

Abstract
The pore-forming protein perforin is synthesized as an inactive precursor in natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), and becomes active when a short C-terminal peptide is cleaved within acidic lysosome-like cytotoxic granules. Although it was shown more than a decade ago that this cleavage is pH dependent and can be inhibited by the generic cysteine cathepsin inhibitor E-64d, no protease capable of processing the perforin C terminus has been identified. Neither is it known whether a single protease is responsible or the processing has inbuilt redundancy. Here, we show that incubation of human NK cells and primary antigen-restricted mouse CTLs with the cathepsin L (CatL) inhibitor L1 resulted in a marked inhibition of perforin-dependent target cell death and reduced perforin processing. In vitro, CatL preferentially cleaved a site on full-length recombinant perforin close to its C terminus. The NK cells of mice deficient in CatL showed a reduction but not a complete absence of processed perforin, indicating that cysteine proteases other than CatL are also able to process perforin. We conclude that granule-bound cathepsins are essential for processing perforin to its active form, and that CatL is an important, but not exclusive, participant in this process.
Keywords: cathepsin L, cytotoxic T cells, cytotoxicity, natural killer cells, perforin deficiency
Introduction
Natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) recognize virus-infected or transformed cells and destroy them through two independent mechanisms. Apoptotic target-cell death can be triggered by engagement of death receptors1 or by release of cytotoxic granules.2 Cytotoxic granules gather at the site of contact between the effector cells (NK cells or CTLs) and the target cell where the immunological synapse is formed. At the immunological synapse, the cytotoxic granule membrane fuses with the cell membrane and releases its contents.3 Cytotoxic granules have secretory lysosome characteristics, including an electron-dense core found in secretory organelles and a vesicular surrounding region typical of lysosomes.4 They contain perforin and serine protease granzymes, as well as several lysosomal proteins [including lysosomal-associated membrane proteins 1 and 2, CD63 and cathepsins C, D and L (CatL)].5,6
Perforin, a calcium-dependent pore-forming protein, facilitates the entry of granzymes into the cytoplasm of a target cell, where they access their substrates to trigger cell death.7 Recently, a structure of perforin was modelled on the basis of homology with the Plu-MACPF domain (a membrane attack complex/perforin region domain containing protein from Photorhabdus luminescens).8 The C2 domain of perforin is crucial for calcium-dependent membrane binding and pore formation.7 The final 12–20 residues of the C terminus are cleaved from the perforin, together with a bulky N-linked glycan group located six residues from the C terminus. After the cleavage, the C2 domain can bind to the membrane. The cleavage is inhibited by incubation with E-64, leupeptin and ammonium chloride, indicating a possible role of cysteine cathepsins in this process.9 The low pH of cytotoxic granules (pH 5·1–5·5) is believed to favour the interaction of perforin with the proteoglycans.2,10 Perforin deficiency has been shown to play a role in the pathogenesis of familial haemophagocytic lymphohistiocytosis (HLH), as 30–40% of familial HLH patients have mutations in both copies of the perforin gene.11,12 Several other known mutations associated with familial HLH affect genes involved in the cytotoxic-granule exocytosis pathway.11 Perforin-deficient mice have decreased ability to clear bacterial and viral infections, fail to reject transplanted tumours and, as they age, spontaneously develop B-cell lymphoma.13,14 Symptoms similar to familial HLH develop in perforin-deficient mice that are infected with lymphocytic choriomeningitis virus.15
Despite more than a decade having passed, no protease capable of processing the perforin C terminus has been described. An early study reported that after cytotoxic granule exocytosis, enzymatically active cathepsin B (CatB), but not CatL, was found on the surface of degranulating CTLs and it was proposed that CatB could degrade perforin to prevent it from harming the killer cell following degranulation.16 Baran et al.17 reported that CTLs isolated from CatB-deficient mice were able to induce normal death of target cells both in vitro and in vivo and survive the encounter with target cells as efficiently as the wild-type CTLs. This indicated that CatB plays no significant part in either activating or inactivating perforin in vivo. Furthermore, recent studies showed that cathepsin C is not essential for the C-terminal perforin processing in human peripheral blood-derived NK cells18 or in murine allospecific CTLs.19
The aim of our study was to identify proteinases that activate perforin through processing of its C terminus, and to determine whether any single proteinase is responsible. Our initial studies showed that CatL processed the C-terminal part of perforin in vitro and that cathepsin inhibition significantly decreased target cell killing by human NK cell lines as well as by primary mouse CTLs. However, target cell killing by CTLs and NK cells from CatL-deficient mice was not diminished despite a reduction in the amount of processed perforin detected in these killer cells. We conclude that CatL is an important participant in perforin activation, but that additional cathepsins can partially compensate in its absence.
