

Abstract
Maternal microbial infections cause adverse fetal developmental outcomes including embryonic resorption, intrauterine fetal death, and preterm labor. Recent studies demonstrated that oxidative-stress plays an important role in chorioamniotitis pathogenesis. Herein we investigated the effect of N-acetyl cysteine (NAC) on lipopolysaccharide-induced preterm labor and fetal demise in murine model. Lipopolysaccharide exposure at embryonic day 18 demonstrated an increase in the abortion rate and fetal demise in pregnant dams. This was associated with increase in an inflammatory response (cytokines, chemokines and iNOS expression) and infiltration of leukocytes (monocytes and polymorphonuclear cells) in the placenta. There was increased expression of cytosolic and secretary phospholipase A2 with increased secretion of prostaglandin-2 and leukotriene B4 in the placenta, suggestive of increased metabolism of phospholipids. In addition, expression of cycloxygenase-2 and malondialdehyde production (oxidative-stress marker) was increased in the placenta. Conversely, NAC pretreatment abolished these effects of lipopolysaccharide in the placenta. Collectively, these data provide evidence that LPS-induced increased inflammation and metabolism of phospholipids at the feto-maternal interface (placenta) is critical for preterm labor and fetal demise during maternal microbial infections which could be blocked by antioxidant-based therapies.
Keywords: Preterm labor, placenta, maternal microbial infection, Lipopolysaccharide and N-acetyl-cysteine
Maternal microbial infections are one of the main causes of preterm labor and adverse fetal outcome, which persist as major and unresolved challenges. Pro-inflammatory cytokines are hypothesized to contribute to these adverse effects due to microbial infections. Pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) are known to stimulate arachidonic acid metabolism in the human reproductive system (1). During normal pregnancy, secretion of these cytokines from uterine tissues is important for the induction of prostaglandin production to initiate labor (1, 2).Whereas microbial infections induce excessive release of these cytokines in the uterus resulting in preterm labor and delivery via disproportionate release of prostaglandins (3, 4).
In studies exploring the mechanisms of preterm labor caused by microbial infections, lipopolysaccharide (LPS) has been often used as a bacterial pathogen model (3, 5). Humans are constantly exposed to low levels of LPS through microbial infections. Systemic maternal LPS exposure has been previously shown to be associated with adverse fetal developmental outcomes such as embryonic resorption, intrauterine fetal death, and preterm labor in animals (6). In addition, microbial invasion of the placenta is reported to cause chorioamniotitis and preterm labor in human patients (1). LPS is reported to induce inflammation in human gestational tissues, resulting in an increase in release of pro-inflammatory cytokines (3) and phospholipid metabolites i.e., prostanoids (1, 2). LPS induces the release of type II phospholipase A2 (PLA2) in the human placenta which promotes the formation of phospholipids metabolites (4). These LPS-induced increase in expression of pro-inflammatory cytokines and phospholipase isoenzymes in the placenta are mediated by NF-κB activity (7).
Compounds that have antioxidant properties, such as N-acetyl-cysteine (NAC)—a glutathione precursor and direct antioxidant—have been shown to inhibit upstream events leading to NF-κB activation (8–10). NAC decreases the generation of free radicals by increasing glutathione synthesis (8, 11). NAC has been shown to attenuate inflammation in various disease models such as those that mimic ischemia-reperfusion injury in brain (12), lethal endotoxemia (13) and hypoxia-ischemic brain injury in neonatal brains (14). Based upon there reports, we hypothesized that maternal microbial infection induced oxidative-stress in the placenta may be responsible for preterm labor. So, in this study, we investigated the therapeutic efficacy of NAC to limit preterm labor and fetal demise in a murine model of maternal microbial infection.
MATERIALS AND METHODS
Chemicals & Reagents
LPS (Escherichia coli, serotype 055:B5), NAC, lysis buffer and other chemicals were purchased from Sigma-Aldrich (Saint Louis MO, USA). Anti-IL-6, -COX-2, -COX-1 and -CD45 antibodies including secondary anti-rabbit HRP conjugated antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz CA, USA). IL-6 and TNF-a detection BD OptEIA ™ Reagent ELISA kits were purchased from BD biosciences (San Jose CA, USA). Prostaglandin E2 (PGE2) and leukotriene B4 (LTB4), enzyme-immunosorbant assay (EIA) kits were purchased form Assay designs Inc (Ann Arbor Michigan, USA). Malondialdehyde (MDA) detection kits were purchased from BIOXYTECH® MDA-586 (Oxis International Inc. Foster City, CA USA).
Animals
All experiments were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH publication number 80-23) and were approved by the Medical University of South Carolina Animal Care and Use Committee. Animals were provided with food and water ad libitum. Timed-pregnant SD rats at embryonic day (E16) were purchased from Harlan laboratory (Harlan, IN USA).
Induction of Preterm labor & Treatments
For induction of preterm labor, an intra-peritoneal injection of LPS (0.5 mg/kg) was administered to pregnant mothers on gestation day E18 (15). Preterm delivery was defined by delivery of at least one pup within 48 h of LPS exposure. For treatment studies, an intra-peritoneal injection of NAC (50 mg/kg) suspended in physiological saline (pH 7.4) was administered to pregnant mothers 2 h prior to LPS administration on E18 as reported earlier (15). To evaluate the clinical relevance of NAC treatment, it was administered to pregnant mothers at 0, 2, 4 and 6 h post LPS administration. Normal control animals treated with saline and NAC alone were also included in the study.
