


Abstract
This report documents the error rate in a commercially distributed subset of the IMAGE Consortium mouse cDNA clone collection. After isolation of plasmid DNA from 1189 bacterial stock cultures, only 62.2% were uncontaminated and contained cDNA inserts that had significant sequence identity to published data for the ordered clones. An agarose gel electrophoresis pre-screening strategy identified 361 stock cultures that appeared to contain two or more plasmid species. Isolation of individual colonies from these stocks demonstrated that 7.1% of the original 1189 stocks contained both a correct and an incorrect plasmid. 5.9% of the original 1189 stocks contained multiple, distinct, incorrect plasmids, indicating the likelihood of multiple contaminating events. While only 739 of the stocks purchased contained the desired cDNA clone, agarose gel pre-screening, colony isolation and similarity searching of dbEST allowed for the identification of an additional 420 clones that would have otherwise been discarded. Considering the high error rate in this subset of the IMAGE cDNA clone set, the use of sequence verified clones for cDNA microarray construction is warranted. When this is not possible, pre-screening non-sequence verified clones with agarose gel electrophoresis provides an inexpensive and efficient method to eliminate contaminated clones from the probe set.
INTRODUCTION
The Integrated Molecular Analysis of Genomes and their Expression (IMAGE) Consortium was initiated in 1993 as a collaborative effort among academic groups to share high-quality arrayed cDNA clones, mapping and expression data for use in the public domain (1). These clones are distributed free of royalty by authorized vendors, such as Research Genetics (http://www.resgen.com), Incyte Genomics (http://www.incyte.com) and the American Type Culture Collection (http://www.atcc.org). The ultimate goal of the IMAGE Consortium is to form a master repository of arrayed clones containing a representative cDNA for each unique human and mouse gene. This collection is the largest public collection of cDNA clones in the world and is used by the international scientific community as the foundation for many biological applications, including gene discovery and gene expression studies. The extent of their use in these and other applications worldwide underscores the need for the collection to accurately represent the sequence that is submitted to the database. Unfortunately, hearsay reports suggest a 10–30% error rate; that is, the sequence of 10–30% of the clones in the collection are not what they are reported to be in dbEST (2). The IMAGE Consortium is aware of this and does list problematic clones on its web site based on user input, however there is no consensus as to the actual error rate or the source of the errors. In addition to sequencing errors, there have also been instances of bacteriophage contamination in sections of the clone bank. Badly contaminated sections of the bank have been withdrawn and clones are routinely tested for contamination before shipment.
The determination of the actual error rate in commercially available stocks is of value to assist investigators in clone selection, and provides a more accurate estimation of potential error rates in experiments if sequence verification is not implemented. Due to the supposed error rate, sequence verification of a set of clones obtained from Research Genetics was performed prior to the construction of a microarray. In addition to incorrect clones, many of the clones were found not to return useable sequence data due to the presence of contaminating plasmids. Here we report on the error rate of a commercially available subsample of the IMAGE cDNA clone collection, including the proportion containing contaminating plasmids.
MATERIALS AND METHODS
Plasmid preparation
Bacterial stocks grown in LB-AMP (+8% glycerol, +50 µg/ml ampicillin) were received from Research Genetics (Huntsville, AL) in 96-well polystyrene plates on dry ice. DNA isolation and sequencing was performed at the Michigan State University DNA Sequencing Facility. Overnight, 2.5 ml cultures were grown in 6-well Autogen culture vessels (Autogen, Framingham, MA) at 37°C in a shaking incubator and plasmids isolated using an Autogen PI-50α automated DNA isolation system. Plasmid DNA was analyzed electrophoretically at 6 V/cm for 90 min in a 1% agarose gel containing 0.4 µg/ml ethidium bromide.
Isolation of individual clones from contaminated wells was performed by streaking out the bacterial stocks on LB-agar plates containing 50 µg/ml ampicillin. Five individual colonies were picked and DNA isolated from 2.5 ml overnight cultures. Approximately 1 µg of plasmid DNA was digested with 10 U of EcoRI (Roche/Boehringer Mannheim, Indianapolis, IN) for 2 h at 37°C, and analyzed electrophoretically as described above. One clone was sequenced for each distinct molecular weight species identified.