Materials and methods
Antibodies and other reagents
The following antibodies were used: rat anti-perforin monoclonal antibody (mAb) P1-8,20 mouse mAb for human perforin pf-344 (Mabtech, Stockholm, Sweden), mAb specific for the C terminus of perforin, clone 6G7/1F10 (Voskoboinik et al., manuscript in preparation); anti-actin (Sigma Aldrich, St Louis, MO), anti-T-cell markers CD8, Vα2, CD25, CD44 and CD69, and anti-mouse NK marker, NK1.1 (BD-Pharmingen, San Diego, CA). All horseradish peroxidase-conjugated secondary antibodies were from Sigma Aldrich, Bradford reagent was from Bio-Rad (Bio-Rad Laboratories, Hercules CA). The enhanced chemiluminescence detection kit was from Amersham Biosciences (Arlington Heights, IL). Recombinant mouse perforin was purified from baculovirus-infected cells, as described.21 Recombinant mouse CatL was prepared as described previously.22 The cysteine protease inhibitor E-64d was purchased from Peptide Research Institute (Osaka, Japan) and L1 (CatL inhibitor I, z-FF-fmk) was purchased from Calbiochem (San Diego, CA).
Mice
C57BL/6 (B6) mice were purchased from Walter and Eliza Hall Institute of Medical Research, Parkville, Australia. The CatL-deficient mice were generated by gene targeting,23 backcrossed to the C57BL/6 genetic background for 10 generations and finally shipped through Dr Jose A. Villadangos (Walter and Eliza Hall Institute of Medical Research). CatL-deficient mice were crossed with OT-1 (ovalbumin-specific, H-2Kb-restricted T-cell receptor transgenic) B6 mice to generate OT-1 transgenic CatL-deficient mice. We used a polymerase chain reaction screening protocol to confirm disruption of the CatL gene and acquisition of the neomycin-resistance cassette in the knockout animals (data not shown). The expression of OT-1 transgenic T-cell receptor was confirmed using anti-CD8 and anti-Vα2 antibodies by flow cytometry (data not shown). The mice used in all experiments were 5–10 weeks of age and the studies conformed to Peter MacCallum Cancer Centre animal ethics committee guidelines.
Cell cultures
The YT cell line was obtained from DSMZ German Collection of Microorganisms and Cell Cultures,24 YT 5 cell line was obtained from Mark Smyth (obtained from National Cancer Institute, Frederick, MD), KHGY1 (obtained from G. Suck, Toronto, Canada),25 NK-92 (kindly provided by E. Vivier, Marseille, France and P. Bird, Clayton, Australia), K562 (kindly provided by E. Vivier, Marseille, France). All were cultured in RPMI-1640 with 10% fetal calf serum (FCS) at 37° in 5% CO2. The mouse thymoma cell line EL4 (provided by Dr F. Carbone, University of Melbourne, Australia) was maintained in Dulbecco's modified Eagle's minimal essential medium supplemented with 10% FCS. For the generation of mouse CTLs, 106 spleen cells were incubated with lipopolysaccharide (200 ng/ml; Sigma Aldrich) and SIINFEKL peptide (0·1 μg/ml) (ovalbumin amino acids 257–264, Auspep, West Melbourne, Australia) in either 24-well (1 ml) or six-well (up to 8 ml) plates for 3–5 days. Viable CD8+ cells were then purified by Ficoll (GE Healthcare, Buckinghamshire, UK) gradient. Activation of CTLs was confirmed by staining for CD8 and the activation markers CD25, CD44 and CD69 and analysis by flow cytometry using a FACScalibur (BD Biosciences, San Diego, CA; data not shown). Mouse NK cells were generated from splenocytes of OT-1 mice (CatL−/−, CatL+/+, B6). The NK cells were enriched by magnetic negative selection using anti-Vα2-biotinylated antibodies (Sabine Hoves et al., unpublished data) to exclude all OT-1-positive CTLs from the spleen preparation. Cells were grown in RPMI-1640/10% FCS with 1000 U/ml recombinant human interleukin-2. The NK cells were > 95% NK1.1+ by day 3, as determined by flow cytometry (data not shown).