Collection of Samples
Timing for collection of samples was at 4 h (E18), 24 h (E19), and 48 h (E20) post-LPS administration in each group. Briefly, pregnant rats were anesthetized with a cocktail of ketamine (90 mg/kg) and xylazine (10 mg/kg) and peritoneal cavities were exposed via a midline incision. The uterus was removed and each chorioamnion was punctured to collect amniotic fluid. Feto-placental units were removed from the uterus horns by dissociation between implantation sites. Samples were individually, snap frozen in liquid nitrogen and stored at −80 °C. Five placentas or amniotic fluid collected from five gestational sacs/pregnant rat were pooled for various analyses mentioned below and six pregnant rats were included in each group at each time point.
Histology
Histological studies were performed on placentas at time intervals 4 h (E18), 24 h (E19) and 48 h (E20) of gestation after LPS exposure. Placentas were fixed in 10% buffered formalin (Stephens Scientific, Riverdale, NJ) and embedded in paraffin to cut 4-μm-thick sections. Sections were stained with anti-CD 45 antibodies using standard methods or Hoechst dye and analyzed by immunofluorescence microscopy (Olympus BX-60) with an Olympus digital camera (Optronics, Goleta, CA) as described earlier (15). The number of cells (CD45+ or Hoechst+) was counted by Image Pro-Plus 4.0 (Media Cybernetics, Maryland, MO) software. Sections of three placentas from 6 similarly treated animals in a group were examined and images of 7–9 fields/placental section were included to calculate the average number of CD45+ and Hoechst+ cells in the region. Results were then plotted as the fold-change from the control values.
RNA purification and quantitative real-time-PCR (QRT-PCR) analysis
Total RNA was purified from placentas (pooled five placentas/pregnant rat) obtained from pregnant rats (n = 6)/group and used for cDNA synthesis using methods described earlier (16). QRT-PCR was performed using an iCycler IQ Real-Time PCR Detection System (BIO-RAD Laboratories, Hercules CA, USA), IQ SYBR Green Supermix (BIO-RAD Laboratories, Hercules CA, USA) and target gene primers (Table 1) designed and purchased from IDT Inc (Coralville IA, USA). Thermal cycling conditions were as follows: activation of iTaq DNA polymerase (Bio-Rad Laboratories, Hercules, CA USA) at 95 °C for 10 min, followed by 40 cycles of amplification at 95 °C for 30 s and 55–60 °C for 1 min. Then, normalized expression data were generated by dividing the amount of target gene concentration with the amount of reference genes i.e., GAPDH or 18S rRNA as described previously (16).
Table 1.
List of primers forward primer (FP) and reverse primer (RP) used for amplification using QRT-PCR
| Gene Name | Primer |
|---|---|
| GAPDH | FP: 5′-cctacccccaatgtatccgttgtg-3′, RP: 5′-ggaggaatgggagttgctgttgaa-3′ |
| 18S rRNA | FP: 5′-ccagagcgaaagcatttgccaaga-3′, RP: 5′-tcggcatcgtttatggtcggaact-3′ |
| IL-1β | FP: 5′-gagagacaagcaacgacaaaatcc-3′, RP: 5′-ttcccatcttcttctttgggtattg-3′ |
| TNF-α | FP: 5′-cttctgtctactgaacttcggggt-3′, RP: 5′-tggaactgatgagagggagcc-3′ |
| IFN-γ | FP: 5′-atttccctccccactccattag-3′, RP: 5′-ctggtgacagctggtgaatca-3′ |
| IL-6 | FP: 5′-cagaaggagtggctaaggac-3′, RP: 5′-cactaggtttgccgagtaga-3′ |
| iNOS | FP: 5′-ggaagaggaacaactactgctggt-3′, RP: 5′-gaactgagggtacatgctggagc-3′ |
| ICAM-1 | FP: 5′-gtccaattcacactgaatgccacc-3′, RP: 5′-ttaaacaggaactttcccgccacc-3′ |
| VCAM-1 | FP: 5′-gacaccgtcattatctcctgcact-3′, RP: 5′-gtgtacgagccatccacagacttt-3′ |
| MCP-1 | FP: 5′-gaccagaaccaagtgagatca-3′, RP: 5′-gcttcagatttatgggtcaagt-3′ |
| CCR2 | FP: 5′-tctacttcttctggactccataca-3′, RP: 5′-ctaagtgcatgtcaaccacac-3′ |
| cPLA-2 | FP: 5′-cctgatgtggagaaggattcgaca-3′, RP: 5′-ttccttggtttccctcagaacacc-3′ |
| sPLA2 | FP: 5′-gccaaatctcctgctctacaaacc-3′, RP: 5′-agatgtctctttcagcaactgggc-3′ |
| COX-2 | FP: 5′-tgtcccttcgcctctttcaatgtg-3′, RP: 5′-ctcttacagctcagttgaacgcct-3′ |
ELISA
Amniotic fluid obtained from pregnant rats (n = 6)/group was used for quantification of IL-6 and TNF-α using BD OptEIA ™ Reagent kits (sensitivity of assays was as ≤ 2 pg/mL of protein) as per manufacturer’s instructions. Each assay was run in duplicate for each sample. Recombinant proteins of TNF-α and IL-6 were used as standards to calculate their levels in the samples. Plates were read on plate reader at 450 nm.
