

Abstract
Although interleukin-21 (IL-21) potently activates and controls the differentiation of immune cells after stimulation in vitro, the role for this pleiotropic cytokine during in vivo infection remains poorly defined. Herein, the requirement for IL-21 in innate and adaptive host defence after Listeria monocytogenes infection was examined. In the innate phase, IL-21 deficiency did not cause significant defects in infection susceptibility, or in the early activation of natural killer and T cells. In the adaptive phase, L. monocytogenes-specific CD8+ T cells expand to a similar magnitude in IL-21-deficient mice compared with control mice. Interestingly, the IL-21-independent expansion of L. monocytogenes-specific CD8+ T cells was maintained even in the combined absence of IL-12 and type I interferon (IFN) receptor. Similarly, L. monocytogenes-specific CD4+ T cells expanded and produced similar levels of IFN-γ regardless of IL-21 deficiency. Unexpectedly however, IL-21 deficiency caused significantly increased CD4+ T-cell IL-17 production, and this effect became even more pronounced after L. monocytogenes infection in mice with combined defects in both IL-12 and type I IFN receptor that develop a T helper type 17-dominated CD4+ T-cell response. Despite increased CD4+ T-cell IL-17 production, L. monocytogenes-specific T cells re-expanded and conferred protection against secondary challenge with virulent L. monocytogenes regardless of IL-21 deficiency, or combined defects in IL-21, IL-12, and type I IFN receptor. Together, these results demonstrate non-essential individual and combined roles for IL-21, IL-12 and type I IFNs in priming pathogen-specific CD8+ T cells, and reveal IL-21-dependent suppression of IL-17 production by CD4+ T cells during in vivo infection.
Keywords: bacterial, cytokine, infection, T cell
Introduction
Interleukin-21 (IL-21) is a relatively new member of the γ-chain cytokine family that all share the conserved γc subunit for receptor signalling.1,2 Each member of this cytokine family that includes IL-2, IL-4, IL-7, IL-9 and IL-15 mediate unique and defined roles in immune cell activation and differentiation, and collectively these γ-chain cytokines are essential for natural killer (NK) and T-cell development. Mice with targeted defects in the γc subunit are devoid of NK cells, and have ∼ 90% reductions in total lymphocyte numbers.3 Although IL-21 was initially thought to mediate NK and T-cell development based on the ability of purified cytokine to stimulate the maturation of these cells in vitro, the normal absolute number and ratio of NK and T-cell subsets in IL-21 receptor-deficient mice indicate that functionally redundant IL-21-independent pathways preserve normal NK and T-cell development.4–6 More recently, IL-21 has been implicated in the activation and differentiation of NK and specific T-cell subsets. For example, IL-21 boosts the cytotoxicity of NK cells stimulated with poly I:C or IL-15, and primes the proliferation of naive CD8+ T cells stimulated with artificial antigen-presenting cells that provide T-cell receptor and co-stimulation signals.6,7 Moreover, IL-21 together with transforming growth factor-β potently stimulates CD4+ T-cell IL-17 production.8–10 These findings, together with the drastic reductions in IL-17 production by CD4+ T cells from mice with targeted defects in IL-21 or IL-21 receptor, suggest that IL-21 plays an important role in CD4+ T-cell T helper type 17 (Th17) differentiation.8–11 This apparent requirement for IL-21 in CD4+ T-cell IL-17 production has been reinforced by markedly reduced disease severity in specific inflammatory autoimmunity disorders such as experimental autoimmune encephalomyelitis, rheumatoid arthritis and systemic lupus erythematosus in mice with targeted defects in IL-21, IL-21-receptor, or treated with IL-21-receptor neutralization proteins.10,12–14 Collectively, these results demonstrate a critical role for IL-21 in the Th17 differentiation programme for naive CD4+ T cells, and suggest that strategies aimed at IL-21 neutralization are promising and intriguing new therapies for inflammatory autoimmunity.
Unfortunately, therapies that moderate autoimmunity are often associated with reduced host defence against infection. In this regard, recent studies clearly demonstrate the critical requirement for IL-21 in the long-term maintenance and functionality of CD8+ T cells that control persistent lymphocytic choriomeningitis virus (LCMV) infection.15–17 By contrast for other viruses (e.g. vaccinia, influenza, LCMV Armstrong strain) that primarily cause acute infection, IL-21 plays reduced or non-essential roles for the priming and maintenance of antigen-specific CD8+ T cells.15–18 Despite these findings for viral infection, the requirement and specific role for IL-21 in host defence against other types of potential human pathogens remains undefined. However, this is a critically important area because other pleiotropic cytokines [e.g. type I interferons (IFNs)] triggered by infection have the potential to play drastically different roles in host defence against viral compared with bacterial pathogens.19–22
Infection with Listeria monocytogenes in mice is a widely used experimental model for identifying the immune mediators of innate and adaptive host defence against intracellular bacterial pathogens.23–25 Interferon-γ produced by NK and both CD4+ and CD8+ T-cell subsets each play important roles in innate host defence at early time-points after this infection.26–29 At later infection time-points, the expansion of L. monocytogenes-specific CD8+ and CD4+ T cells coincides with bacterial eradication, and thereafter the absolute numbers of pathogen-specific cells contract, and are maintained at ∼ 5 to 10% of peak expansion levels.24,25 During secondary infection, L. monocytogenes-specific T cells re-expand and rapidly confer sterilizing immunity to infection. Although the cellular mediators that confer protection in each phase of L. monocytogenes infection have been identified, the specific cytokine signals that activate and sustain these cells remain largely undefined. Given the potency whereby IL-21 stimulates the activation of NK, CD8+ and CD4+ T cells, and the importance of these cells in host defence against L. monocytogenes, the requirement for IL-21 in innate and adaptive immunity after this acute bacterial infection was examined in this study.