In vitro cleavage of mouse perforin
In vitro cleavage of recombinant mouse perforin (180 nmol) by CatL was performed in acetate buffer pH 5·5, containing 2 mm dithiothreitol. The ratio of CatL to perforin ranged from 0·04 to 1·0, corresponding to a final concentration of 7·2–180 nm. Following 25 min incubation at 37° the reaction was terminated by adding sodium dodecyl sulphate (SDS) loading buffer and boiling for 9 min. Activity of CatL was also blocked by pre-treatment with L1 (10 μm)26,27 for 15 min at 37° before the addition of perforin. The reaction products were then separated by SDS–polyacrylamide gel electrophoresis (PAGE) on 8% gels and transferred to polyvinylidene difluoride membranes. (Millipore, Bedford, MA). Western blots were probed with the anti-perforin antibodies, PI-8 or C-terminus-specific 6G7/IF10, and visualized with enhanced chemiluminescence according to the manufacturer's instructions (Amersham Pharmacia Biotech, Uppsala, Sweden).
Treatment with inhibitors, cell lysis
The CTLs and NK cells (0·8 × 106/ml) were treated with the inhibitors L1 (10–20 μm) or E-64d (20–30 μm) for 24 hr at 37° in 24-well plates. Cells were then used in 51Cr-release assays (see below) or were lysed to examine perforin in Western blots. The inhibitor was also added at the same concentration during the 4-hr reactions in some 51Cr-release assays, as indicated. Cell lysates were prepared using NP-40 lysis buffer (25 mm HEPES, 250 mm NaCl, 2·5 mm ethylenediaminetetraacetic acid , 0·1% volume/volume Nonidet P-40) and total protein concentration was determined using the Bradford assay. Equal amounts of protein were loaded and resolved on 8% SDS–PAGE gels. Human or mouse perforin was detected using the appropriate antibodies as indicated. Anti-actin antibody was used as a loading control.
51Cr-release assays
Cell death of K562 or SIINFEKL-pulsed (1 μg/ml) EL4 cells, induced by the human NK cell lines or OT-1-positive CTLs, respectively, was assessed in 4-hr 51Cr-release assays as described previously.28The percentage of 51Cr release was calculated by the following equation (where c.p.m. is counts/min): [(c.p.m. of 51Cr released from sample – c.p.m. of 51Cr released from untreated cells) / (c.p.m. of 51Cr released from cells treated with 1 m HCl − c.p.m. of 51Cr released released from untreated cells) × 100].19 The % inhibition of cytotoxicity was calculated as (% specific control lysis − % specific lysis sample with inhibitor) / (% specific control lysis) × 100.
