

Abstract
PIM (proviral integration site) kinases are a distinct class of serine/threonine-specific kinases consisting of PIM1, PIM2 and PIM3. PIM2 is known to function in apoptosis pathways. Expression of PIM2 is highly induced by pro-inflammatory stimuli but the role of PIM2 in the expression of pro-inflammatory cytokines is unclear. In this study, we showed that over-expression of PIM2 in HeLa cells as well as in human umbilical vein endothelial cells enhanced interleukin-1β (IL-1β) -induced and tumour necrosis factor-α-induced IL-6 expression, whereas over-expression of a kinase-dead PIM2 mutant had the opposite effect. Studies with small interfering RNA specific to PIM2 further confirmed that IL-6 expression in HeLa cells requires PIM2. To investigate the function of PIM2 further, we generated PIM2-deficient mice. It was found that IL-6 production was significantly decreased from PIM2-deficient spleen cells after stimulation with lipopolysaccharide. Taken together, we demonstrated an important function of PIM2 in controlling the expression of the pro-inflammatory cytokine IL-6. PIM2 inhibitors may be beneficial for IL-6-mediated diseases such as rheumatoid arthritis.
Keywords: cytokines, gene regulation, interleukin-6, kinase, provirus integratin site 2
Introduction
The PIM (proviral integration site) kinase family consists of three members: PIM1, PIM2 and PIM3.1 PIM1 was originally identified as a frequent proviral insertion site in Moloney murine leukaemia virus (MLV) -induced T-cell lymphomas.2 PIM2 was discovered by virtue of sequence conservation with the PIM1 oncogene and was also implicated in lymphomagenesis by its presence as a common pro-viral integration site in MLV-induced lymphomas.3 PIM2 is highly expressed in non-Hodgkin's lymphomas over normal lymphocytes.4 PIM1 and PIM2 are constitutively expressed in haematopoietic tissues and their messenger RNA (mRNA) expression can be further induced by multiple cytokines and growth factors such as interleukin-2 (IL-2), IL-3, IL-4, IL-6, IL-7 and interferon-γ (IFN-γ).1,5 T helper type 1 (Th1) -specific cytokines IL-12 and IFN-α, but not Th2-specific cytokine IL-4, transiently up-regulate PIM1 and PIM2 mRNA expression in human T cells.6 PIM1 mRNA up-regulation occurs rapidly after cytokine or mitogenic stimulation followed by induced PIM2 expression several hours later.7 PIM1 kinase is constitutively active, as shown in the crystal structure of apo-PIM1, indicating that PIM1 adopts a constitutively active conformation without phosphorylation.8 Monocytes express high levels of PIM2 mRNA after lipopolysaccharide (LPS) stimulation.9 LPS also highly induces PIM2 expression via the nuclear factor-κB (NF-κB) pathway in pre-B cells.8 TALL-1/BAFF, a member of the tumour necrosis factor (TNF) family, significantly up-regulates PIM2 expression in primary B lymphocytes and B lymphoma cells.10 The PIM2 mRNA is also up-regulated and remains elevated in latently Epstein–Barr virus-infected B-cell lines.11
One important role of PIM2 is to promote cell survival and suppress cell apoptosis. PIM2 kinase phosphorylates the pro-apoptotic protein Bcl2 antagonist of cell death (BAD) on serine 112 and so reverses BAD-induced cell death.12 Recently, it has been reported that both eukaryotic initiation factor 4B (eIF4B) and apoptosis inhibitor 5 (API-5) are potential PIM2 kinase substrates.13 Thompson and colleagues report that ectopic expression of PIM2 leads to sustained NF-κB activity, suggesting that NF-κB activation contributes to the function of PIM2 in promoting cell survival.14 PIM2 activates NF-κB by inducing phosphorylation of Cot, which leads to enhanced inhibitor of NF-κB kinase activity and reduced expression of p50/p105.14
No in vivo abnormalities were shown in PIM1-deficient or PIM2-deficient mice,15,16 indicating a potential functional redundancy of PIM family members. Indeed, Moloney MLV-induced tumours from Eμ-myc transgenic mice, which are deficient in the pim1 gene, showed compensatory activation of the pim2 gene.3 Berns and colleagues generated mice that are deficient for all PIM kinases.16 These mice are viable and fertile but their body size is reduced at birth and throughout post-natal life. In the absence of PIMs, IL-7-mediated proliferation of late pre-B cells is decreased. Decreased IL-3, colony stimulating factor (CSF) or IL-5-mediated cell growth and differentiation of bone marrow cells were observed in PIM-deficient mice. In addition, PIM2-deficient T cells show reduced T-cell proliferation in response to synergistic T-cell receptors and IL-2 stimulation.16 Thompson and colleagues demonstrated that constitutive expression of catalytically active but not kinase-dead PIM2 confers long-term resistance to apoptosis in the absence of IL-3 or when cells are treated with cytotoxic drugs such as staurosporine and thapsigargin.17 The PIM2-dependent maintenance of cell size and survival correlates with its ability to maintain rapamycin-resistant phosphorylation of the translational repressor 4E-BP1 and phosphorylation of the BH3 protein BAD.17 Furthermore, cytokine-induced PIM1 and PIM2 promote the target of rapamycin inhibitor rapamycin-resistant survival of lymphocytes. The endogenous function of the PIM kinases is not restricted to the regulation of cell survival. Although rapamycin has a minimal effect on wild-type T-cell expansion both in vitro and in vivo, it completely suppresses PIM1−/− PIM2−/− T cells in response to IL-4 or IL-7 stimulation.7
Interleukin-6 is a pleiotropic cytokine. It is an activator of acute-phase responses and a lymphocyte stimulatory factor and is critically involved in resolving innate immunity. Recently, it was discovered that IL-6 is also critical for promoting acquired immunity.18 Interleukin-6 elicits the development of specific cellular and humoral immune responses, including end-stage B-cell differentiation, immunoglobulin secretion and T-cell activation.19 It plays a pro-inflammatory role in chronic inflammation. For example, collagen-induced arthritis is associated with increased expression of IL-6. Interleukin-6-deficient mice showed reduced antibody response to type II collagen, and an absence of inflammatory cells and tissue damage in knee joints. Importantly, the IL-6-deficient mice were protected from collagen-induced arthritis.20 In humans, high levels of IL-6 were detected in serum and synovial fluids from the joints of patients with active rheumatoid arthritis.21 Treatment with the anti-IL-6 receptor monoclonal antibody Tocilizumab led to significant improvement in the signs and symptoms of rheumatoid arthritis patients.22
In this report, we show that over-expression of PIM2 enhanced IL-1β and TNF-α-induced IL-6 expression. Meanwhile, over-expression of kinase-dead PIM2 inhibited IL-1β and TNF-α-induced IL-6 expression. Studies with small interfering RNA (siRNA) specific to PIM2 further confirmed that IL-6 expression requires PIM2. Finally, we generated PIM2-deficient mice. It was found that IL-6 production from PIM2-deficient spleen cells was significantly decreased after stimulation with LPS.