Materials and methods
Mice
Interleukin-21-deficient mice on a C57BL/6 (B6) background were obtained from Dr Matthew Mescher through Lexicon Genetics and the Mutant Mouse Regional Resource Centers. B6 control mice were purchased from the National Cancer Institute (Bethesda, MD). Mice with individual defects in IL-12P40 or type I IFN receptor, and mice with combined defects in both IL-12P40 and type I IFN receptor (i.e. double knockout; DKO) have been described.30,31 Mice with combined defects in IL-21, IL-12, and type I IFN receptor (triple knockout; TKO) were generated by inter-crossing IL-21-deficient mice with type I IFN receptor-deficient mice, and then inter-crossing these mice with DKO mice. All experiments were performed under University of Minnesota Institutional Animal Care and Use Committee approved protocols.
Listeria monocytogenes
The wild-type L. monocytogenes strain 10403s, recombinant L. monocytogenes ovalbumin (Lm-OVA), and recombinant Lm-OVA ΔactA that allow a more precise analysis of the immune response to the surrogate L. monocytogenes-specific H-2Kb OVA257–264 antigen have each been described.30–32 For infections, L. monocytogenes was grown to early log phase (optical density at 600 nm 0·1) in brain–heart infusion medium at 37°, washed, and diluted with saline to 200 μl final volume and injected intravenously. At the indicated time-points after infection, the number of recoverable L. monocytogenes colony-forming units (CFUs) in the organs of infected mice were quantified by homogenization in saline containing Triton-X (0·05%), and plating serial dilutions of the homogenate on agar plates as described.30
Reagents for examining cytokine production and cell proliferation
For direct in vivo intracellular staining, 250 μg brefeldin A (Sigma-Aldrich, St. Louis, MO) diluted in dimethylsulphoxide plus saline was injected intravenously into mice 6 hr before splenocyte harvest, and subjected to cell surface and intracellular cytokine staining as described.33,34 The CD8+ T-cell response to OVA257–264 was examined with H-2Kb dimer X (BD Biosciences, San Jose, CA) loaded with OVA257–264 peptide.30 Antibodies for cell surface and reagents for intracellular cytokine staining were purchased from BD Biosciences. For quantifying cytokine production by L. monocytogenes-specific T cells, splenocytes were plated into 96-well round bottom plates (5 × 106 cells/ml), and stimulated with the H-2Kb major histocompatibility complex (MHC) class I OVA257–264 or I-Ab MHC class II listeriolysin O (LLO)189–201 peptides (1 μm) in media supplemented with brefeldin A (Golgi-plug reagent).30,31 The concentration of IFN-γ in serum was quantified by enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN).
Statistics
The differences in geometric mean CFUs, number and percentage of T cells between groups of mice were evaluated using the Student's t-test with P < 0·05 taken as statistically significant (GraphPad Prism software, La Jolla, CA).
Results
IL-21 plays a non-essential role in innate L. monocytogenes host defence
Based on the potency whereby IL-21 controls the activation and differentiation of NK and T cells,1 and the protective roles for each of these cell types in innate L. monocytogenes host defence, the impact conferred by IL-21 deficiency on early susceptibility to L. monocytogenes infection was enumerated. After infection with 1 50% lethal dose (LD50; 105 CFUs in control B6 mice), both IL-21-deficient and control B6 mice each contained similar numbers of recoverable L. monocytogenes CFUs within the first 72 hr after infection (Fig. 1a). Moreover by 72 hr post-infection, the remaining mice in each group uniformly became moribund. Therefore, no apparent defects in innate susceptibility based on the degree of bacterial proliferation and time to death were found for IL-21-deficient compared with control mice after high-dose L. monocytogenes infection. In similar experiments, the susceptibility of IL-21-deficient mice was also enumerated after infection with reduced L. monocytogenes inocula (103 CFUs) to more precisely characterize the potential requirement for IL-21 in innate host defence. With this reduced L. monocytogenes inocula, IL-21-deficient and control mice both appeared healthy and did not become moribund. Furthermore, no significant differences in L. monocytogenes bacterial burden were identified for IL-21-deficient mice compared with control mice at each time-point within the first 7 days post-infection even with this reduced L. monocytogenes dose (Fig. 1b). In both groups of mice, the bacterial burden was sustained over the first 72 hr after infection, and then declined to levels that approached the limits of detection by day 5 post-infection.
Figure 1.
