophy. A, Western blot for GATA-4 and GATA-6 from cardiac nuclear protein extracts from sham and TAC operated wildtype mice. Lamin A/C was used as a loading control. B, Gel shift assay for total GATA binding activity in sham and TAC operated adult, wildtype mice from heart protein nuclear extracts. Arrow indicates GATA specific binding. C, VW/BW from sham and TAC-operated adult mice after 2 weeks of pressure overload. *P<0.05 vs sham, †P<0.05 vs βMHC-cre and Gata6fl/fl TAC. D. Cardiomyocyte cell surface area measured from histological sections. *P<0.05 vs sham; †P<0.05 vs βMHC-cre and Gata6fl/fl TAC. E, Cardiac fractional shortening percentage (FS%) from sham and TAC operated mice after 2 weeks of pressure overload, measured by M-mode echocardiography. *P<0.05 vs βMHC-cre and Gata6fl/fl TAC. Number of mice analyzed is shown in the bars. F, Taqman qPCR for expression of ANF mRNA in sham and TAC operated mice. G, Taqman qPCR for expression of αMHC mRNA in sham and TAC operated mice. *P<0.05 vs βMHC-cre and Gata6fl/fl sham, †P<0.05 vs βMHC-cre TAC. H, Taqman qPCR for expression of skeletal α-actin mRNA in sham and TAC operated mice. *P<0.05 vs βMHC-cre and Gata6fl/fl TAC.
We also conducted a parallel study in Gata4fl/flβMHC-cre mice subjected to 2 weeks of TAC stimulation (not previously done at this time point) to directly assess the potential difference in magnitude of effect versus Gata6 deletion. Gata4fl/flβMHC-cre mice also showed a significant reduction in the hypertrophic response at both the organ and cellular levels after 2 weeks of TAC, although the reduction was similar to that observed in Gata6fl/flβMHC-cre mice (Figure 3A,B). Gata4fl/flβMHC-cre mice also showed a similar reduction in ventricular performance by echocardiography after 2 weeks of TAC to that observed in Gata6fl/flβMHC-cre mice (Figure 3C). These results begin to suggest that GATA-4 and GATA-6 may have redundant functions in controlling the cardiac hypertrophic response.
Figure 3.

GATA-4 is required for pressure overload-induced cardiac hypertrophy. A, VW/BW from sham and TAC-operated adult mice after 2 weeks of pressure overload. *P<0.05 vs sham; †P<0.05 vs control (Gata4fl/fl) TAC. B, Cardiomyocyte surface area measured from histological sections. *P<0.05 vs sham; †P<0.05 vs control TAC. C, Cardiac FS% from sham and TAC-operated mice after 2 weeks of pressure overload. †P<0.05 vs control TAC. Number of mice analyzed is shown in the bars.
The results discussed above were obtained in mice subjected to pressure overload stimulation, but it was also of interest to determine if loss of GATA-6 from the heart impacted hypertrophy due to neuroendocrine-like stimuli. To this end we implanted osmotic minipumps in young adult mice for 2 weeks containing angiotensin II/PE (AngII/PE). As expected, the combination of AngII/PE induced hypertrophic growth of hearts from control mice, although Gata6fl/flβMHC-cre mice showed significantly less heart growth (Figure 4A). As with TAC stimulation, this defect in hypertrophic growth in Gata6fl/flβMHC-cre mice also correlated with a significant reduction in cardiac function as assessed by fractional shortening and more ventricular dilation (Figure 5B,C). Similarly, hearts from Gata6fl/flβMHC-cre mice showed a greater induction of mRNA for fetal genes associated with heart failure and stress stimulation after AngII/PE infusion, compared to hearts from control mice (Figure 4D,E,F). Taken together, these results indicate that loss of GATA-6 from the adult heart compromises the hypertrophic program to pathologic stimulation, rendering the heart more susceptible to dysfunction.
Figure 4.