DNA sequencing
Cycle sequencing reactions were carried out on a PTC-200 table top cycler (MJ Research, Watertown, MA) using the ABI Prism™ Dye Terminator Cycle Sequencing Ready Reaction Kit with Amplitaq DNA polymerase (Perkin-Elmer, Foster City, CA) and a T7 5′ sequencing primer. Automated fluorescent DNA sequencing was performed on a 373A DNA Sequencer (Perkin-Elmer) and base calls performed using ABI Prism DNA Sequencing Software version 2.1.2.
Bioinformatics
Sequence verification was performed using the NCBI Blast 2 Sequences (3) engine. Briefly, the full-length sequence information obtained from the MSU sequencing facility and the GenBank accession number of the ordered IMAGE clone were submitted to the Blast 2 Sequences tool (http://www.ncbi.nlm.nih.gov/gorf/bl2.html), and bit score, expect (E) value, identities and other data parsed out of the retrieved results. The length and percentage ambiguity (N) of the aligned region were also calculated.
Contaminating sequences from original wells with two or more clones selected for sequencing were examined for identity. All sequences derived from a given well were compared in a pairwise fashion using the Blast 2 Sequences tool as described above. Sequences were considered to be unique if they had no substantial sequence similarity to other clones derived from the contaminated well. As expected, some sequence identity was observed between many clones in the extreme 5′ region. This represents common vector sequence and was not considered to be evidence of identity within the cDNA insert.
BLAST homology searching was performed on clones derived from contaminated wells. These sequences (unknowns) were submitted to the NCBI BLASTN 2.0.11 engine (4) and compared against murine expressed sequence tags (ESTs) in dbEST. Unknowns were considered to be significantly identical to an EST if their E-value was ≤1e-50. GenBank accession numbers for up to 10 significant ESTs for each unknown were submitted to the NCBI murine UniGene database and a UniGene Cluster ID was retrieved where available. The most identical EST was also compared to the unknown sequence using the Blast 2 Sequences tool as described above.
The above methods were automated using scripts written in Perl version 5.005_03. These scripts are available upon request. All data and analyses presented in this report are available to the public at http://www.bch.msu.edu/~zacharet/dbtest/IMAGE_Verification.htm.
RESULTS
Clone selection
As part of a project designed to create cDNA microarrays for use in studies examining gene expression in the mouse testis, 1189 non-sequence verified murine IMAGE cDNA clones were obtained as bacterial stock cultures from Research Genetics. These clones were selected through examination of the UniGene (http://www.ncbi.nlm.nih.gov/UniGene/) database (5) and represent probes for genes of known function expressed in the mouse testis (http://www.bch.msu.edu/~zacharet/dbtest), none of which were available as sequence verified clones at the time of purchase.
Electrophoretic pre-screening of samples
Sequencing reactions on the first 144 cDNA clones resulted in 23 (16%) unreadable sequences (data not shown). To reduce the incidence of unreadable sequences, an agarose gel electrophoresis pre-screen of plasmid DNA was implemented (Fig. 1). Plasmids electrophoresed in the expected supercoiled and relaxed forms. Note that the preparation method used resulted in a higher than usual proportion of relaxed plasmid, as well as some larger fragments that may represent chromosomal DNA or concatenated plasmids. Even so, it was apparent that several DNA preparations contained more than one plasmid species (Fig. 1, samples 2A8, 2A11 and 2B11). Of the 1189 samples electrophoresed, 361 (30.4%) were withheld for further processing based on the results of the pre-screen. An additional 56 stocks (4.7%) failed to grow or did not yield plasmid DNA after DNA isolation. These results are summarized in Table 1 and Figure 2a.
Figure 1.
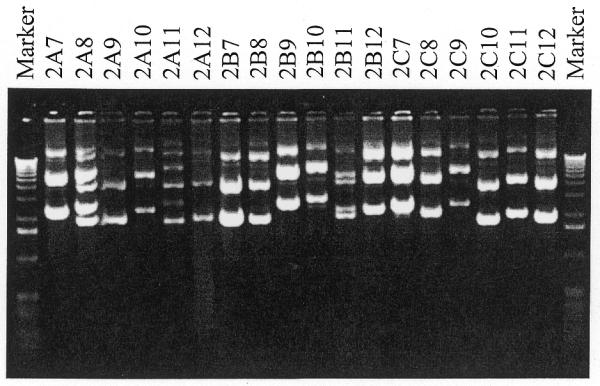
Agarose gel electrophoresis of plasmid DNA isolated from 18 bacterial clones purchased from Research Genetics. Plasmid DNA was isolated and electrophoresed as described in Materials and Methods. Samples 2A8, 2A11 and 2B11 appear to have two distinct plasmid species, and were subjected to clone isolation.
