

Abstract
Mechanism-based inactivators such as bergamottin are useful chemical tools for identifying the roles of specific active-site amino acid residues in the reactions catalyzed by the cytochromes P450 (CYPs or P450s) that are responsible for the metabolism of a wide variety of drugs and endogenous substrates. In clinical settings mechanism-based inactivation of P450s involved in xenobiotic metabolism has the potential to lead to adverse drug-drug interactions and assays to identify and characterize drug candidates as P450 inactivators are important in drug discovery and development. Here we present a quantitative high-throughput protocol for investigating cytochrome P450 mechanism-based inactivators using the example of CYP2B6 and bergamottin to illustrate the finer points of this protocol. This protocol details the adaptation of a 7-ethoxytrifluoromethyl coumarin (7-EFC) O-deethylation fluorescence activity assay to a 96-well microtiter plate format and uses a plate-reader to detect the fluorescence of the product. Compared to previous methods, this protocol requires less P450 and takes significantly less time while greatly increasing throughput. The protocol as written takes approximately two hours to complete. The principles and procedures outlined in this protocol can be easily adapted to other inactivators, P450 isoforms, substrates and plate-readers.
Search terms: Biochemistry, Pharmacology, Drug metabolism, High-throughput analysis, Cytochrome P450, Mechanism-based inactivation, Suicide inhibition, CYP2B6, bergamottin, 7-EFC, 7-HFC
INTRODUCTION
The cytochromes P450 (CYPs or P450s) are a ubiquitous super-family of heme-containing proteins that are responsible for the metabolism of a wide array of endogenous and exogenous compounds including more than 60% of all clinically used drugs (1–3). Most of the reactions catalyzed by P450s generally lead to the formation of more hydrophilic products that can be readily excreted from the body; however, some of these reactions produce highly reactive intermediates, which may target P450 active site amino acid residues for modification and lead to enzyme inactivation. Those compounds are often referred to as “suicide” or “mechanism-based” inactivators (4,5).
The development of quantitative high-throughput assays to assess whether new chemical entities are P450 inactivators is of considerable interest in the pharmaceutical industry and several in vitro assays have been developed over the years (6,7). Ideally, these assays are most useful if they can be designed for use in a high-throughput format while minimizing enzyme consumption. The most widely used method today for the quantitative characterization of a compound for its time- and concentration-dependent inactivation of the catalytic activity of a purified P450 involves initially reconstituting P450 with NADPH-P450 reductase (CPR) and lipid for 60 min at 4 °C. The primary incubation mixtures typically contain 1 nmol of P450, 1 nmol of CPR, 1,2-didodecanoyl-sn-glycero-3-phosphatidylcholine (DLPC) and several different concentrations of the potential inactivator under investigation, in a potassium phosphate buffer at pH 7.4 for a total reaction volume of 1 mL (8,9). These components constitute the primary reaction to which NADPH is added in order to initiate mechanism-based inactivation. At designated times, an aliquot is removed from the primary reaction and diluted into an enzyme assay mixture (secondary reaction) containing saturating concentrations of the substrate. This secondary reaction is used to measure the extent and rate of mechanism-based inactivation incurred in the primary reaction. All of the inactivation experiments are interpreted relative to a control sample, which is treated exactly like the experimental sample except that the solvent in which the inactivating species is dissolved in is added to the primary mixture instead of the inactivating compound. Activity is assessed spectrofluorometrically in the secondary mixture by measuring the rate of O-deethylation of 7-ethoxytrifluoromethyl coumarin (7-EFC) to 7-hydroxytrifluoromethyl coumarin (7-HFC) (Figure 1). This method developed by Buters and co-workers based on earlier work by DeLuca et al. is highly sensitive and has been used as a laboratory standard for investigating mechanism-based P450 inactivation for several years (10,11). However, individual fluorescence measurements at varying concentrations of inactivator and at different time points is slow and renders this protocol impractical for a high-throughput format where large numbers of samples must be tested. Furthermore, to achieve a sufficiently large fluorescence signal-to-background ratio using a standard spectrofluorometer this method requires the use of a considerable amount of protein, which is particularly problematic with some human P450s (e.g CYP2B6) whose expression yields are poor and the low yields may be further exacerbated by site-directed mutagenesis (unpublished observations and 12). These limitations can largely be overcome if the minimal volume required for the secondary reaction and the amount of protein transferred from primary to secondary reaction can be reduced.