Materials and methods
Mice, reagents, cytokines and antibodies
The PIM2 mutant mice were generated in collaboration with Lexicon Genetics, Inc. (The Woodlands, TX). The conditional LoxP targeting vector was derived using the Lambda KOS system.23 The two LoxP sites were targeted before exon I and after exon III of PIM2 (Fig. 4a). The targeted embryonic stem cell clones were microinjected into C57BL/6 (albino) blastocysts to generate a LoxP conditional line. The mice were then subsequently bred with Protamine-Cre transgenic mice generated by Lexicon Genetics. The genetic background of the Protamine-Cre transgenic mice is 129SvEvBrd×C57BL/6. The knockout of PIM2 mice was confirmed by genotyping (Fig. 4a,b).The polymerase chain reaction (PCR) sequences for genotyping are as follows. PCR-F: 5′-CTCCTACATTTGCATACAGG-3′; PCR-R1: 5′-GAGGTGCTGCGTATATTGAGC-3′; PCR-R2: 5′-GAAAGGAGAATCCAGGGCTCTG-3′. The wild-type mice 129SvEvBrd×C57BL/6 F2 were purchased from Jackson Laboratory (Bar Harbor, ME). The animal study protocol was approved by Boehringer Ingelheim Pharmaceutical Animal Care Committee. Murine IL-4 and IL-12 were purchased from Calbiochem (San Diego, CA). Murine IL-2 and monoclonal anti-IL-4 (11B11) were obtained from eBioscience (San Diego, CA). Polyclonal anti-mouse IL-12 was purchased from Cell Sciences (Norwood, MA). Monoclonal anti-mouse CD3ε (500A2) and anti-mouse CD28 (37.51) were purchased from BD Biosciences (San Diego, CA). Ovalbumin (OVA) and Histopaque-1119 were all from Sigma Chemical Co. (St Louis, MO). Human IL-1β and TNF-α were purchased from R&D Systems (Minneapolis, MN).
Figure 4.
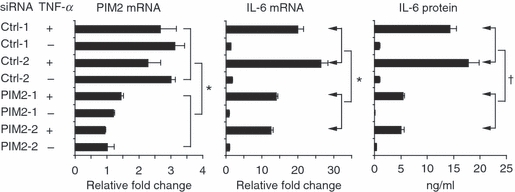
Small interfering RNA (siRNA) against proviral integration site kinase 2 (PIM2) knocked down interleukin-6 (IL-6) expression. HeLa cells were transiently transfected with PIM2-specific and control siRNA for 24 hr. Cell pellets were harvested after the cells were stimulated with tumour necrosis factor-α (TNF-α; 10 ng/ml) for 4 hr. Real-time reverse transcription–polymerase chain reaction was performed to detect the messenger RNA (mRNA) level of PIM2 and IL-6. The culture supernatants were harvested after the cells were stimulated with TNF-α (10 ng/ml) for 18 hr. The IL-6 protein concentration was detected by enzyme-linked immunosorbent assay. Data are representative of three separate experiments. †P < 0·05; *P < 0·01 versus PIM2 siRNA-treated group. For the PIM2 mRNA group, the comparison is between control siRNA and PIM2 siRNA treated with and without TNF-α. For IL-6 mRNA and protein groups, the comparison is between samples treated with TNF-α only.
Cell culture
HeLa cells and human umbilical vein endothelial cells (HUVECs) were cultured in 10% fetal bovine serum (FBS) /RPMI-1640 in the presence of penicillin and streptomycin. Red blood cells from mouse spleen cells were removed using Histopaque-1119 by spinning down at 600 g for 15 min. Cells were cultured in 10% FBS/Iscove's modified Dulbecco's medium with 2 mm l-glutamine (Invitrogen, Carlsbad, CA), 0·1 mm non-essential amino acids (Invitrogen), 1 mm sodium pyruvate (Invitrogen) and 0·05 mm 2-mercaptoethonol (Sigma). The spleen cells were stimulated with LPS for 4 and 24 hr. The cells were harvested for reverse transcription (RT-) PCR analysis after they were stimulated for 4 hr. The supernatants were harvested for cytokine production assay after the cells were stimulated for 24 hr.