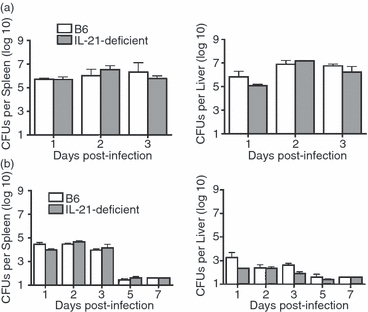
Interleukin-21 (IL-21) plays a non-essential role in Listeria monocytogenes innate host defence. Number of recoverable L. monocytogenes colony-forming units (CFUs) in the spleen and liver at the indicated time-point after infection with either (a) 105 CFUs, 1 LD50 or (b) 103 CFUs wild-type L. monocytogenes strain 10403s in B6 control and IL-21-deficient mice. These results are from six to eight mice per group, and are representative of two independent experiments each with similar results. Bar represents one standard error.
Given the critical requirement for IFN-γ in innate resistance, and the ability of IL-21 to dictate IFN-γ production by the immune cells that produce this cytokine early after L. monocytogenes infection,6,7,18,27,35 we compared the levels of IFN-γ primed by infection in IL-21-deficient and control mice (Fig. 2a). For both groups of mice, the serum concentration of IFN-γ peaked sharply 24 hr post-infection, and although there was a trend towards reduced levels in IL-21-deficient mice, these differences did not reach statistical significance. Thereafter, IFN-γ levels declined rapidly to baseline levels in both groups of mice. As IL-21 can directly stimulate and control IFN-γ production by NK and T cells,6,7,18 and IFN-γ production by these specific cell types has been directly implicated in innate L. monocytogenes host defence, the relative levels of IFN-γ produced by each cell type was also enumerated. The NKp46 marker that specifically identifies NK cells was used because IL-21 directly controls the expression of other NK cell surrogate antigens (e.g. NK1.1).36,37 Interestingly, this analysis revealed no significant difference in percentage or absolute number of IFN-γ-producing NK and T cells 24 hr after infection, which corresponds to the peak serum concentration for this cytokine (Fig. 2b,c). Together, these results demonstrate that IL-21 plays a non-essential role in innate host defence and early IFN-γ production after L. monocytogenes infection.
Figure 2.
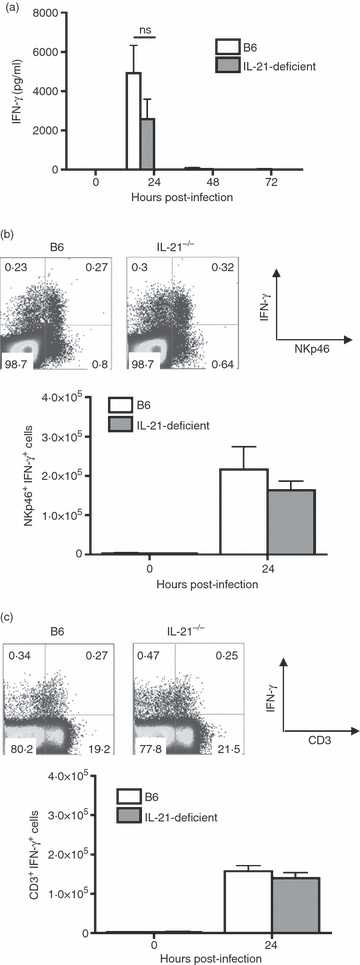
Interferon-γ (IFN-γ) production by innate immune cells in the absence of interleukin-21 (IL-21). (a) IFN-γ serum concentration at the indicated time-points after infection with 105 colony-forming units (CFUs) of Listeria monocytogenes 10403s in B6 control and IL-21-deficient mice. (b) Percent (top), and total numbers (bottom) of IFN-γ-producing natural killer (NK) cells (NKp46+) in B6 control and IL-21-deficient mice 24 hr post-infection. (c) Percentage (top), and total numbers (bottom) of IFN-γ-producing T cells (CD3+) in B6 control and IL-21-deficient mice 24 hr post-infection. These results are from six to nine mice per group, and are representative of three independent experiments each with similar results. Bar represents one standard error; ns, not significant.