Figure 1.

Schematic representation of the O-deethylation of 7-EFC to produce 7-HFC (ex. 410 nm and em. 510 nm), which is catalyzed by a number of human P450s including CYP 1A2, 2B6, 2C19, 2D6, 2E1, and 3A4.
The example demonstrated in this protocol uses CYP2B6 and bergamottin. Human CYP2B6 preferentially metabolizes a variety of widely used pharmaceuticals including bupropion, efavirenz, sertralin, and cyclophosphamide and knowledge concerning the inactivation of this P450 by new drug candidates is important for effective drug development (13,14). Bergamottin (BG), a furanocoumarin component of grapefruit juice and one of the major active ingredients responsible, in part, for the interaction of grape-fruit juice with drugs (15,16), has been shown to be a potent inactivator of CYP2B6 (9) (Figure 2). The protocol described here details the application of a quantitative high-throughput microtiter plate-based fluorescence screen for the inactivation of CYP2B6 by BG. This protocol represents an improvement on the previous methods of Buters and co-workers (10) and DeLuca et al. (11) since it measures fluorescence emission of O-deethylation reactions performed in microtiter wells where the minimal volume required for a fluorescence reading is significantly less than that required in a cuvette. Consequently, the minimal molar amount of fluorescent product needed to produce measurable fluorescence emission is also much less. This characteristic facilitated the design of an assay that uses considerably less P450 and reductase than methods commonly used for these types of studies and is significantly faster. In addition, the ability to perform this assay using microtiter plates allows this to be readily adapted to a high-throughput format. We also show how this assay can readily be used to determine the KI, kinact and t1/2 for the inactivation of CYP2B6 by BG in a single experiment.
Figure 2.
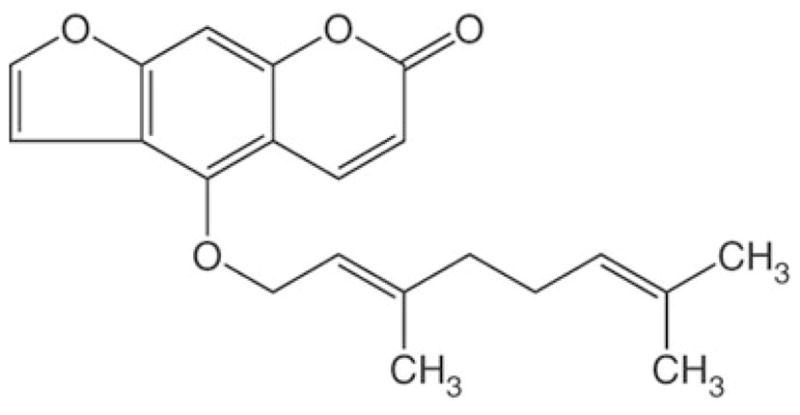
Chemical structure of BG. BG is abundant in grapefruit and easily absorbed through the intestinal wall (17). The mechanism-based inactivation of P450s by BG, which may account for increased drug bioavailability, results in modification of the P450 heme and apoprotein (9).
Adaptation of this method to other systems
Although this protocol was optimized for application to high-throughput studies of the mechanism-based inactivation of purified recombinant CYP2B6 by BG in a reconstituted system, since the principles underlying this method are broad, it can be used as a template for screening many other P450s and inhibitors in other systems. For example, the use of cDNA-expressed P450s in the form of Baculosomes or Supersomes is very common in the pharmaceutical industry and the protocol described here can readily be extended to the use of these commercially available P450 preparations. Our suggested times for assay completion and the results anticipated may vary somewhat according to the P450 and inactivator being tested and will also be influenced by the resources available.
This method is readily adaptable to study any enzyme system which undergoes mechanism-based inactivation by substituting the enzyme of interest, the mechanism-based inactivator, and a probe substrate that shows a significant increase in fluorescence or absorbance upon metabolism. Requirements for use with other systems will include optimization of several experimental parameter such as: 1) the protein and inactivator concentrations in the primary reaction mixture, 2) the incubation times for the primary and secondary reaction mixtures, 3) the volume of the sample transferred from the primary to secondary reaction, and 4) the concentration of the probe substrate used in the secondary reaction.