Table 1. Summary of sequencing data.
| Number | Match length (bases) | Ambiguity (%) | |||||
| |
|
Average ± SD |
Range |
Median |
Average ± SD |
Range |
Median |
| Stocks received | 1189 | n/a | n/a | n/a | n/a | n/a | n/a |
| Failed to grow | 56 | n/a | n/a | n/a | n/a | n/a | n/a |
| No sequence read | 10 | n/a | n/a | n/a | n/a | n/a | n/a |
| Correct (pre-screened)a | 613 | 383 ± 73 | 54–549 | 399 | 2.0 ± 2.5 | 0–17.0 | 1.2 |
| Correct (post-screen) | 126 | 363 ± 96 | 94–500 | 389 | 2.0 ± 2.0 | 0–11.3 | 1.4 |
| Correct (isolated from contaminated stocks) | 84 | 372 ± 79 | 75–467 | 396 | 1.6 ± 2.1 | 0–14.3 | 1.2 |
| Incorrect (pre-screened) | 152 | ||||||
| Accession no. in UniGene cluster | 97 | 386 ± 56 | 186–490 | 399 | 2.4 ± 2.2 | 0–9.3 | 1.7 |
| Accession no. not in UniGene cluster | 15 | 380 ± 100 | 182–476 | 427 | 1.7 ± 2.1 | 0.5–8.6 | 0.9 |
| No significant identity | 40 | n/d | n/d | n/d | n/d | n/d | n/d |
| Incorrect (colony isolation)b | 332 | ||||||
| Accession no. in UniGene cluster | 247 | 369 ± 76 | 111–525 | 383 | 1.7 ± 1.6 | 0–9.0 | 1.2 |
| Accession no. not in UniGene cluster | 61 | 395 ± 85 | 149–546 | 424 | 1.4 ± 1.6 | 0–7.0 | 0.8 |
| No significant identity | 24 | n/d | n/d | n/d | n/d | n/d | n/d |
| Total clones identifiedc | 1243 | 378 ± 76 | 54–549 | 394 | 1.9 ± 2.2 | 0–17.0 | 1.2 |
| Total clonesd | 1303 | n/a | n/a | n/a | n/a | n/a | n/a |
From 1189 stock cultures ordered, the number of entities in each category is noted. Correct is defined as sequence that has identity to the published sequence for the desired clone. Incorrect sequences do not have identity to the desired clone. Incorrect sequences were assigned GenBank accession numbers where possible by comparison to the murine sequences in dbEST. Match length (the length of the region of identity between derived and published sequence) is presented with standard deviation, range and median values. Percent ambiguity (the number of ambiguous base calls divided by the total number of bases over the region of identity) is also presented with standard deviation, range and median values. n/a, Not applicable; n/d, not determined.
aPre-screened, derived from those stocks which passed agarose gel electrophoretic pre-screening.
bColony isolation, derived from those stocks which failed pre-screening.
cIncludes correct clones, and incorrect clones which could be assigned a GenBank accession number cluster. This number is higher than the number of stocks ordered, due to the isolation of more than one incorrect clone from several stock cultures.
dNumber of clones examined, inclusive of clones which could not be identified.
Figure 2.

Summary of sequence verification and clone identification results. Correct clones or stocks are defined as those from which sequence information was obtained which had identity to the published cDNA sequence for the ordered clone. Incorrect clones are defined as those which yielded no identity to the published cDNA sequence. (a) Summary of verification results on a per stock basis. Number and percent of the original 1189 ordered stocks is indicated. Stocks that were subjected to clone isolation (center) can contain a correct clone, one or more incorrect clones, or a combination of correct and incorrect clones. (b) Summary of clone identification results for incorrect stocks (left) or contaminating clones (center) from (a). Clone identification was performed as indicated in Materials and Methods.