L. monocytogenes-specific CD8+ T-cell priming and expansion in the absence of IL-21
Given the importance of IL-21 in priming virus-specific CD8+ T cells in the adaptive immune phase,15–18 additional experiments interrogated the requirement for IL-21 in the priming and expansion of L. monocytogenes-specific CD8+ T cells. Although IL-21, IL-12 and type I IFNs can each independently provide the ‘third signal’ for naive CD8+ T-cell expansion after stimulation in vitro, our recent studies also indicate that IL-12 and type I IFN receptor are simultaneously dispensable for the priming and expansion of L. monocytogenes-specific CD8+ T cells after infection in vivo.7,30,31,38 Accordingly, we hypothesized that IL-21 may mediate the IL-12-independent and type I IFN receptor-independent priming of L. monocytogenes-specific CD8+ T cells. To test this hypothesis, the expansion of L. monocytogenes-specific CD8+ T cells was enumerated for IL-21-deficient mice, and directly compared with the L. monocytogenes-specific CD8+ T-cell response in mice with combined defects in both IL-12 and type I IFN receptor (DKO mice), and mice with combined defects in IL-21, IL-12 and type I IFN receptor (TKO mice). For these experiments, the attenuated strain Lm-OVA ΔactA was used. The expression of the immune dominant H-2Kb OVA257–264 peptide antigen in this recombinant L. monocytogenes allows the antigen-specific CD8+ T-cell response to this surrogate L. monocytogenes-specific antigen to be readily quantified, whereas the ΔactA mutation renders this strain defective in intercellular spread, and these mutants are rapidly cleared even after infection even in IFN-γ-deficient mice, or mice with combined defects in both IL-12 and type I IFN receptor.27,30 Accordingly, the highly attenuated nature of ΔactA L. monocytogenes mutants in both immune competent and mice with innate host defects normalizes the L. monocytogenes antigen load and bypasses the potential limitations imposed by comparing groups of mice with differences in innate susceptibility.27,39
Remarkably, at the peak T-cell response (day 7 post-infection), the expansion magnitude for L. monocytogenes-specific CD8+ T cells quantified using H-2Kb OVA257–264 dimer staining was indistinguishable between IL-21-deficient mice, mice with combined defects in IL-12 and type I IFN receptor (DKO), mice with combined defects in IL-21, IL-12, and type I IFN receptor (TKO) and B6 control mice (Fig. 3a,b). Similarly after stimulation with OVA257–264 peptide, the percentage and total number of IFN-γ-producing CD8+ T cells was also similar between each group of mice (Fig. 3c). Together, these results demonstrate a non-essential role for IL-21 in the priming and expansion of L. monocytogenes-specific CD8+ T cells in both immune competent mice and in mice with combined defects in both IL-12 and type I IFN receptor. Therefore, although IL-21, IL-12 and type I IFNs can each independently provide the ‘third signal’ required for priming and expansion of naive CD8+ T cells in vitro,7,38 these three cytokine are simultaneously non-essential for the expansion of antigen-specific CD8+ T cells in vivo after L. monocytogenes infection.
Figure 3.
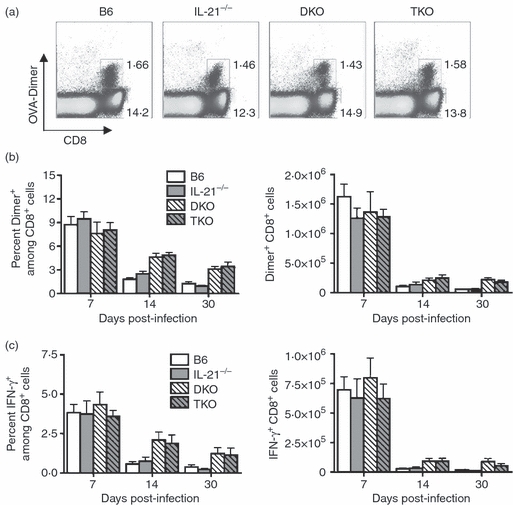
Listeria monocytogenes-specific CD8+ T-cell expansion in the absence of interleukin-21 (IL-21), or combined absence of IL-21, IL-12, and type I interferons (IFNs). (a) Percentage ovalbumin (OVA257–264) dimer-positive CD8+ T cells in control B6, IL-21-deficient, IL-12 and type I IFN receptor-deficient (DKO), and IL-21, IL-12 and type I IFN receptor-deficient (TKO) mice day 7 post-infection with 106L. monocytogenes-OVA ΔactA. (b) Percentage and total numbers of OVA257–264 dimer-positive CD8+ T cells for each group of mice at the indicated time-points after infection. (c) Percentage and total numbers of IFN-γ-producing CD8+ T cells after stimulation with OVA257–264 peptide (1 μm) for each group of mice at the indicated time-points after infection. These data represent 10–12 mice per experimental group, and are representative of four independent experiments each with similar results. Bar represents standard error.
Given the more significant role for IL-21 in sustaining pathogen-specific CD8+ T cells at later time-points after infection recently demonstrated during persistent viral infection,15–17 we extended these experiments to determine the potential requirement for IL-21 for sustaining antigen-specific CD8+ T cells at later time-points during acute bacterial infection (Fig. 3b,c). Compared with the levels on day 7, the percentage and total number of L. monocytogenes-specific CD8+ T cells was significantly reduced by day 14 in B6 mice, IL-21-deficient mice, and in mice with combined defects in either IL-12 and type I IFN receptor (DKO), or IL-21, IL-12 and type I IFN receptor (TKO) (Fig. 3b,c). Importantly, although the magnitude of CD8+ T-cell contraction was reduced in mice with combined defects in IL-12 and type I IFN receptor, which is consistent with previous studies in mice with defects in IL-12,30,40 IL-21-deficiency either alone or combined with defects in IL-12 and type I IFN receptor did not significantly alter the kinetics of L. monocytogenes-specific CD8+ T-cell contraction. Hence, IL-21 is required for neither the expansion nor the contraction of L. monocytogenes-specific CD8+ T cells after in vivo infection.