Furthermore, the high-throughput protocol described below is based on a 96-well plate format using a hand-held multi-channel pipettor, but could be easily adapted to a larger density format (384- or 1538-wells) using automated liquid handlers or a robotic platform and may also be extended to non-fluorescence-based assays. If these alternatives are pursued, then the times required for preparation and completion of the assay may differ significantly from those indicated here. We present the REAGENT SETUP section in a practical order which facilitates the subsequent steps in the PROCEDURE section.
MATERIALS
REAGENTS
Purified recombinant human CYP2B6 stored at −80°C in 0.1M potassium phosphate buffer and 20% glycerol (v/v) (stock concentration of CYP2B6 is 6.5 μM)
Purified recombinant human NADPH-P450 reductase (CPR) in 0.1M potassium phosphate buffer and 20% glycerol (v/v) (stock concentration of CPR is 12 μM)
Potassium phosphate dibasic and monobasic (Fisher cat. no. P288 and cat. no. P285 respectively). Prepared each as 1 M stocks in water, stored at 4°C.
Nicotinamide adenine dinucleotide phosphate (NADPH, Sigma-Aldrich, cat. no. N7505), prepared at 20 mM in water, stored at −20 °C. CRITICAL Since repeated freeze-thawing will contribute to NADPH degradation, it is recommended that 20 mM NADPH stock solutions be stored as several low volume aliquots with only enough NADPH for a single series of assays. Alternatively, vials of pre-weighed solid NADPH can be used.
-
Bergamottin (BG, Indofine Chemical Company, cat. no. 020550S), prepared at 10 mM in methanol, stored at −20 °C. Caution! Toxic! CRITICAL It is advantageous to prepare fresh stocks of BG as solvent evaporation over extended periods of time may result in erroneous stock concentrations.
? TROUBLESHOOTING
1,2-didodecanoyl-sn-glycero-3-phosphatidylcholine (DLPC, Sigma-Aldrich, cat. no. P1263), prepared at 1 mg/ml in water, stored at 4 °C.
Catalase from bovine liver (Sigma-Aldrich, cat. no. C40 lyophilized powder 10,000 units/mg protein), prepared at 1 mg/ml in water, stored at −20 °C.
7-EFC (Invitrogen, Molecular Probes, cat. no. E2882), prepared at 25 mM in DMSO, stored in an amber glass vial at 4 °C.
Methanol (Fisher, cat. no. A452-4) Caution! Flammable and toxic!
Dimethyl sulfoxide (DMSO, Sigma-Aldrich, cat. no. D8418) stored at room temperature. Caution! Readily absorbed through skin and combustible!
Acetonitrile (Fisher, cat. no. A998-4) Caution! Flammable and toxic!
EQUIPMENT
Multichannel pipettors (Fisherbrand)
Sonicator equipped with a 10mm flat tip probe (Branson)
96-well black, low-volume microtiter plates (ThermoFisher)
Fluorescence plate reader – Victor II 1420-042 (PerkinElmer)
Shaking water bath – Precision (ThermoFisher)
Stainless steel block chamber. CRITICAL When the 96-well plate is set over the stainless steel block chamber, uniform heat is provided to all sample wells regardless of their position.
Digital timer
REAGENT SETUP
-
Bergamottin stocks. To achieve final concentrations of 1, 2, 4, 6, 10 and 15 μM BG in 50 μL primary reactions without varying the volume of BG stock added make the following corresponding BG working stocks in methanol by serial dilution: 18.5, 37, 74.1, 111.1, 185.2, 277.8 μM.
CRITICAL Prescreening of the inactivator being tested should be performed to ensure that the compound itself does not exhibit fluorescence properties that would interfere with the fluorometric detection of 7-HFC.
? TROUBLESHOOTING
-
Reconstitution mixture. The reconstituted system will contain 20.4 pmol purified CYP2B6, 40.8 pmol purified CPR and 0.07 mg/ml DLPC per reaction well. In this assay approximately 3.14 μL of 6.5 μM CYP2B6, 3.4 μL of 12 μM CPR and 3.66 μL of sonicated 1 mg/ml DLPC are reconstituted for each inactivator concentration. CRITICAL If more than one inactivator concentration is to be tested with a particular P450, simply multiply the volumes used by the number of inactivator concentrations being tested and reconstitute P450, CPR and DLPC in a single Eppendorf tube to minimize variation between separately reconstituted samples and then aliquot appropriate volumes. It is best to perform triplicate runs for each inactivator concentration and time point and to determine the average and standard deviation of the results.