Sequence verification of pre-screened samples
The remaining 772 samples were sequenced and compared to the published sequence using the Blast 2 Sequences tool (3). For this and subsequent analyses, no attempt was made to remove vector sequences from the 5′- or 3′-ends of the determined sequence. These sequences were compared to published GenBank sequence information, which has vector sequence removed. In this case, the presence of cloning vector derived sequence in the query samples is irrelevant, as it was not used to determine the percent identity to the published EST sequence. Also, N was only calculated for the region of identity (match) within the published sequence information, minimizing the effects of artifactual ambiguities present in the extreme 5′ and 3′ regions of a query sequence.
613 of the 773 samples returned an alignment (Fig. 2a). The length of the match varied widely by clone, reflecting the variability in length of the published sequence information and the length of in-house sequencing reads. The ambiguity in the matched region varied between 0.0 and 17.0%, with an average of 1.98 ± 2.49% and a median of 1.16%. The length of a match ranged between 54 and 549 bases, with an average length of 383 ± 73 bases and a median of 399 bases. Considering the improbability of randomly matching a sequence with a given accession number and the variable lengths of sequences submitted, any sequence returning an alignment was initially considered to be a correct clone. This represents 51.7% of the initial 1189 clones.
152 clones (12.8% of the initial clone set) had readable sequence, yet did not align with the published sequence for the ordered clone. These are considered to be incorrect clones. Seven clones (0.6%) did not return readable sequence information.
Colony isolation and analysis from contaminated stocks
Individual bacterial clones from the 361 stock wells that were flagged as suspect were subsequently isolated, under the hypothesis that at least one of the plasmid species would contain the correct (desired) cDNA insert. Plasmid species were distinguished based on their apparent molecular weights after linearization with EcoRI (Fig. 3). For example, 6A1 and 6A4 contained two distinct species, while 5H7 contained two distinct species, one of which had an EcoRI site in the insert. A representative clone for each phenotype observed was sent for DNA sequencing and sequence verification, as described above.
Figure 3.
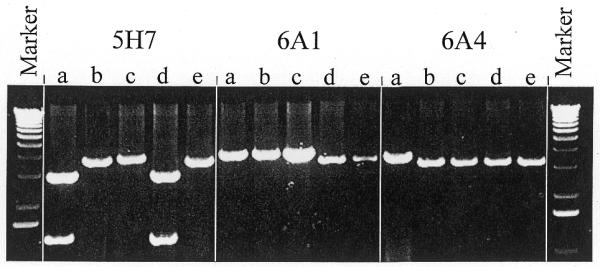
Agarose gel electrophoresis of clones isolated from contaminated bacterial stocks. Plasmid DNA was isolated from five colonies derived from the original stock, digested with EcoRI and electrophoresed as described in Materials and Methods. Two distinct phenotypes are observed in the DNA isolated from each of the original stocks depicted.
126 of the 361 suspect stocks examined (10.6% of the original clone set) contained only a correct plasmid (Fig. 2a). This indicates that wells were marked as being suspect based on the gel electrophoresis pre-screen conservatively, and that 739 of the initial 1189 stocks (62.2%) were correct as ordered. The ambiguity in the matched region for the 126 correct stocks ranged between 0.0 and 11.3%, with an average of 1.95 ± 2.0% and a median of 1.42%. The length of a match ranged between 94 and 500 bases, with an average length of 363 ± 96 bases and a median of 389 bases. Eighty-four stocks contained both a correct and one or more incorrect plasmids (7.1% of the original clone set). Therefore, this clone isolation strategy identified an additional 84 correct clones that would have otherwise been discarded as contaminated or unreadable sequence. The 84 correct clones manually isolated from cross-contaminated stocks had an ambiguity ranging between 0.0 and 14.3%, with an average of 1.60 ± 2.14% and a median of 1.17%. The length of a match ranged between 75 and 467 bases, with an average of 372 ± 79 bases and a median of 396 bases. However, these stocks are considered to be incorrect (failures), as they are contaminated and not useable as received. Three stocks failed to return readable sequence information. The remaining 148 suspect stocks contained only one or more incorrect clones.
Interestingly, this clone isolation strategy revealed that the contamination problem in this IMAGE clone set is significantly more severe than originally suggested. Seventy stock wells (5.9% of the original clone set) contained more than one distinct contaminating plasmid species. Of these, 50 contained two, 17 contained three, and three contained four contaminating species. Since only five colonies for each contaminated stock well were examined, it is possible that more than four distinct contaminating species were present in some stocks. This indicates that the contamination problem is severe and that in some cases more than one contaminating incident occurred.