Retroviral constructs, transfection and infection
Human PIM2 complementary DNA (cDNA) insert was generated by PCR using the IMAGE clone (clone ID: 3913552) as a template. The ATP binding site of the human PIM2 kinase was identified by a homology search using prosite. The predicted active site lysine residue, K61, was mutated to an alanine to produce a kinase-dead form of the protein. The PCR products were then ligated into the BglII/XhoI site of internal ribosome entry site (IRES) green fluorescence protein-retroviral vector (GFP-RV) as described previously.24,25 The final PIM2-retrociral vector (-RV) was confirmed by sequencing. A Phoenix-A packaging cell line (licensed from Dr G. Nolan, Stanford, CA) was transfected to generate the retroviral supernatants according to Dr Nolan's protocol as described previously.26 The viral supernatant and 6 μg/ml polybrene (Sigma) were added to HeLa cells or HUVEC in six-well plates and centrifuged at 600 g for 30 min at room temperature. Cells were then cultured for 24 hr before a one-third split. Infected cells expressed GFP and were purified by cell sorting on day 5 after infection. Cells were further cultured for 5–10 days before the cells were stimulated with IL-1β or TNF-α for RT-PCR analysis.
siRNA transfection
Selection of siRNA was based on the characterization of siRNA described previously.27 The sequences of siRNA duplexes are as follows.
PIM2-1: sense: 5′-GUGAUUCCCCGGAAUCGUGdTdT-3′, anti-sense: 5′-CACGAUUCCGGGGAAUCACdTdT-3′; PIM2-2: sense: 5′-CAUCCUGAUAGACCUACGCdTdT-3′, anti-sense:5′-GCGUAGGUCUAUCAGGAUGdTdT-3′; Ctrl-1: sense: 5′-GUGCUAAGGCCCCUUAGUGdTdT-3′, anti-sense:5′-CACUAAGGGGCCUUAGCACdTdT-3′; Ctrl-2: sense: 5′-CGCAUCCAGAUAGUCCUACdTdT-3′, anti-sense:5′-GUAGGACUAUCUGGAUGCGdTdT-3′. The control sequences were designed to be the inverted PIM2 sequences. HeLa cells were seeded in six-well plates at a density of 2 × 105 cells/well and incubated in 10% FBS RPMI-1640 without antibiotics at 37° overnight. For each siRNA transfection, 10 μl of 20 μm siRNA duplex was mixed with 100 μl Opti-MEM (serum-reduced medium; Invitrogen) in tube A for 5 min at room temperature. In tube B, 4 μl OligofectAMINE Reagent (Invitrogen) was mixed with 100 μl Opti-MEM for 5 min at room temperature. The mixtures in tubes A and B were then combined and were incubated at room temperature for 20 min. The culture medium in the HeLa cell culture well was then replaced with Opti-MEM (800 μl/well). The siRNA mixture was finally added into the HeLa cell culture well. After the cells were incubated at 37° for 4 hr, 2 ml 10% FBS RPMI-1640 without antibiotics was added to each well and the cells were incubated at 37° for another 2 days. Cells were then replaced with fresh medium and stimulated with TNF-α (10 ng/ml) for 2 hr for total RNA extraction or overnight for enzyme-linked immunosorbent assay.
RNA extraction, TaqMan quantitative RT-PCR
Total RNA was extracted using an RNeasy Mini kit (Qiagen, Valencia, CA). The possible remaining DNA was digested with the RNAse-Free DNAse I (Qiagen) during the extraction of total RNA. Complementary DNA synthesis and TaqMan quantitative RT-PCR were described previously.28,29 The principle of the TaqMan real-time detection is based on the fluorogenic 5′ nuclease assay. A thermally stable AmpliTaq Gold DNA polymerase was used for the PCR amplification. Real-time PCR was performed in a MicroAmp Optical 96-Well Reaction Plate from Applied Biosystems (Foster City, CA). Each well contained 2 μl of each RT product (40 ng total RNA), 1× TaqMan buffer A, 5·5 mm MgCl2, 200 μm dATP/dCTP/dGTP, 400 μm dUTP, 200 nm forward and reverse primers, 100 nm TaqMan probe, 0·01 U/μl AmpErase, and 0·025 U/μl AmpliTaq Gold DNA polymerase in a total volume of 25 μl. Each well was closed with MicroAmp Optical caps from Applied Biosystems. Amplification conditions were 2 min at 50° for AmpErase UNG incubation to remove any uracil incorporated into the cDNA, 10 min at 95° for AmpliTaq Gold activation, and then 40 cycles at 95° for 15 seconds, 60° for 1 min. All reactions were performed in the Abi Prism 7700 Sequence Detection System for the test samples, standards, and no template controls. They were run in triplicates using the Sequence Detector V 1.6 program. The Rn and Ct were averaged from the values obtained in each reaction. A standard curve was constructed by plotting Ct against the known copy numbers of the template in the standard. All TaqMan data shown were normalized based on the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA expression level. The TaqMan copy numbers were calculated by using a standard curve and normalized to endogenous GAPDH.30 The siRNA sample TaqMan data were analysed to obtain fold changes by using the relative quantification 2−ΔΔCt method. The PCR primers and TaqMan probes were designed using Primer Express 1.5 software (Applied Biosystems). The TaqMan probes were labelled with a reporter fluorescent dye, FAM (6-carboxyfluorescein), at the 5′ end and a fluorescent dye quencher TAMRA (6-carboxy-tetramethyl-rhodamine) at the 3′ end. TaqMan primers and probes are as follows. Human GAPDH: forward primer: CCAGGTGGTCTCCTCTGACTTC; reverse primer: GTGGTCGTTGAGGGCAATG; and TaqMan probe: 5′-FAM-:AGCGACACCCACTCCTCCACCTTTG-TAMRA-3′. Human PIM2: forward primer: GTGCCAAACTCATTGATTTTGG; reverse primer: CCTTGTCCCATCAAAGTCAGTGT; and TaqMan probe: 5′-FAM-TCTGGTGCCCTGCTTCATGATGAACC-TAMRA-3′. Murine GAPDH: forward primer: GCTACACTGAGGACCAGGTTGTCT, reverse primer: ACCAGGAAATGAGCTTGACAAAGT, and TaqMan probe: 5′-FAM-CAACAGCAACTCCCACTCTTCCACCTTC-TAMRA-3′. Human IL-6 and murine PIM2, TNF-α TaqMan primers and probes were purchased from Applied Biosystems.