? TROUBLESHOOTING
Primary reaction mixture. 1 M potassium phosphate buffer pH 7.4 is prepared by mixing 1 M potassium phosphate dibasic with the same concentration of potassium phosphate monobasic such that the final percentage composition by volume is 81% and 19% respectively. Verify pH with pH meter and adjust by adding appropriate acid or base. Prepare a 245.8 μL reaction buffer solution by mixing 17.5 μL 1 M potassium phosphate buffer pH 7.4, 0.75 μL of 1 mg/ml catalase and add water to 245.9 μL. Since 35.1 μL of this buffer will be added to a final primary reaction volume of 50 μL, the final potassium phosphate concentration will be 50 mM.
Secondary reaction mixture. This protocol determines both the concentration-and time-dependent inactivation of CYP2B6 at seven different concentrations of BG at four different time points. For a sufficient supply of secondary reaction mixture, add 225 μL of 1 M potassium phosphate buffer (pH7.4), 67.5 μL of 20 mM NADPH, 18 μL of 25 mM 7-EFC and then add water to a final volume of 4500 μL. CRITICAL Shield the secondary reaction from light whenever possible since 7-EFC is photoreactive. Prepare NADPH in a transparent colorless 1 ml Eppendorf tube to allow visual assessment of its degradation to a yellow product. The NADPH present in the secondary reaction mixture is sufficient for the reaction time in this protocol (10 min). However, if significantly longer reaction times are required, an NADPH regenerating system is recommended.
EQUIPMENT SETUP
An appropriate excitation filter (405 nm) and emission filter (505 nm) are installed in the filter wheels of the Victor II plate-reader. Although the optimal wavelengths for this assay are 410 nm (excitation) and 510 nm (emission), filters are not available at those wavelengths for this instrument and the filters indicated above work well.
Set sonicator to 40% maximal output with pulsed sonication (1Hz) for 1 min total duration.
Place metal heating block in water bath and set temperature at 37 °C
PROCEDURE
Preparation of primary reaction (TIMING ~1 h)
1. Prepare the stock solutions described above (NADPH, BG, DLPC, catalase and 7-EFC) and keep on ice until use during the experiment.
2. Thaw purified CYP2B6 and CPR by placing them on ice until they are fully thawed.
-
3. Sonicate DLPC stock solution on ice using the settings described above for 1min.
CRITICAL STEP Thorough DLPC sonication will ensure that preformed micelles are disrupted into monomer units and will facilitate the proper formation of liposomes and the efficient incorporation of P450 and CPR into the liposomes.
-
4. Pipette 71.4 μL of the P450, CPR and DLPC reconstitution mixture into a 1ml Eppendorf tube and keep at room temperature for 30 min.
If the use of Supersomes or Baculosomes is desired, the reconstitution step can be bypassed and, once the Supersomes or Baculosomes are thawed, they can be dispensed directly into the primary reaction wells in place of the reconstituted mixture.
5. Using a multichannel pipettor, dispense 35.1 μL of primary reaction mixture into each of seven different inactivator concentration wells as illustrated in Figure 3.
-
6. Using a multichannel pipettor, dispense 2.7 μL of the BG stocks into their respective wells. The control well receives the same volume of methanol instead of BG.
CRITICAL STEP The effect of methanol, as well as any other solvents that may be used to dissolve the inactivator, on the enzymatic activity must be assessed in the control sample since inactivation is calculated as a percent of the control and some organic solvents are known to decrease some P450 activities. In general, it is best to keep the amount of organic solvent added to the primary reactions less than 1 or 2 % of the total volume.
7. When step 3 is complete, dispense 10.2 μL of the reconstitution mixture to the primary reaction wells.
Figure 3.


Layout for the 96-well micro-titer plate used in this assay. Columns 1 and 7 are used for the primary reaction (1° reactions) wells for the BG concentrations indicated and are represented in blue. Columns 2–5 and 8–11 are used for the secondary reaction (2° reactions) wells and are depicted in orange. At the indicated time points, 12.5 μL aliquots from columns 1 and 7 are transferred to the designated wells using a hand-held multichannel pipettor. Wells H2,3,8,9 in yellow are used for background fluorescence measurements. Activity is determined fluorometrically using a plate-reader. This assay can be performed in duplicate as depicted in columns 7–11.