GATA6 is required for the AngII/PE induced cardiac hypertrophy response. A, VW/BW after 2 weeks of vehicle (Veh) or AngII/PE infusion using osmotic minipumps. *P<0.05 vs Gata6fl/fl Veh.; †P<0.05 vs Gata6fl/fl AngII/PE. B, Cardiac FS% from vehicle and Ang/PE infused mice. *P<0.05 vs Gata6fl/fl AngII/PE. C, Left ventricular end diastolic dimension (LVED) after 2 weeks of Veh or AngII/PE infusion. *P<0.05 vs Gata6fl/fl AngII/PE. D, Taqman qPCR for expression of ANF mRNA in Veh and AngII/PE infused mice. *P<0.05 vs Gata6fl/fl AngII/PE. E, Taqman qPCR for expression of αMHC mRNA in Veh and AngII/PE infused mice. *P<0.05 vs Gata6fl/fl Veh. F, Taqman qPCR for expression of skeletal α-actin mRNA in Veh and AngII/PE infused mice. *P<0.05 vs Gata6fl/fl AngII/PE.
Figure 5.

Gata6-deleted mice are susceptible to heart failure. A, Cardiac FS% at 0, 2, 4, and 6 weeks of TAC stimulation in control (βMHC-cre and Gata6fl/fl) and Gata6fl/flβMHC-cre mice. *P<0.05 vs Control (βMHC-cre). B, Lung weight to body weight ratio (LW/BW) 6 weeks after TAC stimulation. *P<0.05 vs Control. C, Left ventricular end diastolic pressure (LVEDP) from sham and TAC operated animals 6 weeks after surgery, measured by invasive hemodynamics. *P<0.05 vs Sham; †P<0.05 vs control TAC. D, Contractility (+dP/dt) by invasive hemodynamics 6 weeks after a sham or TAC procedure. *P<0.05 vs Gata6fl/flβMHC-cre sham; †P<0.05 vs control TAC. E, Left ventricular free wall stress from control and Gata6-deleted mice 6 weeks after TAC. *P<0.05 vs Control. F, Hydroxy-proline content as a measure of fibrosis from sham and TAC operated hearts 6 weeks after surgery. *P<0.05 vs sham; †P<0.05 vs control TAC. G, VW/BW 6 weeks after TAC in the indicated groups of mice. H, Left ventricular end diastolic dimension (LVED) 6 weeks after TAC, measured by echocardiography. *P<0.05 vs Control. I, H&E-stained heart sections from sham and TAC operated mice 6 weeks after surgery from control and Gata6fl/flβMHC-cre mice. Original magnification x0.75. J, Cardiomyocyte surface areas measured from cardiac histological sections 6 weeks after TAC. *P<0.05 vs Control. The number of animals measured is shown in the bars of the various panels.
Gata6 heart-deleted mice are more susceptible to heart failure
The observation of reduced ventricular performance in Gata6fl/flβMHC-cre mice after only 2 weeks of TAC suggested a predisposition to heart failure. To examine this effect in more detail we subjected Gata6fl/flβMHC-cre mice to 6 weeks of TAC stimulation (Supplemental Table II). These mice continued to show reduced ventricular performance at 2, 4, and 6 weeks after induction of TAC, compared with no significant reduction in βMHC-cre or Gata6fl/fl mice (Figure 5A, Supplemental Table II, groups combined as “control” in the figure). Moreover, after 6 weeks of TAC only Gata6fl/flβMHC-cre mice showed pulmonary edema and increases in left ventricular end diastolic filling pressures, two signs of heart failure (Figure 5B,C). Assessment of contractility by invasive hemodynamics with a Millar catheter showed a trend towards a baseline reduction in Gata6fl/flβMHC-cre mice, with a significant reduction after TAC stimulation compared with no deficit in control mice (Figure 5D). Indeed, Gata6fl/flβMHC-cre mice showed greater increases in left ventricular wall stress compared with control mice after 6 weeks of TAC, as well as significant elevations in fibrotic content in the heart by hydroxy-proline quantitation (Figure 5E,F). Interestingly, after 6 weeks of TAC stimulation the significant decrease in organ-level cardiac hypertrophy observed at 2 weeks was lost (Figure 5G). However, hearts from Gata6fl/flβMHC-cre mice showed a robust dilatory response as measured by echocardiography and histological methods (Figure 5H,I). Increases in heart weight during heart failure can occur through a dilatory process associated with only an addition of sarcomeres in series.26 Indeed, measurement of cellular hypertrophy still showed a significant reduction in myocyte cross-sectional surface areas in hearts from Gata6fl/flβMHC-cre mice after 6 weeks of TAC, suggesting that cellular hypertrophy was still inhibited in the absence of GATA-6 (Figure 5J).