EIA
Placentas obtained from pregnant rats (n = 6)/group were homogenized in ethanol, centrifuged and supernatants were used for quantification of PGE2 and LTB4. Concentration of PGE2 in the supernatant was determined by using PGE2, EIA kits as per manufacturer’s manual and run in duplicate for each sample. Similarly, LTB4 was measured in the supernatants of placental tissues using LTB4, EIA kits as per manufacturer’s manual and run in duplicate for each sample, and read at 405nm. Sensitivity of PGE2 and LTB4 assays are 13.4 and 5.63 pg/mL, respectively.
Detection of lipid peroxidation
Placentas obtained from pregnant rats (n = 6) were homogenized in PBS, centrifuged at 10,000 × g for 60 minutes and supernatants were used for analysis. MDA was measured in the supernatants by using BIOXYTECH® MDA-586 as per manufacturer’s instructions. Plates were read at 586 nm and compared with MDA standards provided in the kit. Results are presented as nmol/mg protein for MDA equivalent. Protein concentration in the supernatants was measured by using Bradford’s reagent.
Immunoblotting
Placentas obtained from pregnant rats (n = 6) were homogenized in a cell lysis buffer at 4°C and centrifuged. Proteins in supernatants were fractionated on 12 % polyacrylamide-SDS gels followed by transfer of protein bands to nitrocellulose membranes. After blocking, membranes were incubated with anti-IL-6 or -COX-2 antibodies and then with respective secondary antibodies. Immunoblots were detected using chemiluminescence’s detection kits (Amersham, Arlington Heights, IL, USA).
Data analysis
Statistical analysis was performed using GraphPad software (GraphPad Software Inc. San Diego, CA USA). Effects were measured by comparing with student’s t-test for two data points or by ANOVA (Student-Newman-Keuls) for multiple data points. The criterion for statistical significance was p <0.05.
RESULTS
NAC blocks LPS-induced preterm labor and inflammatory response in placenta
Systemic LPS injection (E18) provoked preterm labor in 10 of 14 treated pregnant mothers with full emptiness of the two uterine horns within 48 h LPS exposure and viability of delivered pups was 60 ± 12.7%. Despite maintaining pregnancies, the remaining four mothers experienced massive in utero fetal demise (72%) and decrease in survival of pups on normal delivery day (E21) following LPS exposure (Fig. 1A). However, no maternal mortality was observed. In contrast, NAC pretreatment significantly attenuated this LPS-induced preterm labor and fetal demise thus normal delivery (Fig. 1A). Furthermore, the evaluation of clinical relevance of NAC therapy showed that it attenuates preterm labor 100%, 93%, 88% and 73.80% when treated at 0, 2, 4 and 24 h following LPS exposure, respectively. QRT-PCR indicated a significant increase in LPS-induced transcripts for pro-inflammatory cytokines i.e., TNF-α, IL-1β and IFN-γ in the placental tissues at 4, 24 and 48 h post-LPS exposure and this was significantly attenuated by NAC (Fig. 1B–D). Expression of these pro-inflammatory cytokines peaked at 24 h post-LPS administration (Fig. 1B–D). Likewise, LPS induced a marked increase in IL-6 transcripts; an important pro-inflammatory cytokine at 24 and 48 h post-LPS exposure and that was significantly blocked by NAC (Fig. 1E). In addition, iNOS transcripts produced because of inflammatory response peaked at 24 h post-LPS exposure and this was significantly attenuated by NAC (Fig. 1F).
Figure 1. NAC attenuates LPS-induced fetal demise via inhibition of inflammation in the placenta of pregnant rats.

Plot depicts number of live (■) and dead pups (□) delivered by pregnant mothers following LPS exposure in the presence/absence of NAC (A). Plots depict transcripts of TNF-α (B), IL-1β (C), IFN-γ (D), IL-6 (E) and iNOS (F) by QRT-PCR in the placentas obtained from pregnant rats (n = 6) following LPS exposure in the presence/absence of NAC. Results in plots are expressed as Mean ± SD. Statistical significance ** p<0.001 and non-significant (§).
NAC attenuates LPS-induced protein expression of the pro-inflammatory cytokine IL-6 and TNF-α in the amniotic fluid
To assess that the onset of preterm labor in LPS-exposed dams was associated with an intrauterine inflammation and that this phenomenon was prevented in the presence of NAC, we measured TNF-α and IL-6 cytokines in amniotic fluids. Maternal LPS exposure at gestation E18 induced a significant increase in TNF-α and IL-6 in amniotic fluid at 4 and 24 h compared with saline treated controls (Fig. 2A–B). Conversely, NAC pretreatment attenuated the LPS-induced increase in TNF-α and IL-6 at 4 and 24 h post-LPS exposure (Fig. 2A–B). Cytokines in NAC-treated control animals were similar to saline-treated controls. Corresponding with QRT-PCR data (Fig. 1E), IL-6 protein was also elevated in the placental tissue following LPS exposure at 24 and 48 h and this was attenuated by NAC (Fig. 2C).