Preparation of secondary reaction (TIMING ~10 min)
8. Prepare the stock solution described above and ensure proper mixing by vortexing.
9. Using a multichannel pipettor, dispense 150 μL into each secondary reaction well and an additional two wells which will serve as background fluorescence measurements (Figure 3)
10. When not making additions, the 96-well plate should be shielded from light to prevent the photodegradation of 7-EFC.
P450 activity assay (TIMING ~30 min)
11. Allow the primary and secondary reactions to reach thermal equilibrium by placing the 96-well plate over the steel block chamber in the 37 °C water bath and let them incubate for 10 min.
12. When the reaction mixtures are equilibrated, add 2 μL of stock NADPH to each primary reaction well using a multichannel pipettor. Steps 12–16 are summarized in Table 1.
13. Using a multichannel pipettor, transfer 12.5 μL from the primary reaction wells to the secondary reaction wells of the 0 min time point and start the timer.
14. At 3 min use a multichannel pipettor to transfer 12.5 μL from the primary reaction wells to the secondary reaction wells of the 3 min time point column.
15. Similarly, at 6 and 9 min transfer 12.5 μL from the primary reaction wells to the secondary reaction wells of the corresponding time point columns.
-
16. Stop the secondary reactions 10 min after each transfer by using 50 μL of ice-cold acetonitrile and pipetting briskly 6–8 times to obtain thorough mixing. For example, the 0 min timepoint is stopped at 10 min and the 3 min timepoint is stopped at 13 min etc.
? TROUBLESHOOTING
17. Add 50 μL of ice-cold acetonitrile to the two designated background wells.
18. Immediately begin measuring the fluorescence intensity of the secondary reactions using a fluorescence plate reader set at an excitation wavelength of 405 nm and emission at 505 nm.
19. Analyze the results by subtracting the average background reading intensity from the fluorescence intensity measurements of the secondary reaction wells. Take the control sample at the 0 min time-point to indicate the maximal activity and calculate the log of percentage activity remaining for the remainder of the samples. Plot the log of percentage activity remaining as a function of time.
-
20. Linear regression analysis of the time course data is used to estimate the initial rate constants (kobs) for the inactivation of CYP2B6 by BG. kobs is then plotted against the BG concentration and the data are fit to Eq 1 from which KI and kinact, are determined. The inactivation half-life (t1/2) can be calculated from Eq 2
(1) (2)
Table 1.
Summary of steps 12–16 under P450 activity assay in the PROCEDURE section. 1° rxn and 2° rxn denote the primary and secondary reaction mixtures respectively.
| Time, min | Action |
|---|---|
| 0 | Add NADPH to 1° rxn, transfer 12.5 μL from 1° rxn to 0 min 2°rxn |
| 3 | Transfer 12.5 μL from 1° rxn to 3 min 2°rxn |
| 6 | Transfer 12.5 μL from 1° rxn to 6 min 2°rxn |
| 9 | Transfer 12.5 μL from 1° rxn to 9 min 2°rxn |
| 10 | Quench 0 min 2° rxn with 50 μL acetonitrile |
| 13 | Quench 3 min 2° rxn with 50 μL acetonitrile |
| 16 | Quench 6 min 2° rxn with 50 μL acetonitrile |
| 19 | Quench 9 min 2° rxn with 50 μL acetonitrile |
TROUBLESHOOTING
See Table 2 for troubleshooting advice.
Table 2.
Troubleshooting table.