Overexpression of GATA-6 enhances cardiac hypertrophy
We have previously shown that Gata4 overexpression in the heart with the αMHC promoter produced baseline hypertrophy.13, 14 To compare these results with GATA-6, and to model the known increase of GATA-6 protein in the heart following TAC stimulation (Figure 2A), here we generated heart-specific GATA-6 expressing transgenic mice using the tet-inducible system (Figure 6A). We generated 3 lines of transgenic mice that were classified as high (8.5-fold), medium (5.5-fold) and low (3.5-fold) overexpression based on Western blotting (Figure 6B). The low and medium overexpressing lines approximated the known increase in GATA-6 protein observed after TAC stimulation. At 8 weeks of age none of the 3 lines of mice showed cardiac hypertrophy, although by 40 weeks of age the medium and high overexpressing lines showed an increase in heart weight normalized to body weight (Figure 6C,D). This profile of cardiac hypertrophy in GATA-6 transgenic mice is reminiscent of data obtained in GATA-4 transgenic mice in which hypertrophy was not observed until 6 months of age.13 More importantly, TAC stimulation for 2 weeks in 8 week-old GATA-6 transgenic mice showed enhanced cardiac hypertrophy at the organ and cellular level in medium and high expressing lines (Figure 6E,F). Echocardiographic assessment of cardiac dimensions and function showed no significant changes between the different groups after TAC (Supplemental Table III). Although wall dimensions were higher in the transgenic mice after TAC, this did not reach statistical significance. Taken together, these results indicate that overexpression of GATA-6 in the heart can enhance cardiac hypertrophy, although it did not appear to worsen heart disease.
Figure 6.

Gata6 transgenic mice develop greater cardiac hypertrophy. A, Schematic of the dual transgenic system used to generate GATA-6 transgenic lines. B, Western blot for GATA-6 and lamin A/C (loading control) from cardiac nuclear protein extracts of 8 week-old mice. Arrows indicate the long and short isoforms of GATA-6. C, Heart weight to body weight ratio (HW/BW) from 8 week-old mice of the indicated groups. D, HW/BW from 40 week-old mice. *P<0.05 vs tTA control. E, VW/BW in sham and TAC operated tTA control or the three lines to GATA-6 transgenic mice 2 weeks after TAC. *P<0.05 vs sham; †P<0.05 vs tTA TAC. F, Cardiomyocyte surface areas measured from cardiac histological sections 2 weeks after TAC. *P<0.05 vs sham; †P<0.05 vs tTA TAC. The number of animals measured is shown in the bars of the various panels.