Figure 2. NAC attenuates LPS-induced inflammatory response in amniotic fluid of pregnant rats.

Plots depict TNF-α (A) and IL-6 (B) protein in the amniotic fluid obtained form gestational sacs at 4 h (■) and 24 h (□) by ELISA and representative immunoblot depicts IL-6 protein in the placental tissue (C) of pregnant rats (n = 6) following LPS exposure in the presence/absence of NAC. Results in plots are expressed as Mean ± SD. Statistical significance ** p<0.001 and non-significant (§).
NAC limits LPS-induced cellular infiltration of leukocytes and chemokines expression in placenta
To further investigate the attenuation of LPS-induced infiltration of leukocytes and expression of chemokines in the placenta by NAC, placental tissues were analyzed for cellular infiltration and expression of MCP-1 and CCR2. MCP-1 belongs to a β-chemokine family, which is pivotal in the pathogenesis of inflammatory diseases. Hoechst staining of placental sections revealed that LPS induces infiltration of leukocytes in the placenta, which was significantly attenuated by NAC (Fig. 3A). Likewise, Leukocyte (CD45+) immunostaining corroborated these data (data not shown). Histological examination showed that NAC attenuates LPS-induced cellular infiltration into the placenta (Fig. 3B). Corroborating these results, LPS-induced increase in MCP-1 transcripts at 24 and 48 h after LPS exposure was significantly attenuated by NAC (Fig. 3C). Likewise, LPS induced an increase in CCR2 transcripts at 24 and 48 h after LPS exposure, which was significantly attenuated by NAC (Fig. 3D).
Figure 3. NAC attenuates LPS-induced leukocyte infiltration and chemokine expression in the placenta of pregnant rats.

Plot depicts Hoechst+ nuclei (A), representative image of placental sections show cellular-infiltration [control (i), LPS (ii) and NAC + LPS (iii)] (B). Arrowhead indicates infiltrates in the placental tissue. Plots depict MCP-1 (C) and CCR2 (D) transcripts in the placentas of pregnant rats (n = 6) following LPS exposure in the presence/absence of NAC. Results in plots are expressed as Mean ± SD. Statistical significance * p<0.05, ** p<0.001 and non-significant (§). Magnifications at 400 ×s.
NAC attenuates LPS-induced increase in cellular adhesion molecule expression in placenta
To further characterize the effect of NAC on LPS-induced increases in expression of cell adhesion molecules (CAMs) associated with cellular infiltration, we measured critical CAMs i.e., intracellular-CAM (ICAM-1) and vascular CAM (VCAM-1) in the placental tissue. LPS induced a significant increase in ICAM-1 and VCAM-1 transcripts in the placental tissue compared with controls (Fig. 4A–B). Conversely, NAC pretreatment significantly attenuated this LPS-induced increase in ICAM-1 and VCAM-1 in placental tissues (Fig. 4A–B).
Figure 4. NAC attenuates LPS-induced expression of CAMs in the placenta of pregnant rats.
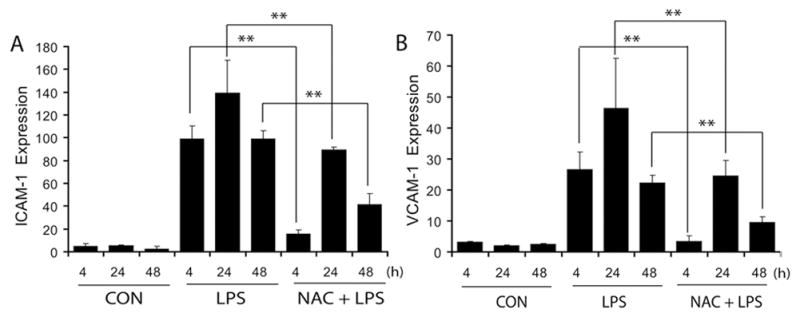
Plots depict ICAM-1 (A) and VCAM-1 (B) transcripts in the placenta of pregnant rats (n = 6) following LPS exposure in the presence/absence of NAC. Results in plots are expressed as Mean ± SD. Statistical significance ** p<0.001.
NAC inhibits LPS-induced up-regulation of phospholipids metabolism in placenta
Since inflammation increases phospholipids metabolism responsible for oxidative-stress, we next determined the level of enzymes i.e., cytoplasmic-PLA2 (cPLA2) and secretary-PLA2 (sPLA2) transcripts responsible for phospholipids metabolism and synthesis of lipid by-products i.e., PGE2 and LTB4. LPS induced an increase in the expression of cPLA2 and sPLA2 in the placental tissue at 24 and 48 h following LPS exposure and this was significantly attenuated by NAC (Fig. 5A–B). Corresponding with these findings, LPS induced an increase in PGE2 in the placenta at 24 and 48 h after LPS exposure, which was attenuated by NAC (Fig. 5C). In addition, LPS induced an increase in LTB4 at 48 h after LPS exposure in the placental tissue, which was attenuated by NAC (Fig. 5D).