Proliferation assay
The proliferation assays were performed using a CellTiter-Glo® Luminescent Cell Viability Assay kit obtained from Promega (Madison, WI) as described previously.31 Briefly, HeLa cells or mouse spleen cells were seeded in 100 μl/well in a 96-well plate and cultured for 24 hr. One hundred microlitres of CellTiter-Glo® Reagent was added to each well. The cells were lysed for 2 min in a shaker. The plates were incubated at room temperature for another 10 min before the luminescence was recorded.
NF-κB luciferase assay
We first established a HeLa cell line stably expressed NF-κB-luciferase reporter. The construct, which was purchased from Stratagene (Cedar Creek, TX), has a minimal promoter containing five NF-κB sites with firefly luciferase reporter in a neo-resistance plasmid. The cell line was then transfected with PIM2 siRNA and the NF-κB luciferase activity was determined using the Bright-Glo™ Luciferase Assay System from Promega.
Cytometric bead array
The cytokine titres in the supernatants were analysed using the BD cytometric bead array (CBA) as described previously.32 The mouse inflammatory cytokine CBA kits were used (BD Biosciences). Briefly, 25 μl of each sample or standard dilutions were mixed with 25 μl of mixed capture beads and 25 μl of the mouse phyoerythrin (PE) detection reagent. After the samples were incubated at room temperature for 2 hr in the dark, they were washed once and resuspended in 200 μl wash buffer before acquisition on a FACScan. Data were analysed using the CBA software. The concentration of each cytokine in the supernatants was calculated with the corresponding standard curve.
Statistical analysis
Statistical analysis was performed using Student's t-test or one-way analysis of variance by Microsoft Excel. P-values < 0·05 were considered significant.
Results
Over-expression of PIM2 enhanced TNF-α-induced and IL-1β-induced IL-6 expression in both HeLa cells and HUVEC while kinase-dead PIM2 mutant inhibited IL-6 expression
Although PIM2 is highly expressed in haematopoietic cells, we have also confirmed its expression in other cell types including HeLa and HUVEC. Since both HeLa cells and HUVEC respond well to pro-inflammatory stimuli and produce inflammatory cytokines, we investigated the function of PIM-2 in regulating cytokine production in these cells. We first generated the wild-type PIM2 retroviral vector (WT PIM2-RV) and the kinase-dead PIM2 (K61A) retroviral vector (KD PIM2-RV) (Fig. S1a). The control retroviral vector was GFP-RV. These retroviral vectors were transiently transfected into Phoenix-A cells and the retroviral supernatants were harvested for further infection studies. The infected cells expressed GFP and were purified by cell sorting. Post-sorting results showed that 97% of cells were GFP-positive (Fig. S1b). Real-time PCR showed that both WT and KD PIM2-RV-infected HeLa cells expressed very high levels of PIM2 (Fig. S1c). Similar expression levels were observed in HUVEC (data not shown). We then stimulated HeLa cells with different concentration of TNF-α for 4 hr before cells were harvested for RNA extraction and RT-PCR analysis. It was found that over-expression of WT PIM2 enhanced TNF-α-induced IL-6 expression (Fig. 1a, compare WT PIM2-RV with GFP-RV). The KD PIM2-RV-infected cells expressed less TNF-α-induced IL-6 mRNA than GFP-RV-infected cells (Fig. 1a, compare KD PIM2-RV with GFP-RV), indicating a dominant negative effect of the mutant. Similar results were also observed in human primary cell line HUVEC (Fig. 1b). These results indicate that the kinase activity of PIM2 is required for TNF-α-induced IL-6 expression. Furthermore, TNF-α-induced IL-6 expression was dose dependent and the effect of PIM2 over-expression on IL-6 expression was confirmed at different doses of TNF-α (Fig. 1a,b). Next, we asked whether IL-6 expression induced by other stimuli also requires PIM2 expression. HeLa cells were stimulated with different doses of IL-1β and the IL-6 mRNA was detected by quantitative real-time RT-PCR. The WT PIM2-RV-infected cells expressed high levels of IL-6 mRNA compared with KD PIM2-RV- or GFP-RV-infected cells (Fig. 1c). Both KD PIM2-RV- and GFP-RV-infected cells expressed very low levels of IL-6 mRNA (Fig. 1c). Similar results were also observed in HUVEC (Fig. 1d). As the above observation was based on the IL-6 mRNA, we next asked whether IL-6 protein is differentially affected by WT PIM2 and KD PIM2. From Fig. 1(e) (HeLa cells) and Fig. 1(f) (HUVEC), we can see that WT PIM2 enhanced IL-6 protein production while KD PIM2 inhibited IL-6 protein production after the cells were stimulated with TNF-α or IL-1β. Hence, PIM2 is required for TNF-α- or IL-1β-induced IL-6 mRNA expression and protein production in both HeLa and primary HUVEC cells. The kinase activity of PIM2 plays a critical role in the induction of TNF-α- or IL-1β-induced IL-6 expression.
Figure 1.