Deletion of Gata6 from the heart alters homeostatic gene expression
To begin to understand the transcriptional regulatory mechanisms whereby loss of GATA-6 from the heart leads to heart failure, dilation, and less cellular hypertrophy we performed an analysis of global gene expression in hearts from 9 week-old Gata6fl/flβMHC-cre mice. We identified 21 genes that were significantly altered in these hearts compared with hearts from Gata6fl/fl andβMHC-cre mice. A select group of these genes that had mechanistic implications in the hypertrophy or failure response were confirmed by qPCR (Figure 7). For example, deletion of GATA-6 protein from the heart resulted in upregulation of βMHC, Lrrcc1, Lamc2, BNP, and downregulation of Adamts3, Pde1c, and Egf (Figure 7A-G). BNP and βMHC genes are activated by stress to the heart, although both also contain important GATA DNA binding sites in their promoters. Pde1c is a major dual specificity cAMP and cGMP diesterase implicated in cardiac contractility23 while Egf has been implicated in heart failure through development of cardiotoxicity after trastuzumab treatment (epidermal growth factor receptor blocker) in breast cancer patients.24 In addition, we found a number of upregulated and downregulated genes for which the importance in the heart is less clear, although they are clearly central regulatory proteins. For example, the centrosomal protein Lrrcc1 is required for spindle pole integrity during cell division,25 and Adamts3 encodes a protein that contains both metallopeptidase domains and thrombospondin motifs.26 These results suggest that loss of GATA-6 from the heart dramatically alters the expression of diverse regulatory genes that could impact heart failure propensity and the hypertrophic response.
Figure 7.
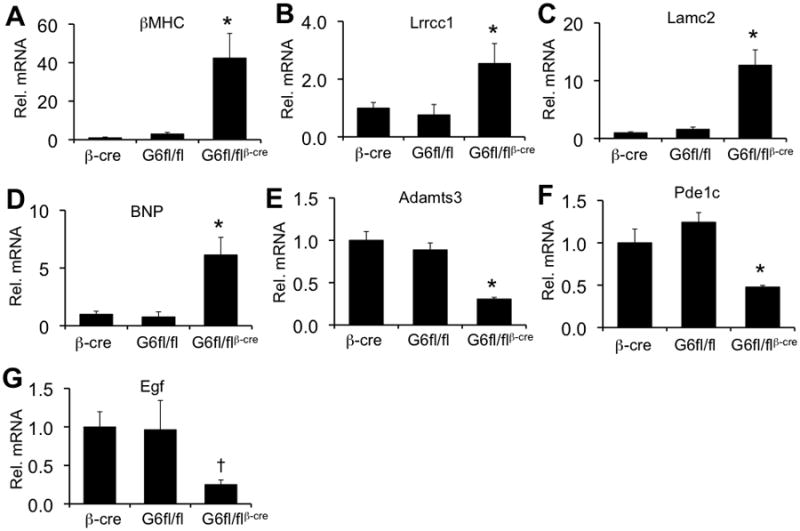
Gata6-deleted hearts show changes in basal gene expression. The panels show qPCR quantitation and confirmation for selected genes identified in the Affymetrix array. Each panel shows the average level of mRNA expression normalized to Ppia and relative to the level of mRNA in hearts from βMHC-cre mice. At least 3 mice per group were analyzed. *P<0.05 vs βMHC-cre and Gata6fl/fl, †P<0.05 vs βMHC-cre.
Combined deletion of Gata4 and Gata6 results in heart failure and death
To further to investigate the issue of redundancy between GATA-4 and GATA-6 in the adult heart we generated mice with combined deletion of both Gata4 and Gata6 using the βMHC-cre transgene, which surprisingly produced offspring at expected Mendelian frequencies (data not shown). However, these mice began to show signs of heart failure and by 16 weeks of age all Gata4fl/flGata6fl/flβMHC-cre mice had died (Figure 8A). Examination of mice and hearts just prior to this lethality time point showed overt heart failure by echocardiography with extremely thin myocardial walls and dilation (Figure 8B,D). Echocardiographic assessment of cardiac function showed significantly reduced fractional shortening at 12 weeks of age (Figure 8C). Although these experiments do not prove redundancy, they do suggest that GATA-4 and GATA-6 may have an additive effect in the adult heart.
Figure 8.