Granzyme activity assay
Whole cell lysates of NK-92, YT 5 and KHGY1 NK cell lines were normalized for protein content using the Bradford assay and analysed for granule serine protease activity by hydrolysis of synthetic peptide thiobenzyl ester substrates: ASPase (GrB) activity, Boc-Ala-Ala-Asp-S-Bzl (a gift from J. Powers, Georgia Institute of Technology, Atlanta, GA); tryptase (GrA) activity, N-α-carbobenzyloxy-l-lysine thiobenzyl ester (BLT; Sigma Aldrich); chymotrypsin-like (chymase) activity for Suc-Phe-Leu-Phe-SBzl (Enzyme system products Dublin, CA).19,29
Cysteine cathepsin activity assay
Whole cell lysates were normalized for protein content with Bradford assay and analysed for z-Phe-Arg-aminomethylcoumarin (Bachem Torrance, CA) activity, as described previously.30,31
Results
It is now many years since evidence was first presented in support of the hypothesis that perforin requires cleavage at a point close to its C terminus to acquire cytotoxic activity. This cleavage was shown to occur in an acidic compartment, almost certainly the lysosome-like cytotoxic granules in which mature perforin is stored together with granzymes and other CTL/NK cell toxins.9
CatL is capable of processing perforin in human NK cell lines
To further characterize this potentially key post-translational step, several perforin-expressing human NK cell lines were incubated for 24 hr with various protease inhibitors and Western immunoblotting was performed to determine whether this would affect perforin processing. L1, a membrane-permeable cysteine protease inhibitor that preferentially blocks human and mouse CatL activity,26,27 significantly influenced the migration of perforin molecules on SDS–PAGE (Fig. 1a). Previous studies have suggested that unprocessed perforin appears as a slowly migrating band on SDS–PAGE, and that removal of the peptide tail and an attached bulky N-linked glycan at Asn529/530 (mouse/human perforin numbering) means that ‘mature’ perforin migrates as a faster species. KHYG-1 cells incubated with dimethylsulphoxide buffer alone contained perforin that was largely processed and mature (Fig. 1a, lane 1); however, L1 inhibitor resulted in two perforin bands (lane 2). One of the bands co-migrated with processed perforin, while the second, slower-migrating band co-migrated with perforin whose processing was blocked by neutralizing the acid granule pH with NH4Cl (lane 3). Although exemplified most clearly in KHYG1 cells, similar results were seen with human NK92 and YT cells treated in the same fashion (Fig. 1a). As previously reported, addition of the cell-permeable generic cysteine protease inhibitor E-64d to YT cells also inhibited perforin processing.9 This resulted in a similar reduction in the ratio of processed/unprocessed perforin as was seen with L1 (Fig. 1a, lanes 7–10). As the YT 5 human NK cell line expresses significantly more perforin than the other NK cell lines, the signal on the Western blot was stronger than with the other cell lines. Some additional perforin bands thought to be degradation products were also evident in YT5 cells (even in the untreated samples), but these were not reduced by either the L1 or E64-d inhibitor and probably arose because of a post-lysis cleavage artefact.
Figure 1.
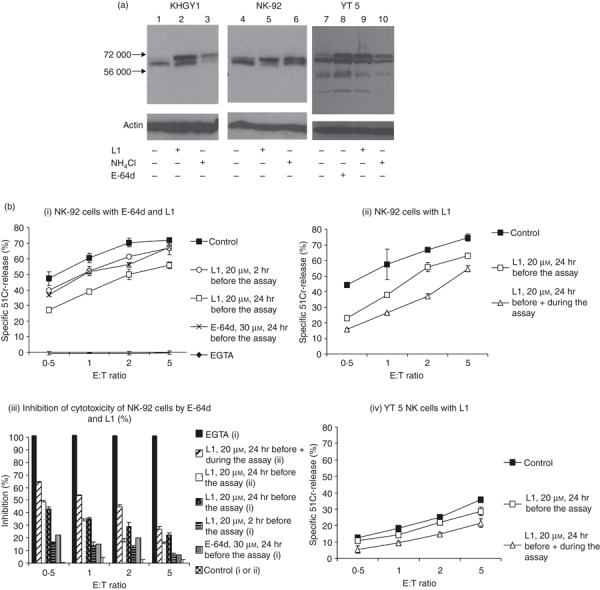