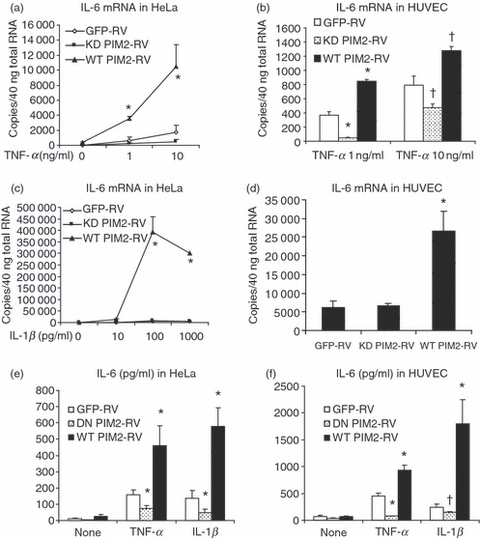
Over-expression of proviral integration site kinase 2 (PIM2) enhanced tumour necrosis factor-α (TNF-α) - and interleukin-1β (IL-1β) -induced IL-6 expression in both HeLa cells and human umbilical vein endothelial cells (HUVEC) while kinase-dead (KD) PIM2 inhibited IL-6 expression. (a) HeLa cells infected with different retroviruses were stimulated with TNF-α for 4 hr and TaqMan real-time reverse transcription–polymerase chain reaction (RT-PCR) was performed for the expression of IL-6 messenger RNA (mRNA). (b) HUVEC infected with different retroviruses were stimulated with TNF-α for 4 hr and TaqMan real-time RT-PCR was performed for the expression of IL-6 mRNA. (c) Retrovirus-infected HeLa cells were stimulated with IL-1β for 4 hr and TaqMan real-time RT-PCR was performed for the expression of IL-6 mRNA. Both KD PIM2-RV-infected and green fluorescent protein (GFP) –RV-infected cells expressed very low levels of IL-6 mRNA. (d) Retrovirus-infected HUVEC were stimulated with IL-1β at 100 pg/ml for 4 hr and TaqMan real-time RT-PCR was performed for the expression of IL-6 mRNA. Data are representative of three separate experiments. HeLa cells (e) or HUVEC (f) infected with different retroviruses were stimulated with TNF-α (10 ng/ml) or IL-1β (100 pg/ml) for 24 hr and the IL-6 protein titres in the supernatants were determined by enzyme-linked immunosorbent assay. Data are representative of two separate experiments. †P < 0·05; *P < 0·01 versus GFP-RV-infected cells.
The enhanced IL-6 production by over-expression of WT PIM2 could be the result of enhanced proliferation of retrovirus-infected cells. However, we did not observe different proliferation after HeLa cells were infected with different retroviruses (Fig. 2). We next asked whether TNF-α expression was enhanced by PIM2. TNF-α mRNA expression was induced by TNF-α itself and IL-1β (Fig. 3). However, WT PIM2 or KD PIM2 had no significant effect on TNF-α mRNA expression. Hence, the enhanced IL-6 production by over-expression of WT PIM2 was not the result of enhanced cell proliferation or TNF-α expression. In addition, we found that there was no different mRNA expression of IL-1β, IL-11, IL-15 among GFP, WT PIM2 and KD PIM2 RV-infected HeLa cells (Fig. S3).
Figure 2.
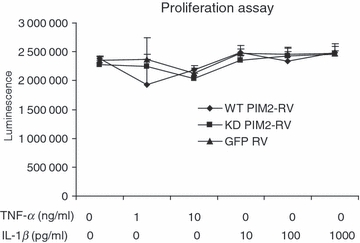
Over-expression of wild-type proviral integration site kinase 2 (PIM2) (WT PIM2) or kinase-dead (KD) PIM2 did not affect HeLa cell proliferation. Retroviruses infected HeLa cells were stimulated with tumour necrosis factor-α (TNF-α) or interleukin-1β (IL-1β) for 24 hr and the proliferation of cells was determined. Data are representative of three separate experiments.
Figure 3.
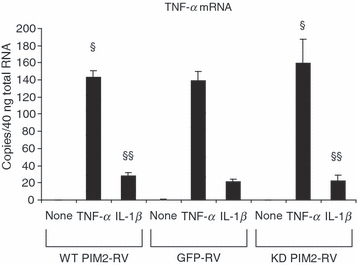
Tumour necrosis factor-α (TNF-α) expression was not affected by proviral integration site kinase 2 (PIM2). Retrovirus-infected HeLa cells were stimulated with TNF-α (10 ng/ml) or interleukin-1β (IL-1β; 1 ng/ml) for 4 hr and the TNF-α messenger RNA expression was determined by TaqMan reverse transcription–polymerase chain reaction. Data are representative of two separate experiments. §P > 0·05 versus green fluorescent protein retrovirus vector (GFP-RV) -infected cells stimulated with TNF-α. §§P>0·05 versus GFP-RV-infected cells stimulated with IL-1β.
siRNA against PIM2 knocked down IL-6 expression
We next asked whether knocking-down PIM2 expression by PIM2-specific siRNA could inhibit IL-6 expression. HeLa cells were transiently transfected with PIM2 siRNAs or control siRNAs for 24 hr. Cells were then stimulated with TNF-α for 4 or 18 hr. Cells were harvested after stimulation for 4 hr and quantitative real-time RT-PCRs were performed. The supernatants were harvested after stimulation for 18 hr and enzyme-linked immunosorbent assay for IL-6 was performed. Figure 4 shows that the two PIM2-specific siRNAs knocked down 50–60% of PIM2 mRNA. The PIM2 siRNA-treated cells showed 40–50% less IL-6 mRNA expression compared with control siRNA-treated cells. Levels of IL-6 protein were 65–70% less in the supernatants of PIM2 siRNA-treated cells than in those of control siRNA-treated cells. Hence, the siRNA studies further confirmed that PIM2 is required for IL-6 expression.
siRNA against PIM2 knocked down NF-κB luciferase activity
As IL-6 is known to be regulated by NF-κB,33 we asked whether PIM2 plays a role in the NF-κB pathway. We knocked-down PIM2 expression by PIM2 siRNAs in a HeLa cell line stably transfected with an NF-κB promoter luciferase reporter. Indeed, PIM2 siRNA-treated cells showed significantly less luciferase activity after cells were treated with TNF-α (Fig. 5a) or IL-1β (Fig. 5b). This study suggested that PIM2 plays a role in activating the NF-κB pathway.