| Problem | Possible reason | Solution |
|---|---|---|
| Low enzyme activity | Low concentration of protein in the 2° reaction mixture | Add more enzyme to 1° reaction and increase amount transferred from 1° to 2° reaction |
| P450:redox-partner ratio | Vary P450:CPR ratio Add b5 to stimulate metabolism in the 2° reaction |
|
| Poor reconstitution | Reconstitute longer Sonicate DLPC more thoroughly |
|
| CPR inhibition by NADP+ | Use an NADPH regenerating system for longer secondary reaction incubations | |
| Enzyme activity of control increases at different time-points | Enzyme activity not fully stopped | Mix acetonitrile more thoroughly with the 2° reaction mixtures to kill the reaction Use trifluoro- or triacetic acid instead of acetonitrile to stop the reaction |
| High inter-day variability | Inactivator solvent evaporation Inactivator decomposition |
Prepare fresh inactivator solutions |
| Inconsistent reconstitution | Scale up reconstitution and aliquot equal volumes of reconstitution into 1° reaction wells | |
| Reaction components not thermally equilibrated | Use a longer incubation time for the 1° reaction mixture |
ANTICIPATED RESULTS
Previous reports from our lab have established that BG is a potent inactivator of human P450s. Specifically, Lin et al (9) have found that BG inactivates CYP2B6 in a concentration- and time-dependent manner and that this inactivation requires the presence of NADPH. They proposed that BG-dependent inactivation of CYP2B6 occurs by heme destruction and apoprotein modification as a result of covalent binding of a reactive intermediate of BG (9). It is well known that a number of P450s O-deethylate 7-EFC to 7-HFC (Figure 1) and this serves as an easy and sensitive method to assay mechanism-based inactivators of these purified P450s. The fluorescence emission of the product (7-HFC) is different from 7-EFC and NADPH, thus allowing product formation to be monitored spectrofluorometrically in a 1 ml cuvette. Although very popular, this approach typically requires transferring 10–25 pmol from the primary to the secondary reaction mixture to generate sufficient fluorescent product. This approach is time-consuming and unrealistic in a high-throughput setting as one must measure fluorescence intensity of all secondary reactions individually.
In this protocol we detail the application of a widely used and highly reproducible 7-EFC activity assay to determine CYP2B6 inactivation in a quantitative high-throughput format that reduces the amount of enzyme required by 50–70%. This improvement in enzyme consumption is substantial since characterizing the maximal rate of inactivation (kinact) of a P450 by an inactivator and determining the concentration of inactivator required for half-maximal inactivation (KI) requires assessing secondary substrate turnover across a number of different time points and inactivator concentrations. As shown in Figure 3 and Table 1, after P450 mechanism-based inactivation is initiated by the addition of NADPH, aliquots of the primary reaction are transferred into the secondary reaction mixtures where activity is assessed spectrofluorometrically by measuring 7-EFC to 7-HFC conversion. As shown in Figure 4a, increasing the BG concentration increases the rate of inactivation. Thus, the inactivation follows pseudo-first order kinetics and the apparent kinetic constants are determined by plotting kobs as a function of increasing BG concentration (Figure 4b). In this example the kinact for the inactivation of CYP2B6 by BG was 0.06 min−1, with a KI of 2.32 μM, and the half-time for inactivation (t1/2) was 11.5 min.
Figure 4.

Representative plots for the time- and concentration-dependent inactivation of the catalytic activity of CYP2B6 by BG. Results are averages of two trials. (a) Inactivation of the 7-EFC O-deethylation activity of CYP2B6 in the reconstituted system incubated with 0, 1, 2, 4, 6, 10 and 15 μM BG. Aliquots were removed from the primary reactions at the indicated time and assayed for 7-EFC activity. (b) From the slope of the initial linear phase of the inactivation reactions an apparent inactivation rate constant (kobs) is determined. The value of kobs is then plotted against the concentration of the inactivator to determine KI and kinact using GraphPad Prism 5.0 from GraphPad Software.
It is worth noting that Figure 4 represents the results of an optimized experiment. If, for example, a very potent inactivator is used, then measuring the rates of inactivation at the desired time points can be challenging. In this case, particular experimental conditions like the concentration of the inactivator, the pH and temperature need to be shifted from their optimal values to reduce the rate of P450 inactivation (6). Conversely, if inactivation is slow then identifying the optimal conditions including increasing the ratio of the redox partners (CPR and b5) to P450, performing a longer reconstitution to ensure a maximal concentration of functional P450-redox partner complexes and increasing the length of the primary incubation is recommended (Table 2). Increasing the incubation temperature for the primary reaction mixture will also increase the rate of reaction but may result in some instability of the enzyme’s activity. In both cases, experimental optimization may change the KI and will definitely change the kinact thereby precluding the comparison of kinetic data regarding the same inactivator that was examined under different experimental conditions.