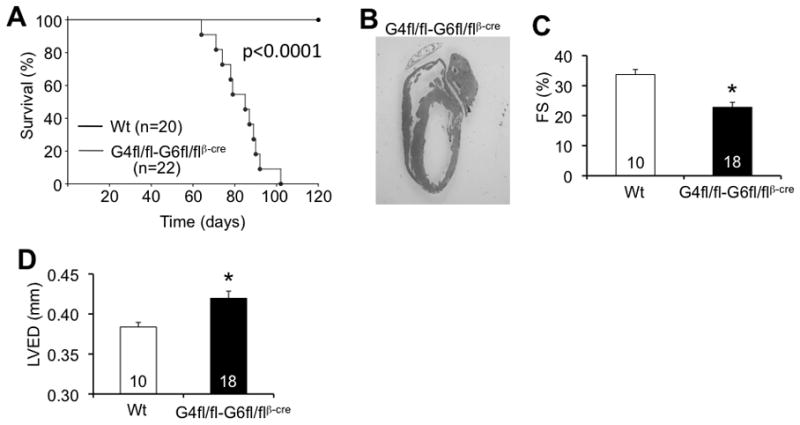
Gata4-Gata6-double deleted mice develop spontaneous dilated cardiomyopathy leading to premature death. A, Survival curve for wildtype control and Gata4-Gata6-double heart-deleted mice. P-value <0.05 for difference in survival. B, H&E-stained heart section from Gata4fl/fl-Gata6fl/flβMHC-cre mouse in overt heart failure. Original magnification x0.75. C, Cardiac FS% from Wt and Gata4fl/fl-Gata6fl/flβMHC-cre mice at 12 weeks of age. *P<0.05 vs wt. D, Left ventricular end diastolic dimension (LVED) from Wt and Gata4fl/fl-Gata6fl/flβMHC-cre mice at 12 weeks of age. *P<0.05 vs wt. The number of animals measured is shown in the bars of the various panels.
Discussion
In the early developing embryo Gata4 and Gata6 were shown to be of critical importance for the establishment of the entire cardiac gene program, as loss of both factors, but not either alone, results in acardia.27 This result demonstrates the importance of GATA transcription factors as necessary regulators of cardiac differentiation specific gene expression, hence it was not entirely unexpected when GATA-4 was shown to be necessary for cardiac hypertrophy in the adult heart, as this process requires re-establishment of the fetal gene program. While almost nothing is known about the role of GATA-6 in regulating hypertrophy or differentiation specific gene expression in the adult heart, GATA-4 and GATA-6 are each capable of inducing hypertrophy when overexpressed in neonatal rat cardiomyocytes, suggesting for the first time that GATA-6 might function similar to GATA-4 in this respect.13, 28 Indeed, GATA-4 can functionally and physically interact with GATA-6 in activation of the ANF and BNP promoters.28 While GATA-4 and GATA-6 have been shown to positively regulate BNP and MYH promoters in cultured cardiomyocytes13, 28, we actually observed upregulation of BNP and βMHC expression in the absence of GATA-6. It is likely that gene regulation in vivo is distinctly different from conditions that occur with transfection of minimal promoters and co-overexpression of GATA-4 and/or GATA-6 in cultured cells. Also, these minimal promoter constructs that were transfected into neonatal myocytes could also easily lack inhibitory GATA binding sites or other negative regulatory sites that can bind secondary transcription factors induced by GATA-4/6.
Here we showed for the first time, that in addition to GATA-4, GATA-6 protein expression is dramatically enhanced in mouse hearts subjected to pressure overload stimulation, which correlated with a dramatic increase in total GATA DNA binding activity. It was intriguing that deletion of Gata6 from the neonatal mouse heart, but not Gata4, severely reduced total GATA DNA binding activity assayed with a canonical GATA binding site from the αMHC promoter. This suggests that GATA-6 protein levels may be higher than GATA-4 in the postnatal heart, although the overall transcriptional potency of both proteins may differ such that GATA-4 may still be just as critical. In addition, we found a significant reduction in GATA-4 protein in the Gata6-deleted hearts, which could contribute to the reduced DNA binding activity. Intriguingly, this result hints at a positive regulatory relationship between GATA-6 and GATA-4 in the adult heart.