Assigning identity to contaminating or incorrect clones
All incorrect clones derived from the individual colony isolation procedure were submitted for BLAST (4,6) similarity searching against the murine section of dbEST. Matches were considered to be significant if the E-value was ≤10e-50. The GenBank accession numbers for a maximum of 10 significant hits per clone were queried against the UniGene murine database (build #73). UniGene cluster identification numbers (IDs) were assigned where possible. In all but two cases, all GenBank accession numbers submitted for a given clone were associated with a single UniGene cluster ID or were not present in the UniGene database. This allowed for greater confidence in the assignment of a given clone to a cluster. 332 clones were examined (Fig. 2b). Of these, 247 UniGene cluster IDs could be assigned, resulting in 239 unique clusters. An additional 61 clones had significant homology to murine ESTs which could not be assigned to UniGene clusters. The percent ambiguity in the match was determined for the 308 clones that could be identified, by comparison of their sequence to the full-length published sequence for the most similar cDNA in dbEST. Ambiguity ranged between 0.0 and 9.0%, with an average of 1.59 ± 1.60% and a median of 1.11%. The length of a match ranged between 111 and 546 bases, with an average length of 376 ± 76 bases and a median of 387 bases. Twenty-four clones did not yield significant hits in dbEST, although 18 of these did have best hits ranging in E-value between 1e-46 and 6.4. Only two clones had significantly better matches to a non-murine EST: one matched a human clone with an E-value of 4e-34, and the other matched a Zea maize clone with an E-value of 1e-31. The remaining six clones had sequence data of very poor quality, and did not yield hits with any degree of similarity in dbEST, or in a VecScreen (http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html) search of UniVec, a non-redundant database of vector sequences.
A similar analysis was performed on the 152 clones that returned incorrect sequences in the initial sequencing run (Fig. 2b). Of these, 97 could be assigned to 94 unique UniGene clusters, and an additional 15 had significant homology to murine ESTs not yet assigned to UniGene clusters. For the clones with significant matches, the ambiguity in the matched region ranged between 0.0 and 9.32%, with an average of 2.31 ± 2.19% and a median of 1.55%. The length of a match ranged between 182 and 490 bases, with an average length of 385 ± 63 bases and a median of 404 bases. Forty clones could not be assigned a significant match in the murine section of dbEST, although 39 had best hits ranging in E-value between 3e-46 and 8.5. Only two of the 40 clones had significantly better matches to a non-murine EST, although neither of these matches, one human and one rabbit, matched with an E-value <1e-24. Most of the 40 clones that could not be assigned a significant hit in dbEST had sequence data with a high degree of ambiguity (total ambiguity 14.25 ± 5.34%). Preliminary data suggest that many of these stocks contain more than one plasmid species, however the entire set has not yet been examined.
Percent ambiguity is a predictor of contamination
A histogram plot of the percent identity and percent ambiguity of all clones that matched their published sequence indicates that the majority of these clones exhibit >90% identity and <4% ambiguity (data not shown). As expected, the greater the ambiguity in the matched region, the lower the percent identity to the published sequence. There are some interesting observations to be made in the subset of sequences that had <90% identity. Eight of these clones, with an average identity of 77.6 ± 8.2%, were subjected to clone isolation as described above. Of these eight, all contained at least two distinct plasmid species and one contained three. In all cases, it was possible to isolate one correct clone, with the average identity of these correct clones being 95.6 ± 2.9%.
Error rate is related to vector used in cDNA library construction
One source of error in our clone set was related to the plasmid vector. Libraries were categorized by vector used. Soares and Barstead cDNA libraries, using vector pT7T3D-Pac, were well represented in our clone set (970 clones). Stratagene, Beier and Schiller libraries, using the vector pBluescript SK-, were grouped together (172 clones), and a third group contained the remaining libraries made with the vectors pBluescribe, pSPORT1, pCMV-SPORT2 and pME18S-FL3 (47 clones). Clones in the vector pBluescript SK- had a very high failure rate of 78.5% (Fig. 4). 27.3% of the clones in this vector failed to grow or yield sufficient plasmid DNA to sequence. An additional 42.4% yielded incorrect sequence, and 8.7% were contaminated. In contrast, clones in the vector pT7T3D-Pac showed an overall failure rate of 31.8%: 1.5% failed to grow, 17.4% were incorrect and 12.8% were contaminated. Clones from other libraries had an overall failure rate of 21.3%, with 6.3% failing to grow, 10.6% incorrect and 4.3% contaminated.