Perforin processing is inhibited by the generic cysteine protease inhibitors E-64d and L1. (a) KHGY1, NK-92 and YT 5 cells were cultured in the presence or absence of 20 μm L1 inhibitor or 10 mm NH4Cl for 24 hr. In addition, YT 5 cells were incubated with 20 μm E-64d. Western blots were probed with the anti-perforin antibody, pfn-344 (Mabtech); 100 μg of NK-92 and KHYG1 or 50 μg of YT5 cell lysates were loaded. An anti-actin antibody was used as a loading control. Migration of molecular weight markers is indicated. (b) Cytotoxic activity of NK-92 and YT 5 is inhibited by L1 and E-64d. The effect of the inhibitors L1 and E-64d on NK-92 (i, ii and iii) and L1 on YT 5 (iv) cytotoxicity was determined in 51Cr-release assays. Before the assay the effector cells were pre-incubated with inhibitors for the times indicated, and then incubated with the 51Cr-labelled K562 cells for 4 hr. The % specific 51Cr-release induced by NK-92 (i. and ii.) and YT 5 (iv.) was calculated. The data represent the mean of triplicate data points ± SD and are representative of three similar experiments. The P-values have been calculated to determine the significance of differences in the level of lysis for each treatment (Table 1). The percentage of inhibition of cytotoxicity of NK-92 from (i) and (ii) was calculated from values of % specific 51Cr-release at each effector : target ratio (% specific control lysis − % specific lysis sample with inhibitor)/(% specific control lysis) × 100. (iii.).
Next, we examined the effect of L1 pre-incubation on NK cell cytotoxicity. Exposure of NK-92 cells to L1 for 24 hr before the assay moderately reduced their capacity to kill K562 target cells in a standard 51Cr-release assay (Fig. 1bi). This level of inhibition was also seen when the effector cells were pre-incubated with E-64d. However, the inhibition was maximized when L1 was also added throughout the 4-hr cytotoxicity assay at the same concentration (Fig. 1bii). Under these conditions, specific 51Cr-release at an effector : target (E : T) ratio of 5 was less than with buffer alone at an E : T ratio of 1, signifying > 80% inhibition of cytotoxicity (Fig. 1bii,iii). Similar data were also obtained with YT 5 killer cells (Fig. 1biv) and KHYG1 cells (data not shown). Given that NK cells constitutively synthesize and store large amounts of perforin, this level of inhibition of target cell death was seen as quite significant. This result clearly indicated that sufficient active perforin can be regenerated in as little as 4 hr (the duration of the cytotoxicity assay) following washout of the inhibitor. The P-values have been determined for the differences at 2 E : T ratios for each of the inhibitors compared with controls (Table 1). This emphasized that treatment before and during the assay was most effective. The death of K562 cells was completely dependent on perforin, as complexing calcium ions with EGTA completely abrogated target cell death (Fig. 1bi,iii). Neither L1 nor E-64d (or dimethylsulphoxide) addition had any deleterious effect on the viability of any of the NK cell lines studied (data not shown). Furthermore, the observed reductions in cytotoxicity could not be put down to accidental inhibition of granzymes. As demonstrated in Fig. 2(a), L1 and E-64d (which specifically block cysteine proteases, but not serine proteases such as granzymes) were able to completely block turnover of the CatL substrate Z-Phe-Arg-aminomethylcoumarin, when pre-incubated with NK-92 (Fig. 2ai) or YT 5 (Fig. 2aii) cells. However, there was no significant effect on the turnover of peptide substrates cleaved by the serine protease granzymes A, B or H (Fig. 2b). NK-92 lysate was used to compare the BLT esterase (tryptase) activity (Fig. 2bi) as these cells express high levels of GrA but minimal amounts of GrB. Conversely YT 5 cells express high levels of GrB, resulting in significant Aspase activity (Fig. 2bii) but tryptase activity is not detectable in these cells.
Table 1.