We modeled the observed increase in GATA-6 protein levels after pressure overload stimulation using a transgenic approach. The low and medium expressing line approximated this increase in endogenous GATA-6 with hypertrophy, suggesting that our transgenic approach closely model this known increase. Finally, and perhaps more importantly, deletion of Gata6 from the mouse heart significantly reduced the hypertrophic growth response, both after TAC and after stimulation with AngII/PE, together suggesting that GATA-6 functions as a necessary and sufficient mediator of adult cardiac hypertrophic growth.
That GATA-6 appears to be equally important to GATA-4 in regulating the cardiac hypertrophic response might suggest a degree of functional redundancy between these 2 transcription factors. Indeed, we previously demonstrated that Gata4+/− Gata6+/− (double heterozygotes) are not viable and perish during mid-gestation, while single heterozygotes for either gene were viable.9 In the adult heart, the phenotype of reduced cardiac hypertrophy after 2 weeks of TAC was nearly identical between Gata4 and Gata6 heart-deleted mice, and each succumbed to heart failure and reductions in cardiac function to a roughly similar extent. Moreover, GATA-4 and GATA-6 overexpressing transgenic mice each developed similar levels of spontaneous hypertrophy at about the same time in middle adulthood.13 Finally, deletion of both Gata4 and Gata6 specifically from the heart resulted in spontaneous heart failure and death by the age of 16 weeks. While these collective observations suggest functional redundancy between GATA-4 and GATA-6, some differences were noted that might suggest unique functions for each. First, Gata4fl/flβMHC-cre mice showed spontaneous deterioration of cardiac function with aging, but Gata6fl/flβMHC-cre mice showed more preserved cardiac function up to one year of age as assessed by echocardiography (although invasive hemodynamics showed a trend towards reduced baseline function with Gata6 deletion). Similarly, we found a slight but significant reduction in heart size at baseline in Gata6-deleted mice due to decreased cardiomyocyte content in the heart, while we did not observe this effect in Gata4-deleted mice. However, Pu and colleagues did observe a decrease in myocyte proliferation in the hearts of Gata4 heart-specific hypomorphic mice (their targeted allele had partial expression), suggesting that there could still be functional similarity between GATA-4 and GATA-6 with respect to control of cell number in the heart.29 Thus, our working hypothesis is that GATA-4 and GATA-6 are roughly equivalent in effect in the heart, such that each of the 4 alleles functions in a dosage-dependent manner. However, it will be important to generate combinations of Gata4 and Gata6 allelic deletions, as well as conduct rescue approaches whereby the GATA-4 transgene is crossed into the Gata6 heart-specific deleted background, and vice versa, before we can unequivocally determine if these 2 factors are completely redundant in function in the heart or if some unique regulatory actions persist between them.
Novelty and significance.
What is known?
GATA-6 can induce hypertrophy in cultured cardiomyocytes
Upon pressure overload, GATA binding activity increases in the murine heart
GATA-4 has been shown to be both required and sufficient for cardiac hypertrophy in vivo.
What new information does this article contribute?
We provide the first description of adult cardiac deletion of GATA-6 showing its requirement for both cardiac hypertrophy in response to various stimuli and for cardiac compensation
We provide the first proof that cardiac-specific overexpression of GATA-6 is sufficient to induce cardiac hypertrophy in vivo
We provide the first description of cardiac-specific deletion of both GATA-4 and GATA-6 from the adult heart, which results in spontaneous dilated cardiomyopathy.