Figure 4.

Failure rate as a function of cloning vector. 1189 bacterial stock cultures were categorized based on the cloning vector used to create the cDNA library containing the desired clone. %Failure (Total) is the percentage of stocks which failed due to any reason, presented as the total number of failed stocks divided by the total number of stocks for a given vector. %Incorrect is the percentage of clones that yielded sequence data that did not match the desired clone. %Contaminated is the percentage of stocks that contained one or more distinct plasmids. %No Growth is the percentage of stocks that did not grow in culture, or those that did not yield sufficient plasmid DNA for sequencing. Data presented for the pT7T3D-Pac vector represents an analysis of 970 stocks. Data presented for ‘Other’ represents 47 stocks derived from libraries made with pBluescribe, pSPORT1, pCMV-SPORT2 or pME18S-FL3. Data presented for the pBluescript SK- vector represents an analysis of 172 stocks.
DISCUSSION
It is well known that the IMAGE Consortium clone repository contains a number of incorrect or contaminated clones. The latest internal analysis by the IMAGE Consortium reveals a <10% in-house error rate (http://image.llnl.gov). Analysis of a set of 1189 clones ordered from an authorized vendor revealed that only 62.2% of the clones ordered matched the published sequence information for that clone and could be considered to be correct. Although these clones were selected based on our knowledge of gene expression in the mouse testis, they are effectively random in their distribution on the original IMAGE Consortium stock plates. The observed error rate was substantially higher than the internal error rate indicated by IMAGE. This rate is also higher than the anecdotally reported 10–30% error rate noted by researchers using commercially available IMAGE clones, however it is difficult to determine which vendors, clone sets and species were examined. It is possible that the human clone repository is of higher fidelity, however given the large set of clones analyzed in this report, it is likely that commercially available murine clones have an actual error rate well in excess of the <10% error reported by the IMAGE Consortium. It is important to note that the clones examined in this report were purchased from only one authorized vendor. It is possible that clones purchased from other vendors may be significantly more or less error prone, however the essential point of this report is to demonstrate that errors are frequent and that simple validation techniques are useful, and indeed required, when using IMAGE clones.
The source of errors in the clone set was varied. A substantial fraction of wells contained only a single plasmid containing an insert that bore no identity to the published sequence. There are numerous opportunities for the introduction of this type of error. These clones are submitted to the sequence database after a single sequencing run, and could contain sequencing errors (http://genome.wustl.edu/est/est_general/mouse_disclaimer.html). Also, these clones could be plated to the wrong locations on the stock plates. Indeed, analysis of 10 incorrect clones revealed that half had high identity to a clone derived from the same cDNA library which had an accession number that was very close to that of the desired clone (data not shown). Presumably, these clones could have been plated to a location near the desired clone on the same IMAGE Consortium stock plate. This same error phenotype could also be attributed to incorrect annotation, including incorrect assignment of sequence data to neighboring EST clones through lane tracking errors in the automated sequencing process. If IMAGE identifiers are assumed to be assigned sequentially, with the oldest clones having the lowest numbers, our data do not support the hypothesis that older clones are of lesser quality than newer clones. There was a wide variation in the number of incorrect or contaminated wells over the entire data set, however no statistical correlation could be shown between the age of clone and the error observed (data not shown). This sampling only covers a 2.5 year period between IMAGE Clone 303474 (entered into dbEST April 26, 1996) and IMAGE Clone 1974474 (entered into dbEST December 17, 1998).
In addition to possible errors in the preparation or annotation of the IMAGE Consortium stock plates, there could have been errors in picking and replating clones on the part of the distributor. An incremental increase in error through this process has been demonstrated by the IMAGE Consortium, showing that a sampling of a master set of clones at the Lawrence Livermore National Labs (LLNL) had a per well error of 9.71%. A sampling of a set of clones that had been sent to a distributor (Research Genetics), who replicated these clones and sent them back to LLNL, showed a 12.34% per well error (http://image.llnl.gov/image/qc/bin/display_error_rates). This documents an increase in error through a simple replication of plates. It would be expected that transferring clones from stock plates to plates in varying formats for distribution to customers would incur additional error. These errors could also be compounded by the end user of these clones, and it is not our intention to imply that the error rate observed is due solely to the creation and distribution of these clones. Rather, it should be stressed that despite the best efforts of all parties, errors within a clone set accumulate.