Figure 5.
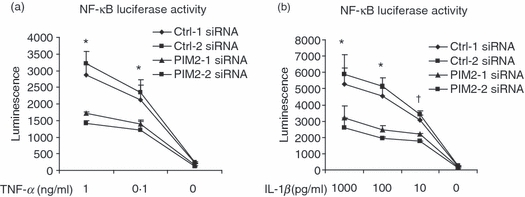
Proviral integration site kinase 2 (PIM2) small interfering RNA (siRNA) knocked down nuclear factor-κB (NF-κB) luciferase activity. HeLa cells stably transfected with the vector containing NF-κB promoter and luciferase reporter were transiently transfected with PIM2-specific and control siRNA for 24 hr. The NF-κB activity was determined by its luciferase expression after cells were stimulated with tumour necrosis factor-α (TNFα) (a) or interleukin-1β (IL-1β) (b) for 24 hr. Data are representative of two separate experiments. †P < 0·05; *P < 0·01 compare the average of Ctrl-1 and Ctrl-2 siRNA-treated groups with the average of PIM2-1 and PIM2-2 siRNA-treated groups.
PIM2-deficient spleen cells produce less IL-6
To study the PIM2 function in vivo, we generated PIM2-deficient mice as described in the Materials and methods (Fig. S2a,b). No PIM2 mRNA was detected in PIM2-deficient cells (Fig. 2c) while PIM1 expression was not affected (data not shown). These mice were fertile and exhibited no gross anatomical abnormalities (data not shown). In the following assays, the PIM2−/− mice were comparable to wild-type mice: the population of CD3+, CD4+, CD8+, CD4+/CD8+, CD45/B220+, CD3+/CD45+, FasL+, Fas+, CD40+, CD40L+, CD69+, CD25+, CD11b+, CD45RB+ cells in lymph nodes, spleens and thymuses; the population of neutrophils, macrophages, mast cells and T cells in peritoneal exudates; cell proliferation of splenocytes induced by LPS, anti-CD3, concanavalin A and phorbol 12-myristate 13-acetate/inonomycin; OVA-specific proliferation, IFN-γ production and immunoglobulin G production by lymph node cells from OVA-immunized mice.
We then tested whether IL-6 expression is impaired in PIM2−/− spleen cells. Spleen cells from wild-type and PIM2-deficient mice were stimulated with different stimuli including LPS, IL-1β and TNF-α. No detectable IL-6 was found when spleen cells from both wild-type and PIM2-deficient mice were stimulated with TNF-α or IL-1β (data not shown). However, IL-6 mRNA could be induced by LPS stimulation. PIM2−/− spleen cells expressed significantly less IL-6 mRNA after being stimulated with LPS for 4 hr (Fig. 6a). Lipopolysaccharide dose-dependently induced the protein expression of IL-6, TNF-α and IL-10 after a 24-hr stimulation (Fig. 6b–d). PIM2-deficient spleen cells produced significantly less IL-6 than wild-type cells (Fig. 6b) whereas the induction of TNF-α and IL-10 were not impaired by PIM2 deficiency (Fig. 6c,d). Finally, there was no significant difference in proliferation between wild-type and PIM2−/− spleen cells after the cells were stimulated with LPS for 24 hr (data not shown).
Figure 6.
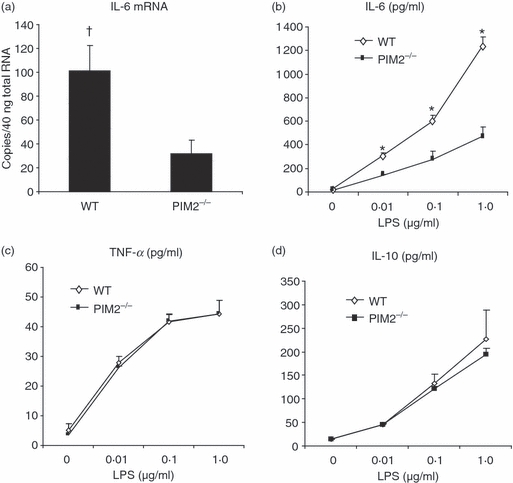
Proviral integration site kinase 2 (PIM2) deficient (PIM2−/−) spleen cells produce less interleukin-6 (IL-6). (a) TaqMan reverse transcription–polymerase chain reaction for IL-6 messenger RNA expression was performed after cells were stimulated with lipopolysaccharide (LPS; 1 μg/ml) for 4 hr. (b–d) Wild-type and PIM2−/− spleen cells were stimulated with different concentrations of LPS for 24 hr and the culture supernatants were harvested to detect the titres of IL-6, tumour necrosis factor-α (TNF-α) and IL-10 by cytometric bead assay as described in the Materials and methods. Data are representative of four separate experiments. †P < 0·05; *P < 0·01 versus PIM2−/− cells.