P-values for Figure 1(b)
| Effector : target ratio | 2 : 1 | 5 : 1 |
|---|---|---|
| (i.) NK-92 cells with E-64d and LI | ||
| LI, 20 μm, 2 hr before the assay | 0.006 | 0.035 |
| L1, 20 μm, 24 hr before the assay | 0.001 | < 0.001 |
| E-64d, 30 μm, 24 hr before the assay | 0.001 | 0.189 |
| (ii.) NK-92 cells with LI | ||
| LI, 20 μm, 24 hr before the assay | 0.004 | 0.002 |
| L1, 20 μm, 24 hr before + during the assay | < 0.001 | 0.002 |
| (iii.) YT 5 natural killer cells with LI | ||
| LI, 20 μm, 24 hr before the assay | 0.086 | 0.005 |
| LI, 20 μm, 24 hr before + during the assay | < 0.001 | 0.001 |
Figure 2.
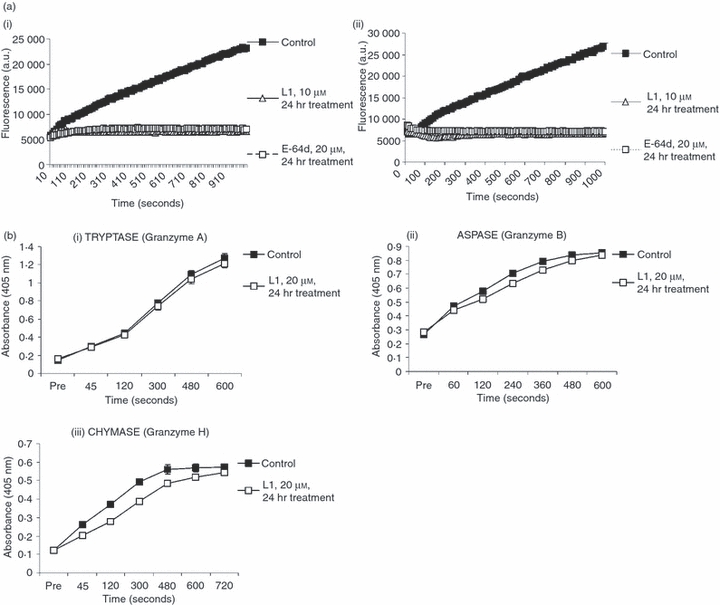
Effect of E-64d and L1 inhibitors on cathepsin (Cat) and granzyme activity. (a) Inhibitors E-64d and L1 block Cat activity. NK-92 (i.) and YT 5 (ii.) Natural killer (NK) cells were left untreated or pre-treated for 24 hr with L1 (10 μm) or E-64d (20 μm). Cell lysates were prepared and assayed for Cat activity using the peptide substrate Z-Phe-Arg-aminomethylcoumarin, as described in the Materials and methods. (b) Granzymes A, B and H activities are not significantly affected by L1. NK-92 and YT 5 cells were left untreated or pre-treated for 24 hr with the L1 (20 μm) inhibitor. The granzyme activity in cell lysates was measured by hydrolysis of specific peptide thioester substrates. Error bars represent SEM between at least three independent experiments (each of which was performed in triplicate). (i) Tryptase activity (granzyme A) in NK-92 was measured by cleavage of Z-L-Lys-SBzl (BLT). (ii) ASP-ase activity (granzyme B) in YT-5 was measured by cleavage of Boc-Ala-Ala-Asp-S-Bzl. (iii) Chymotrypsin-like (chymase) (GrH) activity in NK-92 was measured by cleavage of Suc-Phe-Leu-Phe-SBzl substrate.
Mouse perforin is a potential substrate for CatL
To definitively demonstrate that CatL can cleave perforin, we exposed recombinant, purified mouse perforin to purified CatL in vitro. The recombinant perforin was engineered to be full length, including the N-linked glycosylation site (Asn548) and the residues extending to the C-terminal Trp (residue 554). Following co-incubation with CatL in acid buffer (pH 5·5, approximating the acidic milieu in lysosomal granules), the remaining perforin was analysed by SDS–PAGE and Western blotting with antibodies that react with different domains of the perforin molecule (Fig. 3). When mAb PI-8, which detects an epitope mapping approximately to the centre of the perforin molecule (residues 189–360)20 was used, the perforin signal was maintained as the amount of CatL added was increased (7·2–180 nm). However, when the same samples were probed with a mAb detecting an epitope within the final 12 residues at the extreme C terminus (6G7), the perforin signal was progressively lost as the concentration of protease was increased (Fig. 3). The findings clearly indicated that CatL can cleave mouse perforin resulting in preferential loss of the extreme C terminus. The persistence of the PI-8 signal and concomitant loss of the 6G7 epitope at the C terminus strongly suggested that CatL preferentially removes the C terminus in this process. Addition of the CatL inhibitor L1 completely abrogated the processing of the perforin molecule by CatL.