One very substantial source of error in this clone set was due to a general failure in clones constructed in the pBluescript SK- cloning vector. Approximately 78% of these clones failed, as compared to an average failure rate of 31.3% in clones constructed in other vectors. Since there was a greatly increased percentage of bacterial stocks that failed to grow, it is likely that this plasmid was not well maintained in the host bacteria under our culture conditions. This hypothesis is supported by the observation of an increased percentage of incorrect clones derived from this library. These incorrect clones could be in plasmids that are better maintained under our culture conditions, allowing for the selection of incorrect clones over the desired clones. This potential source of error is avoidable, either by optimizing culture conditions to enable proper maintenance of pBluescript SK- or by selecting clones from other libraries where available.
A second major source of error in this clone set was cross-contaminating plasmid species. 7.1% of the samples contained both a correct and one or more incorrect contaminating plasmids. There was also a substantial percentage of samples which contained multiple incorrect plasmids. In each case, a poor quality or unreadable sequence would have been returned had these samples not been pre-screened with agarose gel electrophoresis and withheld for manual processing. While this is labor intensive, it may be appropriate for clones that are difficult to replace due to rarity of that particular sequence in the IMAGE Consortium cDNA clone collection. The electrophoretic pre-screen represents a simple, inexpensive and effective way to analyze large sets of non-sequence-verified IMAGE clones, and is sufficiently discriminating to eliminate most bacterial stock cultures harboring contaminating plasmids. In addition, it minimizes the expense incurred by failed sequencing reads, and affords the opportunity, if desired, for individual manipulation of contaminated stocks in an effort to isolate pure populations.
The strategy employed had the additional benefit of isolating individual clones from contaminated stocks, which could be used to increase the diversity of the ordered clone set. Many contaminating or incorrect clones with unknown identity could be identified through comparison to sequences contained in dbEST. EST sequences were considered to be significantly identical if the E-value was ≤1e-50. This criterion was arbitrarily selected, and reflects a desire to be conservative in the assignment of identity to cDNA clones that are orphaned with respect to their original cDNA library, tissue of origin and species. The 10 most significantly identical ESTs were used to query the UniGene database of NCBI, an experimental system for partitioning GenBank sequences (including both well characterized genes and ESTs) into a non-redundant set of gene-oriented clusters. It should be noted that the UniGene clustering process can result in a given EST being present in different clusters in different builds of the database, and that this is only a tool for providing tentative identification of an unknown clone.
Of the 332 incorrect clones isolated after agarose gel pre-screening, 247 could be assigned to a mouse UniGene cluster and 61 had significant identity to murine ESTs which are not yet assigned to UniGene clusters. An analysis of the 152 incorrect stock cultures from the initial sequencing run yielded an additional 97 clones in UniGene clusters and 15 clones not yet assigned to UniGene clusters. This results in a total of 420 clones that would otherwise have been discarded as contaminants, but are now identified and can be used in future experiments. Remarkably, only four clones were identified which had significantly greater homology to non-murine ESTs than to murine ESTs. This suggests that contamination within commercially available murine clones is mainly restricted to cross contamination with other murine clones, rather than cross-species contamination events.
In addition, assigning identities to these clones allows for some basic conclusions about the source of contamination in the IMAGE Consortium clone collection. These data do not demonstrate that a single plasmid or group of plasmids is present in multiple wells, which would argue for a single contaminating event such as a contaminated stock solution or aerosolization of bacterial cultures. Rather, given the presence of multiple distinct clones, it is more likely that there have been multiple contaminating events. It is likely that the observed contamination rate is caused by incremental errors throughout the entirety of library creation, original sequence analysis, storage and distribution of stock cultures by vendors, and use by individual investigators, rather than a failure at any single step in the process.