Figure 5. NAC attenuates LPS-induced phospholipids metabolism in the placenta of pregnant rats.
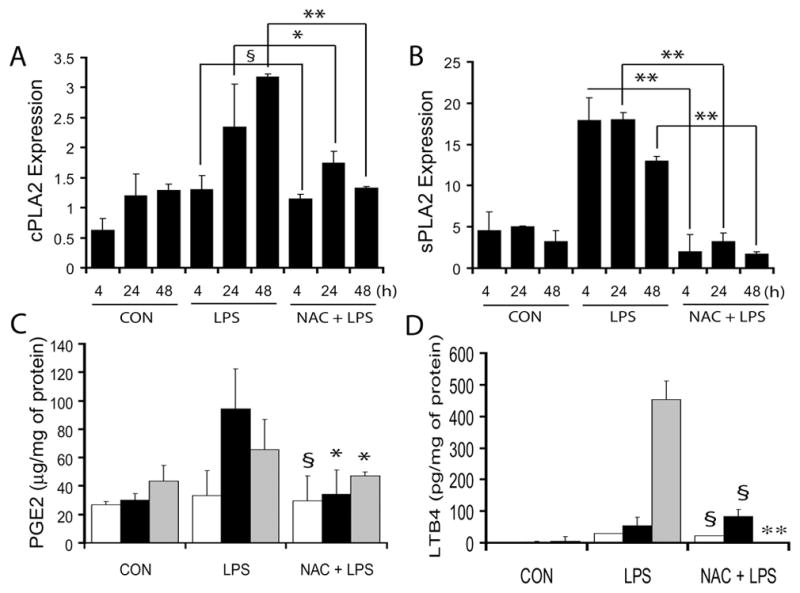
Plots depict cPLA2 (A) and sPLA2 (B) transcripts in the placenta of pregnant rats (n = 6) following LPS in the presence/absence of NAC. Plots depict PGE2 (C) and LTB4 (D) level in the placentas of pregnant rats at 4 h (white bar), 24 h (black bar) and 48 h (grey bar) post-LPS exposure in the presence/absence of NAC. Results in plots are expressed as Mean ± SD. Statistical significance * p<0.05, ** p<0.001 and non-significant (§), and versus LPS for C and D only.
NAC attenuates LPS-induced COX-2 expression and oxidative-stress in placenta
Since synthesis of PGE2 is associated with COX-2, we next measured COX-2 expression and generation of oxidative stress in placental tissue. LPS-induced increase in COX-2 transcripts both at 24 and 48 h in the placental tissue was attenuated by NAC (Fig. 6A). These data revealed that COX-2 expression is quite robust in LPS-injected dams at 24 and 48 h post-administration. Interestingly, NAC was quite potent in inhibiting LPS-induced COX-2 expression confirming transcriptional upregulation of COX-2 in response to the labor-associated inflammatory milieu. Immunoblotting further confirmed that COX-2 protein (72 kDa) was also significantly elevated in the placenta at 48 h after LPS exposure, which was attenuated by NAC (Fig. 6B). No change in COX-1 both at mRNA and protein level was seen among the groups (data not shown). Furthermore, to determine oxidative-stress in the placenta, we measured lipid peroxidation (MDA) in the placental tissue. The effects of LPS exposure at a late gestational stage is presented in Fig. 6C. Results indicate that LPS exposure increases MDA level in the placenta, which was significantly attenuated by NAC (Fig. 6C).
Figure 6. NAC attenuates LPS-induced phospholipids metabolism in the placenta of pregnant rats.

Plot depicts COX-2 transcripts (A) at 24 h (■) and 48 h (□), representative immunoblot depicts COX-2 protein (B) and plot depicts MDA level (C) in the placental tissues of pregnant rats (n = 6) following LPS exposure in the presence/absence of NAC. Results in plots are expressed as Mean ± SD. Statistical significance ** p<0.001.
Altogether, these data suggest that NAC can prevent LPS-induced preterm labor and fetal demise via attenuation of inflammation, cellular infiltration, and release of phospholipids metabolites at the feto-maternal interface of the placenta during maternal systemic/intrauterine microbial infections.
DISCUSSION
Our results indicate the therapeutic efficacy of NAC against LPS-induced preterm labor and fetal demise during maternal systemic microbial infections. LPS-induced effects in pregnant rats were mediated by inflammatory responses and oxidative-stress in the placenta in addition to increase in leukocyte infiltration, CAMs expression, phospholipids metabolism and lipid peroxidation. Conversely, LPS-induced effects in this setting were abolished by NAC.