In conclusion, our protocol is useful for high-throughput analysis of chemical entities to assess their potency as P450 mechanism-based inactivators. Therefore, this protocol might be valuable for the assessment of small molecules including drugs and chemical probes for elucidating P450 catalytic mechanisms. Furthermore, by performing P450-mediated reactions directly in a microtiter plate, this protocol significantly reduces the amount of the P450s and CPR needed. This is particularly important considering the effort and cost to obtain purified recombinant P450s and that the expression levels of certain P450s can be relatively poor, particularly for some of the commonly occurring human genetic polymorphic variants.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grant CA16954 (to P. F. H.).
Footnotes
AUTHOR CONTRIBUTIONS
C.K optimized and performed the assays with the advice of H.Z. C.K wrote the majority of the paper with the assistance of H.Z and P.F.H.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- 1.Williams AJ, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCI/AUC) ratios. Drug Metab Dispos. 2004;32:1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 2.Porter TD, et al. Cytochrome P-450 Multiplicity of isoforms, substrates, and catalytic and regulatory mechanisms. J Biol Chem. 1991;266:13469–13472. [PubMed] [Google Scholar]
- 3.Evans WE, et al. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 4.Kent UM, et al. Mechanism-based inactivators as probes of cytochrome P450 structure and function. Curr Drug Metab. 2001;2:215–243. doi: 10.2174/1389200013338478. [DOI] [PubMed] [Google Scholar]
- 5.Hollenberg PF, et al. Mechanism-based inactivation of human cytochrome P450s: Experimental characterization, reactive intermediates, and clinical implications. Chem Res Toxicol. 2008;21:189–205. doi: 10.1021/tx7002504. [DOI] [PubMed] [Google Scholar]
- 6.Silverman RB. Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology. CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- 7.Mayer RT, et al. A real-time fluorescence assay for measuring N-dealkylation. Drug Metab Dispos. 2007;35:103–109. doi: 10.1124/dmd.106.011601. [DOI] [PubMed] [Google Scholar]
- 8.Blobaum AL, et al. P450 active site architecture and reversibility: Inactivation of cytochromes P450 2B4 and 2B4 T302A by tert-butyl acetylenes. Biochemistry. 2005;44:3831–3844. doi: 10.1021/bi0478953. [DOI] [PubMed] [Google Scholar]
- 9.Lin H, et al. The grapefruit juice effect is not limited to cytochrome P450 3A4: Evidence for bergamottin-dependent inactivation, heme destruction, and covalent binding to protein in P450s 2B6 and 3A5. J Pharmacol Exp Ther. 2005;313:154–164. doi: 10.1124/jpet.104.079608. [DOI] [PubMed] [Google Scholar]
- 10.Buters JT, et al. A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem Pharmacol. 1993;46:1577–1584. doi: 10.1016/0006-2952(93)90326-r. [DOI] [PubMed] [Google Scholar]
- 11.DeLuca JG, et al. A direct, highly sensitive assay for cytochrome P-450 catalyzed O-deethylation using a novel coumarin analog. Biochem Pharmacol. 1988;37:1731–1739. doi: 10.1016/0006-2952(88)90436-4. [DOI] [PubMed] [Google Scholar]
- 12.Scott EE, et al. A truncation of 2B subfamily cytochromes P450 yields increased expression levels, increased solubility, and decreased aggregation while retaining function. Arch Biochem Biophys. 2001;395:57–68. doi: 10.1006/abbi.2001.2574. [DOI] [PubMed] [Google Scholar]
- 13.Rendic S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab Rev. 2002;34:83–448. doi: 10.1081/dmr-120001392. [DOI] [PubMed] [Google Scholar]
- 14.Xie HJ, et al. Role of polymorphic human CYP2B6 in cyclophosphamide bioactivation. Pharmacogenomics J. 2003;3:53–61. doi: 10.1038/sj.tpj.6500157. [DOI] [PubMed] [Google Scholar]
- 15.Schmiedlin-Ren P, et al. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents: decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug Metab Dispos. 1997;25:1228–1233. [PubMed] [Google Scholar]
- 16.Bailey DG, et al. Grapefruit juice–drug interactions. Br J Clin Pharmacol. 1998;46:101–110. doi: 10.1046/j.1365-2125.1998.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey DG, et al. Grapefruit-felodipine interaction: effect of unprocessed fruit and probable active ingredients. Clin Pharmacol Ther. 2000;68:468–477. doi: 10.1067/mcp.2000.110774. [DOI] [PubMed] [Google Scholar]