Interestingly, the presence of contaminating plasmids did not always result in unreadable sequence, making it possible to obtain sequence data that results in a ‘positive’ match from a contaminated stock. A high percent ambiguity in the matched region indicated a high likelihood of contamination. Based on the probability of a chance base match at a given position in the sequence, it is quite likely that any contaminating plasmids in these stocks are maintained at low levels compared to the desired clone. In these cases, however, the percent identity of the sequence obtained to its published sequence is generally low, and the percent ambiguity in the matched region is high. Considering that contaminated stocks can give readable sequence information, rigorous criteria should be established for claiming that a plasmid is sequence verified. The majority of ‘correct’ sequences obtained have at least 90% identity to the published sequence information, and <4% ambiguity in the region of the match. If these cutoffs are used as selection criteria, only 644 of the 739 ‘correct’ clones (53.9% of the 1189 stock cultures) examined in this report can be considered sequence verified. It would be instructive to compare these sequence verification criteria to those employed by commercial distributors of sequence verified clones, however these are as yet unpublished.
The stringency of verification criteria used by a vendor would be quite useful in estimating the fidelity of an ordered clone set. It is common practice to amplify cDNA from purchased clones and to use this material for preparation of microarrays, with the assumption that the incorrect or contaminated clones are relatively rare, or will be detected as false positives upon verification of microarray data with other techniques such as RT–PCR, northern blot or in situ hybridization. However, 10.7% of the PCR amplification reactions performed on a set of 960 sequence verified cDNA stocks ordered from Research Genetics yielded two or more distinctly sized PCR products (unpublished observations). A small subset of these stocks were analyzed for the presence of multiple plasmid species, revealing that five out of 21 contained more than one plasmid. Based on these preliminary data, it can be estimated that ~2.5% of the 960 sequence verified stocks contain contaminating plasmids. This error rate is relatively small and is unlikely to seriously compromise future experiments using these clones, however it is important to note that bacterial stocks that are purchased as sequence verified can harbor plasmids containing both correct and incorrect cDNA inserts.
This report documents the error rate in a commercial subset of the IMAGE Consortium mouse cDNA clone collection. In a set of 1189 purchased stocks, only 62.2% were correctly identified and non-contaminated, and thus directly usable as ordered. The 38.8% failure rate was substantially skewed by a general failure of clones constructed using the pBluescript SK- cloning vector, and if these clones are removed from analysis the failure rate (33.1%) approaches the anecdotal 30% error rate. With substantial manual effort, it was possible to isolate a correct clone from an additional 7.1% of cross-contaminated stocks, however it is unlikely that most researchers who use these clones would invest the time and labor required to do this. The error rate is significant, and requires an informed decision on the part of researchers who use clones distributed from this collection. Of great concern is the fact that a large percentage of stock cultures contain both the correct plasmid and one or more contaminating plasmids. Depending on the relative concentration of these plasmids in the amplified cDNA used to print microarrays, it may be possible to obtain false positives or negatives that may not be easily excluded upon secondary verification.
While sequence verification represents a substantial effort in terms of time and expense, it may serve to reduce the number of false conclusions in future experiments and assist in the interpretation of microarray data. Given the complex nature of array results, sequence verified clones should be purchased if available. However, when sequence verified clones are not available, and sequence verification is not feasible for reasons of cost or labor, gel electrophoresis pre-screening is suggested as an inexpensive and efficient strategy for removing badly contaminated clones from the probe set.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by US Environmental Protection Agency grant # R 827-402-01-0. R.G.H. is supported by grant number T32 ES07255 from the National Institute of Environmental Health Sciences (NIEHS). This publication’s contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS.
References
- 1.Lennon G., Auffray,C., Polymeropoulos,M. and Soares,M.B. (1996) The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics, 33, 151–152. [DOI] [PubMed] [Google Scholar]
- 2.Boguski M.S., Lowe,T.M. and Tolstoshev,C.M. (1993) dbEST–database for “expressed sequence tags”. Nature Genet., 4, 332–333. [DOI] [PubMed] [Google Scholar]
- 3.Tatusova T.A. and Madden,T.L. (1999) BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett., 174, 247–250. [DOI] [PubMed] [Google Scholar]
- 4.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheeler D.L., Chappey,C., Lash,A.E., Leipe,D.D., Madden,T.L., Schuler,G.D., Tatusova,T.A. and Rapp,B.A. (2000) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 28, 10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]