Discussion
Tumour necrosis factor-α induces the expression of several pro-inflammatory cytokines, such as IL-6.33 We have shown that HeLa cells produced IL-6 after TNF-α stimulation (Fig. 1) and that over-expression of WT PIM2 significantly increased IL-6 expression while WT PIM2 significantly inhibited TNF-α-induced and IL-1β-induced IL-6 expression in both HeLa cells and primary HUVEC (Fig. 1). PIM2 is an oncogene that promotes cell survival. It prevents apoptosis of cells induced by withdrawal of growth factors such as IL-3. The PIM2-protected cells can survive for an extended period in the absence of growth factor.17 Therefore inhibition of IL-6 expression by KD PIM2 might be the result of an associated reduction in the numbers of viable cells induced by KD PIM2. However, this hypothesis is not supported by the data showing that the proliferation of KD PIM2-RV-infected cells was similar to GFP-RV-infected cells or WT PIM2-RV-infected cells (Fig. 2). PIM2 has also been shown to regulate cell size in addition to cell proliferation. However, we did not see a significant change of cell size under the microscope by over-expression of PIM2 or PIM2 siRNA. In addition, we found that both IL-6 protein and IL-6 mRNA expression were affected by PIM2 when we normalized IL-6 mRNA to GAPDH mRNA. It is unlikely that the effect on IL-6 level by PIM2 was the result of our normalization to cell numbers. Therefore, PIM2-enhanced IL-6 expression may be the result of its activation of a signalling pathway such as NF-κB. Indeed, NF-κB activation could be induced by PIM2 kinase.14 Knocking-down PIM2 mRNA by PIM2-specific siRNA significantly reduced NF-κB luciferase activity (Fig. 5). Tumour necrosis factor-α activates NF-κB leading to the induction of several pro-inflammatory cytokines including IL-6.33 Both TNF-α and PIM2 could synergistically activate NF-κB, thereby inducing IL-6 expression. The NF-κB signalling could affect the expression of several cytokines but we found that PIM2 was required only for the IL-6 expression. Therefore, the PIM2 may be important for other signalling pathway(s) that are critical specifically for IL-6 expression. PIM1 and PIM2 are closely related. Both PIM1 and PIM2 induced lymphomas alone or in synergy with c-myc.3,34 Mice lacking PIM1 underwent compensatory activation of PIM2.3 Because TNF-α activates PIM1 kinase,1 it could also activate PIM2 kinase. The possible activation of PIM2 kinase by TNF-α could then further activate NF-κB and induce the expression of IL-6. More studies are need to support this hypothesis. Importantly, because IL-6 promotes cell growth and survival in a broad array of cell types, the enhanced IL-6 production induced by PIM2 over-expression provides another aspect of PIM2 function beyond cell growth and survival.
To further support our discovery that PIM2 is required for IL-6 expression, we performed siRNA knocking-down studies. Two PIM2 siRNAs and two control siRNAs, which are the reverse siRNA of the two PIM2 siRNAs, were used in the experiments. Both PIM2 siRNAs showed 50–60% suppression of PIM2 mRNA (Fig. 4) and no suppression of PIM1 mRNA (data not shown). We found 65–70% inhibition of IL-6 production by PIM2 siRNA treatment, indicating that PIM2 plays an important role in controlling IL-6 expression at a physiological level. This is further supported by our finding that PIM2−/− spleen cells expressed significantly less IL-6 after LPS stimulation (Fig. 6a,b). However, the inhibition of IL-6 expression was not complete in PIM2-deficient cells. The PIM kinases (PIM1, PIM2 and PIM3) are closely related and compensatory activation could occur by the deficiency of one PIM member. Indeed, PIM1 deficiency led to compensatory activation of PIM2.3 Therefore, the function of PIM2 in PIM2-deficient mice might be partially compensated for by other PIM kinases. Further studies are needed to support this hypothesis.
Taken together, our current studies used the methods of over-expression of WT PIM2 and KD PIM2, PIM2 siRNA and PIM2 knock-out mice. These studies demonstrated that PIM2 plays an important role in IL-6 expression. Interleukin-6 is a pro-inflammatory cytokine that is involved in chronic inflammation such as rheumatoid arthritis. Neutralizing IL-6 receptor monoclonal antibody significantly improves signs and symptoms in rheumatoid arthritis patients.22 For this reason, small molecule inhibitors of PIM2 might have the potential to be an effective therapy for IL-6-mediated autoimmune diseases such as rheumatoid arthritis.
Acknowledgments
We thank Tina Morwick for critical reading of the manuscript, Carol Stearns for cell sorting, Kevin Barringer for providing PIM2 cDNA, Katrina Catron for providing the HeLa cell line stably expressed NF-κB-luciferase reporter and Amanda Ciaszki for technical help.
Disclosures
The authors declare conflicts of interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Over-expression of PIM2 or kinase-dead PIM2 using GFP-RV retrovirus.
Figure S2. Generation of PIM2-deficient mice.