LPS is reported to induce an increase in expression of TNF-α and IL-1β in both maternal and fetal compartments of pregnant mothers when administered at E18 (17, 18). Clinical studies describe greater pro-inflammatory cytokines TNF-α, IL-1β and IL-6 in the amniotic fluid during intrauterine infections (19). IL-6 secretion in the fetal cavity is considered to be a determining factor of preterm labor (20). We observed a significant increase in the expression of pro-inflammatory cytokines and CAMs (ICAM-1 and VCAM-1) with corresponding increase in infiltration of leukocytes in the placenta of pregnant rats following LPS exposure compared with controls. ICAM-1 and VCAM-1 play crucial role in transmigration of leukocytes from the bloodstream to sites of inflammation. The expression of these CAMs is reported to be up-regulated by pro-inflammatory cytokines (21). In addition, MCP-1 and CCR2 expression was correspondingly increased in the placenta of pregnant rats after LPS exposure compared with controls. MCP-1 activation has been associated with a majority of preterm births (22). The major function of MCP-1 is to recruit monocytes and other leukocytes to sites of inflammation. During parturition, MCP-1 may initiate or propagate labor by serving as a chemo-attractant for leukocytes in gestational tissues. It is noteworthy that MCP-1 expression is up-regulated by several inflammatory mediators i.e., TNF-α, IL-1β and INF-γ (23). In addition, PGE2 has been shown to stimulate the release of MCP-1 in a placenta perfusion model (24).
In the placenta, reactive oxygen species were reported to be involved in the expression of COX-2 and iNOS in trophoblasts via activation of NF-κB (25). COX-2 is a rate-limiting factor in prostaglandin synthesis. The contribution of COX-2 to oxidative damage has been reported to be involved in PGE2 production and hippocampal CA1 neuronal loss in a model of transient global cerebral ischemia (26). We observed that LPS induces a corresponding increase in COX-2 and PGE2 in the placenta at 24 and 48 h after LPS exposure to pregnant rats. This profile of change is consistent with a report describing ischemia–reperfusion (I/R) injury induced COX-2 expression at 24 h after I/R injury in the placenta (27). Inflammation-induced PGE2 production is regulated by the availability of arachidonic acid and by the production of COX-2 (28). TNF-α has been shown to increase the synthesis of PGE2 through COX-2 activity in amniotic and decidual cells (29). Arachidonic acid is cleaved from phospholipids during metabolism by phospholipases i.e., cPLA2 and sPLA2. Similarly, we observed an increase in expression of cPLA2 and sPLA2 in the placenta after LPS exposure. PLA2 activity provides a substrate for peroxidase, COX-2, and PGE synthase responsible for synthesis of PGE2. LPS has been shown to cause the depletion of intracellular glutathione and an increase in MDA, a byproduct of lipid peroxidation (30). An observed increase in MDA in the placental tissue is suggestive of LPS-induced ROS generation in the placenta of pregnant mothers. Lipid peroxidation and a decrease in anti-oxidant mechanisms are reported to be responsible for damage in patients who miscarry (31).
A popular therapeutic approach for the attenuation of LPS-induced ROS generation and lipid peroxidation-mediated preterm labor and fetal demise is the use of antioxidants. NAC is a widely investigated agent with both antioxidant and anti-inflammatory properties. We and others have previously shown that NAC attenuates brain white matter injury in a systemic maternal infection model of periventricular leukomalacia (15, 32). NAC induced beneficial effects are attributed to free radical quenching via its direct anti-oxidant properties and synthesis of intracellular reduced GSH. In addition, anti-inflammatory activities of NAC are attributed to suppression of pro-inflammatory cytokine expression/release (33), inhibition of adhesion molecule expression (34), and inhibition of NF-κB activation (10).
Overall, this study illustrates the underlying mechanism of preterm labor resulting from oxidative stress, increased lipid peroxidation, and metabolism of phospholipids across the feto-maternal interface via activation of the arachidonic acid-cyclooxygenase pathway. It is likely that pro-inflammatory cytokines induce up-regulation of prostaglandin production in gestational tissues contributing to the preterm labor during microbial infections. Based upon these findings, we suggest that NAC may be evaluated for its therapeutic efficacy in the management and limitation of preterm labor in pregnant mothers during systematic microbial infections.
In summary, we conclude that LPS-induced inflammatory response induces leukocyte infiltration, expression of CAMs and chemokines, phospholipids metabolism, and lipid peroxidation in the placenta of pregnant rats resulting in preterm labor and fetal demise. In contrast, a single injection of NAC blocks these LPS-induced adverse effects in pregnant mothers.
Acknowledgments
We thank all members of our laboratory for their valuable comments and help during the course of this study. We thank especially Dr. Shalendra Giri PhD for detection of PGE2 and LTB4 in the placental tissue and Dr. Jennifer G. Schnellmann PhD for critical reading of this manuscript.
Financial Support: This study was supported in part by grants from the National Institutes of Health: NS-22576, NS-34741, NS-37766, NS-40810, and NCRR grants C06 RR018823 and C06 RR015455 from the Extramural Research Facilities Program.