L. monocytogenes-specific CD4+ T-cell differentiation in the absence of IL-21
In addition to stimulating NK and CD8+ T cells, IL-21 also sustains and amplifies CD4+ T-cell IL-17 production, which is the lineage-defining marker for the recently described Th17 CD4+ T-cell subset.8–11 Accordingly, we hypothesized that IL-21 may also be required for the differentiation of pathogen-specific IL-17-producing CD4+ T cells after in vivo infection. Because the Th1-dominated IFN-γ-producing CD4+ T-cell response in control B6 mice is replaced by a Th17-dominated IL-17-producing CD4+ T-cell response in mice with combined defects in IL-12 and type I IFN receptor,30 the relative production of IL-17 and IFN-γ by L. monocytogenes-specific CD4+ T cells in mice with combined defects in IL-21, IL-12 and type I IFN receptor (TKO) was compared with that in DKO mice, IL-21-deficient mice and control B6 mice (Fig. 4). Surprisingly, the additive effect of IL-21 deficiency in mice with combined defects in IL-12 and type I IFN receptor not only did not ablate, but accentuated IL-17 production after stimulation with the L. monocytogenes-specific I-Ab class II peptide LLO189–201 (Fig. 4a,b). Importantly, increased IL-17 production by L. monocytogenes-specific CD4+ T cells, which occurs with IL-21 deficiency, was not restricted only to mice with combined defects in IL-12 and type I IFN receptor because despite sharp reductions in the magnitude of IL-17-producing CD4+ T cells, a similar twofold increase in percentage and total number of IL-17-producing L. monocytogenes-specific CD4+ T cells was found for IL-21-deficient mice compared with B6 control mice (Fig. 4a,b). Interestingly, despite the increased production of IL-17 that occurs in the absence of IL-21, the percentage and absolute numbers of IFN-γ-producing CD4+ T cells were not reciprocally reduced in IL-21-deficient compared with control B6 mice (Fig. 4c). Taken together, these results indicate that IL-21, IL-12 and type I IFNs synergize and play additive inhibitory roles in the differentiation of L. monocytogenes-specific IL-17-producing CD4+ T cells. Interleukin-21 therefore plays dramatically opposing roles in Th17 CD4+ T-cell differentiation under infective and non-infective conditions.
Figure 4.
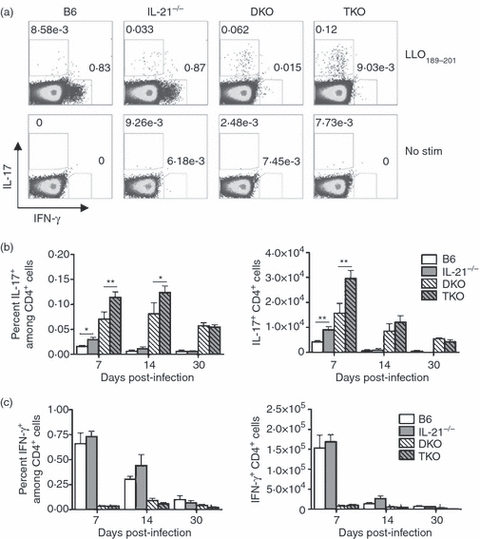
Listeria monocytogenes-specific CD4+ T-cell differentiation in the absence of interleukin-21 (IL-21), or combined absence of IL-21, IL-12 and type I interferons (IFNs). (a) Percentage IL-17- and IFN-γ-producing CD4+ T cells after stimulation with listeriolysin O (LLO189–201) peptide or no peptide for control B6, IL-21-deficient, IL-12 and type I IFN receptor-deficient (DKO), and IL-21, IL-12 and type I IFN receptor-deficient (TKO) mice on day 7 post-infection with 106L. monocytogenes-OVA ΔactA. (b) Percentage and total numbers of IL-17-producing CD4+ T cells after stimulation with LLO189–201 peptide for each group of mice at the indicated time-points after infection. (c) Percentage and total numbers of IFN-γ-producing CD4+ T cells after stimulation with LLO189–201 peptide for each group of mice at the indicated time-points after infection. These data represent 10–12 mice per experimental group, and are representative of four independent experiments, each with similar results. Bar represents standard error. *P < 0·05, **P < 0·01.