Figure 3.
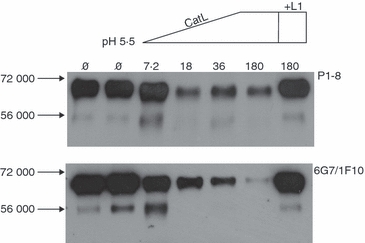
Perforin C-terminal peptide is removed following treatment with cathepsin L (CatL) in vitro. Recombinant full-length mouse perforin (180 nm) was cleaved with recombinant mouse CatL at increasing concentrations of CatL (7.2–180 nm). L1 inhibitor (10 μm) prevented the cleavage. The resultant samples were run on 8% sodium dodecyl sulphate–polyacrylamide gel electrophoresis, under reduced conditions. Western blots were probed with anti-perforin PI-8 antibody (upper panel) or the perforin C-terminus-specific antibody (6G7/IF10) (lower panel). The migration of molecular weight standards is indicated.
We also wished to determine whether, as with human NK cell lines, the CatL inhibitor L1 was capable of inhibiting perforin-dependent target cell death induced by intact CTLs. To test this possibility, CD8+ CTLs from transgenic B6 mice that express the OT-1 T-cell receptor were incubated with inhibitor and tested for their ability to kill syngeneic (EL4) tumour target cells pulsed with the cognate ovalbumin peptide SIINFEKL. As seen with the human NK cell lines above, the death of EL4 cells under these conditions was perforin-dependent because it was significantly abrogated in the absence of free calcium ions (Fig. 4a). Addition of L1 (10 μm) solely during the cytotoxicity assay produced minimal reduction of target cell death, but the inhibition was greatly augmented when the CTLs were pre-incubated in culture with the inhibitor for 24 hr before the cytotoxicity assay (Fig. 4a). This suggested that sufficient processed perforin to be permissive for cytotoxicity was accumulating in the CTL granules during the 24 hr before the assay. As with the NK cell results (above), the data also indicated that processing of newly synthesized perforin was also occurring during the time in which the killer cells were incubated with target cells. The inhibition of cell death mediated by L1 was quite profound, indeed it was comparable to that seen when E-64d was used at similar concentrations in its stead (Fig. 4b) (P values presented in Table 2 also highlight the significance of these differences).
Figure 4.