ABBREVIATIONS
- COX-2
cysloxygenase-2
- ICAM-1
intracellular adhesion molecule-1
- LPS
lipopolysaccharide
- LTB4
leukotriene B4
- MDA
malionaldehyde
- NAC
N-acetyl cysteine
- PLA2
phospholipase 2
- VCAM-1
vascular adhesion molecule-1
References
- 1.Mitchell MD, Edwin SS, Lundin-Schiller S, Silver RM, Smotkin D, Trautman MS. Mechanism of interleukin-1 beta stimulation of human amnion prostaglandin biosynthesis: mediation via a novel inducible cyclooxygenase. Placenta. 1993;14:615–625. doi: 10.1016/s0143-4004(05)80379-0. [DOI] [PubMed] [Google Scholar]
- 2.Dowling DD, Romero RJ, Mitchell MD, Lundin-Schiller S. Isolation of multiple substances in amniotic fluid that regulate amnion prostaglandin E2 production: the effects of gestational age and labor. Prostaglandins Leukot Essent Fatty Acids. 1991;44:253–255. doi: 10.1016/0952-3278(91)90026-2. [DOI] [PubMed] [Google Scholar]
- 3.Fortunato SJ, Menon RP, Swan KF, Menon R. Inflammatory cytokine (interleukins 1, 6 and 8 and tumor necrosis factor-alpha) release from cultured human fetal membranes in response to endotoxic lipopolysaccharide mirrors amniotic fluid concentrations. Am J Obstet Gynecol. 1996;174:1855–1861. doi: 10.1016/s0002-9378(96)70221-1. discussion 1861-1852. [DOI] [PubMed] [Google Scholar]
- 4.Farrugia W, Nicholls L, Rice GE. Effect of bacterial endotoxin on the in vitro release of Type II phospholipase-A2 and prostaglandin E2 from human placenta. J Endocrinol. 1999;160:291–296. doi: 10.1677/joe.0.1600291. [DOI] [PubMed] [Google Scholar]
- 5.Anteby EY, Johnson RD, Huang X, Dryden DK, Nelson DM, Sadovsky Y. Lipopolysaccharide enhances the transcription of prostaglandin H synthase-2 gene in primary human trophoblasts. Am J Obstet Gynecol. 1998;178:469–473. doi: 10.1016/s0002-9378(98)70422-3. [DOI] [PubMed] [Google Scholar]
- 6.O’Sullivan AM, Dore CJ, Boyle S, Coid CR, Johnson AP. The effect of campylobacter lipopolysaccharide on fetal development in the mouse. J Med Microbiol. 1988;26:101–105. doi: 10.1099/00222615-26-2-101. [DOI] [PubMed] [Google Scholar]
- 7.Lappas M, Permezel M, Georgiou HM, Rice GE. Regulation of phospholipase isozymes by nuclear factor-kappaB in human gestational tissues in vitro. J Clin Endocrinol Metab. 2004;89:2365–2372. doi: 10.1210/jc.2003-031385. [DOI] [PubMed] [Google Scholar]
- 8.Neuschwander-Tetri BA, Bellezzo JM, Britton RS, Bacon BR, Fox ES. Thiol regulation of endotoxin-induced release of tumour necrosis factor alpha from isolated rat Kupffer cells. Biochem J. 1996;320:1005–1010. doi: 10.1042/bj3201005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verhasselt V, Vanden Berghe W, Vanderheyde N, Willems F, Haegeman G, Goldman M. N-acetyl-L-cysteine inhibits primary human T cell responses at the dendritic cell level: association with NF-kappaB inhibition. J Immunol. 1999;162:2569–2574. [PubMed] [Google Scholar]
- 10.Pahan K, Sheikh FG, Namboodiri AM, Singh I. N-acetyl cysteine inhibits induction of no production by endotoxin or cytokine stimulated rat peritoneal macrophages, C6 glial cells and astrocytes. Free Radic Biol Med. 1998;24:39–48. doi: 10.1016/s0891-5849(97)00137-8. [DOI] [PubMed] [Google Scholar]
- 11.Song M, Kellum JA, Kaldas H, Fink MP. Evidence that glutathione depletion is a mechanism responsible for the anti-inflammatory effects of ethyl pyruvate in cultured lipopolysaccharide-stimulated RAW 264.7 cells. J Pharmacol Exp Ther. 2004;308:307–316. doi: 10.1124/jpet.103.056622. [DOI] [PubMed] [Google Scholar]
- 12.Sekhon B, Sekhon C, Khan M, Patel SJ, Singh I, Singh AK. N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res. 2003;971:1–8. doi: 10.1016/s0006-8993(03)02244-3. [DOI] [PubMed] [Google Scholar]
- 13.Victor VM, Rocha M, De la Fuente M. N-acetylcysteine protects mice from lethal endotoxemia by regulating the redox state of immune cells. Free Radic Res. 2003;37:919–929. doi: 10.1080/1071576031000148727. [DOI] [PubMed] [Google Scholar]
- 14.Jatana M, Singh I, Singh AK, Jenkins D. Combination of systemic hypothermia and N-acetylcysteine attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr Res. 2006;59:684–689. doi: 10.1203/01.pdr.0000215045.91122.44. [DOI] [PubMed] [Google Scholar]
- 15.Paintlia MK, Paintlia AS, Barbosa E, Singh I, Singh AK. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78:347–361. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- 16.Paintlia AS, Paintlia MK, Singh AK, Stanislaus R, Gilg AG, Barbosa E, Singh I. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by Lovastatin. J Neurosci Res. 2004;77:63–81. doi: 10.1002/jnr.20130. [DOI] [PubMed] [Google Scholar]