Protective immunity to virulent L. monocytogenes challenge in the absence of IL-21
To identify the individual and collective roles of IL-21, IL-12 and type I IFNs in priming protective immunity to secondary L. monocytogenes infection, the susceptibility to re-challenge with virulent L. monocytogenes was enumerated for each group of mice. Thirty days after primary L. monocytogenesΔactA inoculation, groups of B6, IL-21-deficient, DKO and TKO mice were each challenged with 105 CFUs of virulent Lm-OVA.30,32 Compared with naive mice, L. monocytogenesΔactA-primed mice in each group were uniformly highly protected, and by day 3 after re-challenge contained four to five log10 reductions in recoverable L. monocytogenes CFUs (Fig. 5a). Moreover, by day 5 after re-challenge, virulent L. monocytogenes was cleared from both the spleen and liver in L. monocytogenesΔactA-primed mice in each group. The marked reductions in bacterial burden after re-challenge in L. monocytogenesΔactA-primed compared with naive mice in each group was associated with robust secondary expansion of L. monocytogenes-specific CD8+ and CD4+ T cells beginning on day 3 and continuing through to day 5 after re-challenge (Fig. 5b). Consistent with the similar expansion kinetics that occurs after primary infection, L. monocytogenes-specific CD8+ T cells expand with parallel kinetics in B6, IL-21-deficient, DKO and TKO mice. For each group of mice, Lm-OVA257–264-specific CD8+ T cells expanded approximately fivefold, and ∼ 50-fold by days 3 and 5 after re-challenge, respectively. Interestingly, even under re-challenge conditions with virulent L. monocytogenes, the increased IL-17 production that occurs with IL-21 deficiency alone or in mice with combined defects in IL-21, IL-12 and type I IFN receptor is also maintained (Fig. 5c). Hence, despite the increased Th17 differentiation by L. monocytogenes-specific CD4+ T cells that occurs in the absence IL-21 alone, or combined with defects in IL-12 and type I IFN receptor, the protective sterilizing immunity against secondary re-challenge with virulent L. monocytogenes is preserved. Taken together, these results demonstrate previously unanticipated roles for IL-21 in limiting the Th17 differentiation programme for pathogen-specific CD4+ T cells after primary and secondary intracellular bacterial infection.
Figure 5.
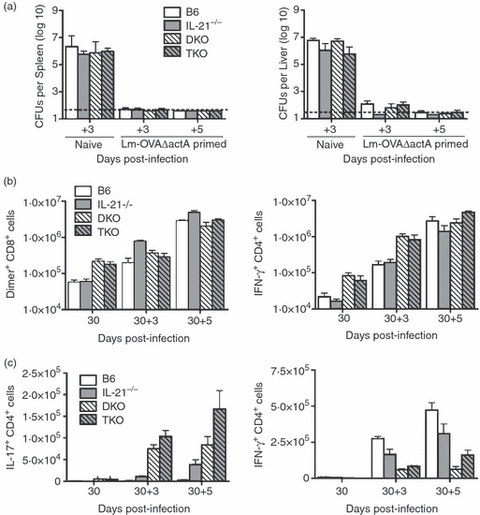
Protective immunity to secondary Listeria monocytogenes infection and re-expansion of L. monocytogenes-specific CD8+ and CD4+ T cells in the absence of interleukin-21 (IL-21), or combined absence of IL-21, IL-12 and type I interferons (IFNs). (a) Number of recoverable L. monocytogenes colony-forming units (CFUs) at the indicated time-points post-infection with 105L. monocytogenes–ovalbumin (OVA) for groups of either naive mice or mice inoculated with L. monocytogenes-OVA ΔactA 30 days before infection. (b) Total numbers of OVA257–264 dimer-positive (left) and IFN-γ-producing right CD8+ T cells after stimulation with OVA257–264 peptide before (D30), and on day 3 (30 + 3) and day 5 (30 + 5) after secondary infection with L. monocytogenes-OVA. (c) Total numbers of IL-17-producing (left) and IFN-γ-producing (right) CD4+ T cells after stimulation with listeriolysin O (LLO189–201) peptide before (D30), and on day 3 (30 + 3) and day 5 (30 + 5) after secondary infection with L. monocytogenes-OVA. These data represent 12 mice per experimental group, and are representative of three independent experiments each with similar results. Bar represents standard error.
Dicsussion
Although in vitro studies using purified cytokine demonstrate that IL-21 has the potential to activate numerous immune cell subsets important for host defence, the requirements for IL-21 in immunity to infection remains uncertain, and has been only recently demonstrated to play an important role for sustaining virus-specific CD8+ T cells during persistent LCMV infection.15–17 In this context, targeted defects in the IL-21 receptor cause virus-specific CD8+ T cells to become ‘exhausted’, as these cells do not produce effector cytokines such as IFN-γ and do not eradicate infection. In contrast to these roles during persistent infection, IL-21 appears to play more modest or functionally redundant roles for priming the expansion of antigen-specific T cells after infection with viruses that primarily cause acute infection.16,18 The experiments described in this study extend these newly identified roles for IL-21 to acute bacterial infection conditions. Mice with targeted defects in IL-21 compared with control mice were equally susceptible to acute L. monocytogenes infection in the innate phase, and NK and innate T cells in these mice produced similar levels of IFN-γ within the first 24 hr after infection (Figs 1 and 2). Similarly in the adaptive phase, L. monocytogenes-specific CD8+ T cells were found to expand to a similar magnitude and with identical kinetics regardless of IL-21 deficiency (Fig. 3). Interleukin-21 therefore plays non-essential roles in the activation of innate and adaptive immune components required for host defence against primary and secondary L. monocytogenes infection.