Summary
This study was designed to evaluate the importance of GATA-6 in vivo for cardiac hypertrophy and compensation. We studied both GATA-6 overexpression and deletion as effectors of pressure overload induced hypertrophy. We also studied the role of GATA-6 deletion in neurohormonal induced hypertrophy. Finally, we also studied GATA-4 deletion as a comparison and both GATA-4 and GATA-6 deletion to begin to address the possible redundancy between these transcription factors. Our results show the requirement of GATA-6 for cardiac hypertrophy and compensation in response to pressure overload and neurohormonal stimulation. In addition, we show the both GATA-4 and GATA-6 are required to maintain normal cardiac function suggesting they may act redundantly.
Supplementary Material
Acknowledgments
Sources of funding
This work was supported by grants from the National Institutes of Health (J.D.M., S.A.D.), the Fondation Leducq (Heart failure network grant to J.D.M), and the Howard Hughes Medical Institute (J.D.M.). J.H.v.B. was supported by an ICIN fellowship and a Rubicon fellowship from the Netherlands Organization for Scientific Research (NWO).
Non-Standard Abbreviations
- ANF
atrial natriuretic factor
- AngII
angiotensin II
- αMHC
α-myosin heavy chain
- BNP
b-type natriuretic peptide
- βMHC
β-myosin heavy chain
- fl
loxP site
- PE
phenylephrine
- TAC
transverse aortic constriction
Footnotes
Disclosures: None
References
- 1.Lloyd-Jones DM, Larson MG, Leip EP, Beiser A, D’Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D. Lifetime risk for developing congestive heart failure: The framingham heart study. Circulation. 2002;106:3068–3072. doi: 10.1161/01.cir.0000039105.49749.6f. [DOI] [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Molkentin JD, Dorn GW., 2nd Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 4.Charron F, Tsimiklis G, Arcand M, Robitaille L, Liang Q, Molkentin JD, Meloche S, Nemer M. Tissue-specific gata factors are transcriptional effectors of the small gtpase rhoa. Genes Dev. 2001;15:2702–2719. doi: 10.1101/gad.915701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang Q, Wiese RJ, Bueno OF, Dai YS, Markham BE, Molkentin JD. The transcription factor gata4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol Cell Biol. 2001;21:7460–7469. doi: 10.1128/MCB.21.21.7460-7469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hautala N, Tenhunen O, Szokodi I, Ruskoaho H. Direct left ventricular wall stretch activates gata4 binding in perfused rat heart: Involvement of autocrine/paracrine pathways. Pflugers Arch. 2002;443:362–369. doi: 10.1007/s004240100699. [DOI] [PubMed] [Google Scholar]
- 7.Hautala N, Tokola H, Luodonpaa M, Puhakka J, Romppanen H, Vuolteenaho O, Ruskoaho H. Pressure overload increases gata4 binding activity via endothelin-1. Circulation. 2001;103:730–735. doi: 10.1161/01.cir.103.5.730. [DOI] [PubMed] [Google Scholar]
- 8.Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor gata4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 9.Xin M, Davis CA, Molkentin JD, Lien CL, Duncan SA, Richardson JA, Olson EN. A threshold of gata4 and gata6 expression is required for cardiovascular development. Proc Natl Acad Sci U S A. 2006;103:11189–11194. doi: 10.1073/pnas.0604604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molkentin JD. The zinc finger-containing transcription factors gata-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275:38949–38952. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- 11.Oka T, Xu J, Molkentin JD. Re-employment of developmental transcription factors in adult heart disease. Semin Cell Dev Biol. 2007;18:117–131. doi: 10.1016/j.semcdb.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pikkarainen S, Tokola H, Kerkela R, Ruskoaho H. Gata transcription factors in the developing and adult heart. Cardiovasc Res. 2004;63:196–207. doi: 10.1016/j.cardiores.