- 17.Bell MJ, Hallenbeck JM, Gallo V. Determining the fetal inflammatory response in an experimental model of intrauterine inflammation in rats. Pediatr Res. 2004;56:541–546. doi: 10.1203/01.PDR.0000139407.89883.6B. [DOI] [PubMed] [Google Scholar]
- 18.Huleihel M, Amash A, Sapir O, Maor E, Levy S, Katz M, Dukler D, Myatt L, Holcberg G. Lipopolysaccharide induces the expression of interleukin-1alpha distinctly in different compartments of term and preterm human placentae. Eur Cytokine Netw. 2004;15:30–36. [PubMed] [Google Scholar]
- 19.Hagberg H, Mallard C, Jacobsson B. Role of cytokines in preterm labour and brain injury. BJOG. 2005;112:16–18. doi: 10.1111/j.1471-0528.2005.00578.x. [DOI] [PubMed] [Google Scholar]
- 20.Romero R, Sepulveda W, Kenney JS, Archer LE, Allison AC, Sehgal PB. Interleukin 6 determination in the detection of microbial invasion of the amniotic cavity. Ciba Found Symp. 1992;167:205–220. doi: 10.1002/9780470514269.ch13. discussion 220-203. [DOI] [PubMed] [Google Scholar]
- 21.Radi ZA, Kehrli ME, Jr, Ackermann MR. Cell adhesion molecules, leukocyte trafficking, and strategies to reduce leukocyte infiltration. J Vet Intern Med. 2001;15:516–529. doi: 10.1892/0891-6640(2001)015<0516:camlta>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 22.Esplin MS, Romero R, Chaiworapongsa T, Kim YM, Edwin S, Gomez R, Mazor M, Adashi EY. Monocyte chemotactic protein-1 is increased in the amniotic fluid of women who deliver preterm in the presence or absence of intra-amniotic infection. J Matern Fetal Neonatal Med. 2005;17:365–373. doi: 10.1080/14767050500141329. [DOI] [PubMed] [Google Scholar]
- 23.Proost P, Wuyts A, van Damme J. The role of chemokines in inflammation. Int J Clin Lab Res. 1996;26:211–223. doi: 10.1007/BF02602952. [DOI] [PubMed] [Google Scholar]
- 24.Denison FC, Kelly RW, Calder AA, Riley SC. Cytokine secretion by human fetal membranes, decidua and placenta at term. Hum Reprod. 1998;13:3560–3565. doi: 10.1093/humrep/13.12.3560. [DOI] [PubMed] [Google Scholar]
- 25.Callejas NA, Casado M, Bosca L, Martin-Sanz P. Requirement of nuclear factor kappaB for the constitutive expression of nitric oxide synthase-2 and cyclooxygenase-2 in rat trophoblasts. J Cell Sci. 1999;112:3147–3155. doi: 10.1242/jcs.112.18.3147. [DOI] [PubMed] [Google Scholar]
- 26.Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Alvarez D, Al-Dalain S, Martinez G, Leon OS, Springer JE. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem. 2003;86:545–555. doi: 10.1046/j.1471-4159.2003.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamazaki K, Endo T, Kitajima Y, Manase K, Nagasawa K, Honnma H, Hayashi T, Kudo R, Saito T. Elevation of both cyclooxygenase-2 and prostaglandin E2 receptor EP3 expressions in rat placenta after uterine artery ischemia-reperfusion. Placenta. 2006;27:395–401. doi: 10.1016/j.placenta.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Miyaura C, Inada M, Matsumoto C, Ohshiba T, Uozumi N, Shimizu T, Ito A. An essential role of cytosolic phospholipase A2alpha in prostaglandin E2-mediated bone resorption associated with inflammation. J Exp Med. 2003;197:1303–1310. doi: 10.1084/jem.20030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perkins DJ, Kniss DA. Tumor necrosis factor-alpha promotes sustained cyclooxygenase-2 expression: attenuation by dexamethasone and NSAIDs. Prostaglandins. 1997;54:727–743. doi: 10.1016/s0090-6980(97)00144-5. [DOI] [PubMed] [Google Scholar]
- 30.Victor VM, De la Fuente M. Immune cells redox state from mice with endotoxin-induced oxidative stress. Involvement of NF-kappaB. Free Radic Res. 2003;37:19–27. doi: 10.1080/1071576021000038522. [DOI] [PubMed] [Google Scholar]
- 31.Biri A, Kavutcu M, Bozkurt N, Devrim E, Nurlu N, Durak I. Investigation of free radical scavenging enzyme activities and lipid peroxidation in human placental tissues with miscarriage. J Soc Gynecol Investig. 2006;13:384–388. doi: 10.1016/j.jsgi.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- 33.Tsuji F, Miyake Y, Aono H, Kawashima Y, Mita S. Effects of bucillamine and N-acetyl-L-cysteine on cytokine production and collagen-induced arthritis (CIA) Clin Exp Immunol. 1999;115:26–31. doi: 10.1046/j.1365-2249.1999.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rahman A, Kefer J, Bando M, Niles WD, Malik AB. E-selectin expression in human endothelial cells by TNF-alpha-induced oxidant generation and NF-kappaB activation. Am J Physiol. 1998;275:L533–L544. doi: 10.1152/ajplung.1998.275.3.L533. [DOI] [PubMed] [Google Scholar]