Despite these apparently negative results for IL-21 on L. monocytogenes susceptibility, several important findings on the specific cytokine requirements for priming pathogen-specific CD8+ T cells and the differentiation programme of CD4+ T cells during in vivo infection are revealed. The activation and expansion of CD8+ T cells using artificial antigen-presenting cells in vitro requires three inter-related stimulation signals.7,38 When only T-cell receptor stimulation (Signal 1) and co-stimulation (Signal 2) are provided, naive CD8+ T cells do not proliferate and produce little to no effector cytokines. By contrast, when exogenous IL-21, IL-12 or type I IFN is provided with signal 1 and 2, CD8+ T cells readily proliferate and expand.7,38 To our knowledge, these are the only known ‘third signals’ that have been identified for priming the expansion of naive CD8+ T cells. Therefore, our results demonstrating the normal expansion magnitude of L. monocytogenes-specific CD8+ T cells in mice with combined defects in all three of these cytokine signals (IL-21, IL-12, type I IFNs) suggest that either ‘third signals’ are not required for the expansion of CD8+ T cells during in vivo infection conditions, or that additional unidentified ‘third signals’ triggered by complex pathogens like L. monocytogenes play functionally redundant roles in priming the expansion of pathogen-specific CD8+ T cells. In this regard, a potential candidate may be the direct effects of IFN-γ stimulation on CD8+ T cells because markedly reduced expansion occurs for adoptively transferred antigen-specific IFN-γ-receptor-deficient compared with receptor-sufficient CD8+ T cells after acute LCMV infection.41 However, these effects were not reproduced after enumerating the relative expansion of virus-specific IFN-γ receptor-deficient compared with receptor-sufficient CD8+ T cells among the polyclonal repertoire in mixed bone marrow chimera mice containing congenically marked populations of both cell types.42 Moreover, purified IFN-γ with artificial antigen-presenting cells does not stimulate naive CD8+ T-cell proliferation or expansion in vitro.38 Therefore, additional in vitro and complementary in vivo studies are required for identifying the requirement, and/or specific other cytokine signals triggered by L. monocytogenes infection that primes pathogen-specific CD8+ T-cell expansion in the absence of all previously identified ‘third signals’.
Equally intriguing to these findings for CD8+ T cells is the sharply contrasting role for IL-21 in regulating IL-17 production by pathogen-specific CD4+ T cells. Compared with recent studies suggesting that IL-21 is required for sustaining and amplifying CD4+ T-cell IL-17 production, our results demonstrating increased IL-17 production by L. monocytogenes-specific CD4+ T cells from IL-21-deficient compared with IL-21-sufficient control mice challenge this requirement, and reveal context-dependent stimulatory and inhibitory roles for IL-21 in Th17 CD4+ T-cell differentiation. Along with two recent studies demonstrating the development of Th17-mediated experimental autoimmune encephalomyelitis and autoimmune myocarditis associated with normal or slightly enhanced IL-17 production by CD4+ T cells in IL-21 and IL-21-receptor-deficient mice, these are the only reports demonstrating IL-21-independent CD4+ T-cell IL-17 production.43,44 Moreover, because each of these studies primed IL-21-independent Th17 CD4+ T-cell differentiation with dead adjuvant, our results represent the first demonstration of these effects after in vivo infection and highlight the generalizability of IL-21-independent CD4+ T-cell IL-17 production for both infective and non-infective inflammatory conditions. Although the specific immune signals that dictate whether IL-21 stimulates or inhibits CD4+ T-cell IL-17 production are presently unknown, an interesting candidate for this ‘switch’ is IL-6 because this cytokine can potently drive CD4+ T-cell IL-17 production even in the absence of IL-21-receptor signalling, and is highly expressed after L. monocytogenes infection.8 Collectively, these results underscore the importance of identifying the immune signals that dictate how IL-21 controls CD4+ T-cell differentiation before therapies aimed at targeting IL-21 are developed and implemented for the treatment of inflammatory autoimmunity.
Acknowledgments
The authors are grateful to Dr Matthew Mescher for providing IL-21-deficient mice, Dr Hao Shen for providing Lm-OVA, and Drs Matthew Mescher, Stephen McSorley and Christopher Wilson for helpful discussions and critical reviews of this manuscript. This work was supported through funding from the following sources: NICHD/NIH-K08HD51584, Vikings Children's Fund, the Minnesota Medical Foundation and a Grant-in-Aid from the University of Minnesota.
Disclosure
The authors each have no conflicts of interest, or financial conflicts to disclose.