Figure S3. Experiments were performed as in Figure 1c. The mRNA level of IL-1b, IL-11 and IL-15 was determined by RT-PCR.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bachmann M, Moroy T. The serine/threonine kinase Pim-1. Int J Biochem Cell Biol. 2005;37:726–30. doi: 10.1016/j.biocel.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Selten G, Cuypers HT, Berns A. Proviral activation of the putative oncogene Pim-1 in MuLV induced T-cell lymphomas. EMBO J. 1985;4:1793–8. doi: 10.1002/j.1460-2075.1985.tb03852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Lugt NM, Domen J, Verhoeven E, et al. Proviral tagging in E mu-myc transgenic mice lacking the Pim-1 proto-oncogene leads to compensatory activation of Pim-2. EMBO J. 1995;14:2536–44. doi: 10.1002/j.1460-2075.1995.tb07251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen AM, Grinblat B, Bessler H, et al. Increased expression of the hPim-2 gene in human chronic lymphocytic leukemia and non-Hodgkin lymphoma. Leuk Lymphoma. 2004;45:951–5. doi: 10.1080/10428190310001641251. [DOI] [PubMed] [Google Scholar]
- 5.Allen JD, Verhoeven E, Domen J, van der valk M, Berns A. Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene. 1997;15:1133–41. doi: 10.1038/sj.onc.1201288. [DOI] [PubMed] [Google Scholar]
- 6.Aho TL, Lund RJ, Ylikoski EK, Matikainen S, Lahesmaa R, Koskinen PJ. Expression of human pim family genes is selectively up-regulated by cytokines promoting T helper type 1, but not T helper type 2, cell differentiation. Immunology. 2005;116:82–8. doi: 10.1111/j.1365-2567.2005.02201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fox CJ, Hammerman PS, Thompson CB. The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp Med. 2005;201:259–66. doi: 10.1084/jem.20042020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Peet GW, Balzarano D, et al. Novel NEMO/IkappaB kinase and NF-kappa B target genes at the pre-B to immature B cell transition. J Biol Chem. 2001;276:18579–90. doi: 10.1074/jbc.M100846200. [DOI] [PubMed] [Google Scholar]
- 9.Jiang H, Van Dv, Satwani P, Baxi LV, Cairo MS. Differential gene expression patterns by oligonucleotide microarray of basal versus lipopolysaccharide-activated monocytes from cord blood versus adult peripheral blood. J Immunol. 2004;172:5870–9. doi: 10.4049/jimmunol.172.10.5870. [DOI] [PubMed] [Google Scholar]
- 10.Xu LG, Wu M, Hu J, Zhai Z, Shu HB. Identification of downstream genes up-regulated by the tumor necrosis factor family member TALL-1. J Leukoc Biol. 2002;72:410–6. [PubMed] [Google Scholar]
- 11.Rainio EM, Ahlfors H, Carter KL, et al. Pim kinases are upregulated during Epstein–Barr virus infection and enhance EBNA2 activity. Virology. 2005;333:201–6. doi: 10.1016/j.virol.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Yan B, Zemskova M, Holder S, et al. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. J Biol Chem. 2003;278:45358–67. doi: 10.1074/jbc.M307933200. [DOI] [PubMed] [Google Scholar]
- 13.Peng C, Knebel A, Morrice NA, et al. Pim kinase substrate identification and specificity. J Biochem (Tokyo) 2007;141:353–62. doi: 10.1093/jb/mvm040. [DOI] [PubMed] [Google Scholar]
- 14.Hammerman PS, Fox CJ, Cinalli RM, et al. Lymphocyte transformation by Pim-2 is dependent on nuclear factor-kappaB activation. Cancer Res. 2004;64:8341–8. doi: 10.1158/0008-5472.CAN-04-2284. [DOI] [PubMed] [Google Scholar]
- 15.Laird PW, van der Lugt NM, Clarke A, et al. In vivo analysis of Pim-1 deficiency. Nucleic Acids Res. 1993;21:4750–5. doi: 10.1093/nar/21.20.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mikkers H, Nawijn M, Allen J, Verhoeven E, Berns A. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol. 2004;24:6104–15. doi: 10.1128/MCB.24.13.6104-6115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox CJ, Hammerman PS, Cinalli RM, Master SR, Chodosh LA, Thompson CB. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003;17:1841–54. doi: 10.1101/gad.1105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175:3463–8. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- 19.Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl. 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alonzi T, Fattori E, Lazzaro D, et al. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–8. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirano T, Matsuda T, Turner M, et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 1988;18:1797–801. doi: 10.1002/eji.1830181122. [DOI] [PubMed] [Google Scholar]
- 22.Smolen JS, Maini RN. Interleukin-6: a new therapeutic target. Arthritis Res Ther. 2006;8(Suppl. 2):S5. doi: 10.1186/ar1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wattler S, Kelly M, Nehls M. Construction of gene targeting vectors from lambda KOS genomic libraries. BioTechniques. 1999;26:1150–6. doi: 10.2144/99266rr02. 1158, 1160. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Zhu H, Murphy TL, Ouyang W, Murphy KM. IL-18-stimulated GADD45 beta required in cytokine-induced, but not TCR-induced, IFN-gamma production. Nat Immunol. 2001;2:157–64. doi: 10.1038/84264. [see comment] [DOI] [PubMed] [Google Scholar]
- 25.Ouyang W, Ranganath SH, Weindel K, et al. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–55. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Castle BE, Hanidu A, et al. Sphingosine kinase 1 is a negative regulator of CD4+ Th1 cells. J Immunol. 2005;175:6580–8. doi: 10.4049/jimmunol.175.10.6580. [DOI] [PubMed] [Google Scholar]
- 27.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Massa PE, Hanidu A, et al. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 2002;277:45129–40. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Wang X. Application of real-time polymerase chain reaction for the quantitation of interleukin-1beta mRNA upregulation in brain ischemic tolerance. Brain Res Brain Res Protoc. 2000;5:211–7. doi: 10.1016/s1385-299x(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Yang M, Htut TM, et al. Epstein–Barr virus-induced gene 3 negatively regulates IL-17, IL-22 and RORgamma t. Eur J Immunol. 2008;38:1204–14. doi: 10.1002/eji.200838145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galkin AV, Melnick JS, Kim S, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci USA. 2007;104:270–5. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stevens L, Htut TM, White D, et al. Involvement of GATA3 in protein kinase C theta-induced Th2 cytokine expression. Eur J Immunol. 2006;36:3305–14. doi: 10.1002/eji.200636400. [DOI] [PubMed] [Google Scholar]
- 33.Ammit AJ, Lazaar AL, Irani C, et al. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smooth muscle cells: modulation by glucocorticoids and beta-agonists. Am J Respir Cell Mol Biol. 2002;26:465–74. doi: 10.1165/ajrcmb.26.4.4681. [DOI] [PubMed] [Google Scholar]
- 34.van Lohuizen M, Verbeek S, Krimpenfort P, et al. Predisposition to lymphomagenesis in pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell. 1989;56:673–82. doi: 10.1016/0092-8674(89)90589-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.