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 13.Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD. The transcription factors gata4 and gata6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem. 2001;276:30245–30253. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 14.Heineke J, Auger-Messier M, Xu J, Oka T, Sargent MA, York A, Klevitsky R, Vaikunth S, Duncan SA, Aronow BJ, Robbins J, Crombleholme TM, Molkentin JD. Cardiomyocyte gata4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J Clin Invest. 2007;117:3198–3210. doi: 10.1172/JCI32573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 16.Aries A, Paradis P, Lefebvre C, Schwartz RJ, Nemer M. Essential role of gata-4 in cell survival and drug-induced cardiotoxicity. Proc Natl Acad Sci U S A. 2004;101:6975–6980. doi: 10.1073/pnas.0401833101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bisping E, Ikeda S, Kong SW, Tarnavski O, Bodyak N, McMullen JR, Rajagopal S, Son JK, Ma Q, Springer Z, Kang PM, Izumo S, Pu WT. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc Natl Acad Sci U S A. 2006;103:14471–14476. doi: 10.1073/pnas.0602543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sodhi CP, Li J, Duncan SA. Generation of mice harbouring a conditional loss-of-function allele of gata6. BMC Dev Biol. 2006;6:19. doi: 10.1186/1471-213X-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 20.Koutsourakis M, Langeveld A, Patient R, Beddington R, Grosveld F. The transcription factor gata6 is essential for early extraembryonic development. Development. 1999;126:723–732. [PubMed] [Google Scholar]
- 21.Brewer A, Gove C, Davies A, McNulty C, Barrow D, Koutsourakis M, Farzaneh F, Pizzey J, Bomford A, Patient R. The human and mouse gata-6 genes utilize two promoters and two initiation codons. J Biol Chem. 1999;274:38004–38016. doi: 10.1074/jbc.274.53.38004. [DOI] [PubMed] [Google Scholar]
- 22.Takeda M, Obayashi K, Kobayashi A, Maeda M. A unique role of an amino terminal 16-residue region of long-type gata-6. J Biochem. 2004;135:639–650. doi: 10.1093/jb/mvh077. [DOI] [PubMed] [Google Scholar]
- 23.Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, Radwanski PB, Florio V, Movsesian MA. Cyclic nucleotide phosphodiesterase pde1c1 in human cardiac myocytes. J Biol Chem. 2007;282:32749–32757. doi: 10.1074/jbc.M703173200. [DOI] [PubMed] [Google Scholar]
- 24.Feldman AM, Lorell BH, Reis SE. Trastuzumab in the treatment of metastatic breast cancer: Anticancer therapy versus cardiotoxicity. Circulation. 2000;102:272–274. doi: 10.1161/01.cir.102.3.272. [DOI] [PubMed] [Google Scholar]
- 25.Muto Y, Yoshioka T, Kimura M, Matsunami M, Saya H, Okano Y. An evolutionarily conserved leucine-rich repeat protein clerc is a centrosomal protein required for spindle pole integrity. Cell Cycle. 2008;7:2738–2748. doi: 10.4161/cc.7.17.6591. [DOI] [PubMed] [Google Scholar]
- 26.Le Goff C, Somerville RP, Kesteloot F, Powell K, Birk DE, Colige AC, Apte SS. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous adamts proteases: Insights on collagen biosynthesis and dermatosparaxis. Development. 2006;133:1587–1596. doi: 10.1242/dev.02308. [DOI] [PubMed] [Google Scholar]
- 27.Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both gata4 and gata6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charron F, Paradis P, Bronchain O, Nemer G, Nemer M. Cooperative interaction between gata-4 and gata-6 regulates myocardial gene expression. Mol Cell Biol. 1999;19:4355–4365. doi: 10.1128/mcb.19.6.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeisberg EM, Ma Q, Juraszek AL, Moses K, Schwartz RJ, Izumo S, Pu WT. Morphogenesis of the right ventricle requires myocardial expression of gata4. J Clin Invest. 2005;115:1522–1531. doi: 10.1172/JCI23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.