References
- 1.Leonard WJ, Zeng R, Spolski R. Interleukin 21: a cytokine/cytokine receptor system that has come of age. J Leukoc Biol. 2008;84:348–56. doi: 10.1189/jlb.0308149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandt K, Singh PB, Bulfone-Paus S, Ruckert R. Interleukin-21: a new modulator of immunity, infection, and cancer. Cytokine Growth Factor Rev. 2007;18:223–32. doi: 10.1016/j.cytogfr.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 3.DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci USA. 1995;92:377–81. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parrish-Novak J, Dillon SR, Nelson A, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 5.Ozaki K, Spolski R, Feng CG, et al. A critical role for IL-21 in regulating immunoglobulin production. Science (New York, NY) 2002;298:1630–4. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 6.Kasaian MT, Whitters MJ, Carter LL, et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. 2002;16:559–69. doi: 10.1016/s1074-7613(02)00295-9. [DOI] [PubMed] [Google Scholar]
- 7.Casey KA, Mescher MF. IL-21 promotes differentiation of naive CD8 T cells to a unique effector phenotype. J Immunol. 2007;178:7640–8. doi: 10.4049/jimmunol.178.12.7640. [DOI] [PubMed] [Google Scholar]
- 8.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–7. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immun. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 10.Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–3. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 11.Awasthi A, Kuchroo VK. Th17 cells: from precursors to players in inflammation and infection. Int Immunol. 2009;21:489–98. doi: 10.1093/intimm/dxp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol. 2007;178:3822–30. doi: 10.4049/jimmunol.178.6.3822. [DOI] [PubMed] [Google Scholar]
- 13.Young DA, Hegen M, Ma HL, et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007;56:1152–63. doi: 10.1002/art.22452. [DOI] [PubMed] [Google Scholar]
- 14.Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC, III, Leonard WJ, Roopenian DC. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci USA. 2009;106:1518–23. doi: 10.1073/pnas.0807309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science (New York, NY) 2009;324:1572–6. doi: 10.1126/science.1175194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frohlich A, Kisielow J, Schmitz I, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science (New York, NY) 2009;324:1576–80. doi: 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- 17.Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science (New York, NY) 2009;324:1569–72. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng R, Spolski R, Finkelstein SE, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201:139–48. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pietras EM, Saha SK, Cheng G. The interferon response to bacterial and viral infections. J Endotoxin Res. 2006;12:246–50. doi: 10.1179/096805106X118799. [DOI] [PubMed] [Google Scholar]
- 20.O’Connell RM, Saha SK, Vaidya SA, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–45. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535–40. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–33. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edelson BT, Unanue ER. Immunity to Listeria infection. Curr Opin Immunol. 2000;12:425–31. doi: 10.1016/s0952-7915(00)00112-6. [DOI] [PubMed] [Google Scholar]
- 24.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–23. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 25.Zenewicz LA, Shen H. Innate and adaptive immune responses to Listeria monocytogenes: a short overview. Microbes Infect. 2007;9:1208–15. doi: 10.10110/2/076/j.micinf.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunn PL, North RJ. Early gamma interferon production by natural killer cells is important in defense against murine listeriosis. Infect Immun. 1991;59:2892–900. doi: 10.1128/iai.59.9.2892-2900.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3:109–17. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 28.Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med. 2003;198:1583–93. doi: 10.1084/jem.20031051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu J, August A. Naive and innate memory phenotype CD4+ T cells have different requirements for active Itk for their development. J Immunol. 2008;180:6544–52. doi: 10.4049/jimmunol.180.10.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orgun NN, Mathis MA, Wilson CB, Way SS. Deviation from a strong Th1-dominated to a modest Th17-dominated CD4 T cell response in the absence of IL-12p40 and type I IFNs sustains protective CD8 T cells. J Immunol. 2008;180:4109–15. doi: 10.4049/jimmunol.180.6.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Way SS, Havenar-Daughton C, Kolumam GA, Orgun NN, Murali-Krishna K. IL-12 and type-I IFN synergize for IFN-gamma production by CD4 T cells, whereas neither are required for IFN-gamma production by CD8 T cells after Listeria monocytogenes infection. J Immunol. 2007;178:4498–505. doi: 10.4049/jimmunol.178.7.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol. 2002;168:1528–32. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Whitton JL. Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J Immunol. 2005;174:5936–40. doi: 10.4049/jimmunol.174.10.5936. [DOI] [PubMed] [Google Scholar]
- 34.Plitas G, Chaudhry UI, Kingham TP, Raab JR, DeMatteo RP. NK dendritic cells are innate immune responders to Listeria monocytogenes infection. J Immunol. 2007;178:4411–6. doi: 10.4049/jimmunol.178.7.4411. [DOI] [PubMed] [Google Scholar]
- 35.Dai WJ, Bartens W, Kohler G, Hufnagel M, Kopf M, Brombacher F. Impaired macrophage listericidal and cytokine activities are responsible for the rapid death of Listeria monocytogenes-infected IFN-gamma receptor-deficient mice. J Immunol. 1997;158:5297–304. [PubMed] [Google Scholar]
- 36.Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol. 2004;172:2048–58. doi: 10.4049/jimmunol.172.4.2048. [DOI] [PubMed] [Google Scholar]
- 37.Walzer T, Blery M, Chaix J, et al. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci USA. 2007;104:3384–9. doi: 10.1073/pnas.0609692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–9. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 39.Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–9. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 40.Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J Immunol. 2007;179:2074–81. doi: 10.4049/jimmunol.179.4.2074. [DOI] [PubMed] [Google Scholar]
- 41.Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201:1053–9. doi: 10.1084/jem.20041463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tewari K, Nakayama Y, Suresh M. Role of direct effects of IFN-gamma on T cells in the regulation of CD8 T cell homeostasis. J Immunol. 2007;179:2115–25. doi: 10.4049/jimmunol.179.4.2115. [DOI] [PubMed] [Google Scholar]
- 43.Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI. Cutting edge: IL-21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J Immunol. 2008;180:7097–101. doi: 10.4049/jimmunol.180.11.7097. [DOI] [PubMed] [Google Scholar]
- 44.Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol. 2008;38:1833–8. doi: 10.1002/eji.200838511. [DOI] [PubMed] [Google Scholar]