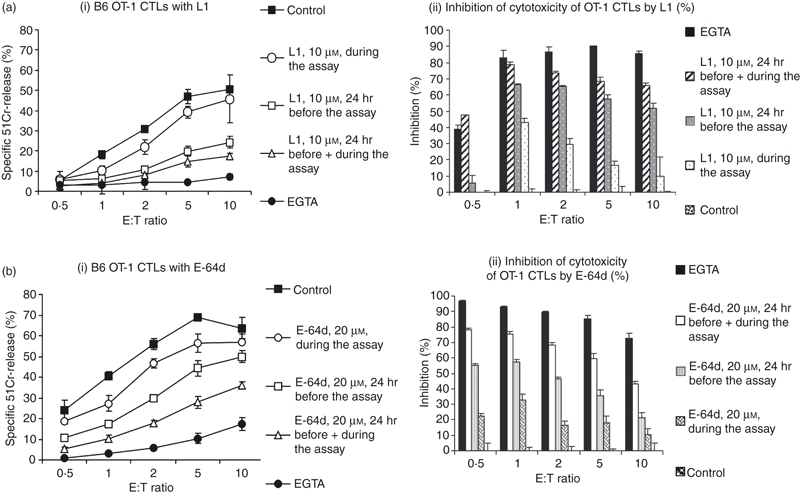
Effect of cathepsin (Cat) inhibitors on mouse cytotoxic T lymphocyte (CTL) cytotoxicity. L1 and E-64d inhibitors decrease target cell killing by wild-type OT-I transgenic CTLs. Lysis of 51Cr-labelled SIINFEKL-pulsed EL4 target cells by CTLs from B6 OT1 CTLs were pre-incubated with L1 (ai) or E-64d (bi) for the time and concentration indicated. The data represent the mean of triplicate data points ± SEM and are representative of three similar experiments. The P-values have been calculated to determine the significance of differences in the level of lysis for each treatment (Table 2). The % of inhibition of cytotoxicity of wild-type B6 OT-1CTLs by L1 (ai) or E-64d (bi) was calculated from values of percentage of specific 51Cr-release at each effector : target (E : T) ratio, or (% specific control lysis − % specific lysis sample with inhibitor) / (% specific control lysis) × 100. (aii, bii).
Table 2.
P-values for Figure 4
| Effector : target ratios | 2 : 1 | 10 : 1 |
|---|---|---|
| Inhibitor | ||
| L1, 10 μm, during the assay | 0.017 | 0.509 |
| LI, 10 μm, 24 hr before the assay | < 0.001 | < 0.001 |
| LI, 10 μm, 24 hr before + during the assay | < 0.001 | < 0.001 |
| E-64d, 20 μm, during the assay | 0.010 | 0.151 |
| E-64d, 20 μm, 24 hr before the assay | < 0.001 | 0.015 |
| E-64d, 20 μm, 24 hr before + during the assay | < 0.001 | < 0.001 |
Redundancy of perforin processing: CatL−/− mice have diminished, but not abolished perforin processing
Given the evidence presented above that CatL is capable of processing perforin, we examined directly the effect of loss of CatL on perforin processing and cytolytic activity in cytotoxic effector cells (CTLs and NK cells) derived from CatL-deficient OT1 (CatL−/−.OT1) mice. Western immunoblotting showed significant changes in the migration of perforin species detected in the NK cells from CatL−/− and CatL+/+ mice. The NK cells were incubated with buffer alone or with either L1 or E-64d before the immunoblot analysis (Fig. 5). Probing of the blots with PI-8 (left hand panel) showed that perforin levels overall were similar. In particular, although both the processed (lower band) and unprocessed (upper band) perforin were present, the relative amounts of these forms were different (Fig. 5 lane 1 versus lane 4). Most of the perforin in the CatL+/+ NK cells was of the lower processed form whereas there was an apparently equal level of processed and unprocessed forms in the CatL−/− mice (compare lanes 1 and 4). This suggested that CatL was indeed responsible for at least some perforin processing; however, an alternative protease(s) was able to partially compensate for the absence of CatL. This was made more evident when the NK cells were incubated with L1. Despite the lack of CatL, pre-incubation of the NK cells with L1 resulted in a marked increase in the unprocessed form in both the CatL−/− (compare lane 1 with lane 3), as well as in the CatL+/+ NK cells as expected (compare lane 4 with lane 6). This suggested that the alternative protease(s) was also inhibited by L1. Likewise, treatment of the NK cells with E-64d had a similar inhibitory effect on the perforin processing in the NK cells from both mice (lane 2 and 5). When the same lysates were probed with 6G7 (right hand panel), which detects the extreme C terminus, only low to negligible levels of unprocessed perforin were detected in both CatL+/+ and CatL−/− NKs (Fig. 5 lanes 7 and 10). Clearly this again suggested that despite the lack of CatL, the perforin had been processed. In both cases, the signal was enhanced when the NKs were pre-incubated with either L1 or E-64d. Again, this result was consistent with the presence of cysteine protease(s) in addition to CatL being capable of processing perforin.
Figure 5.
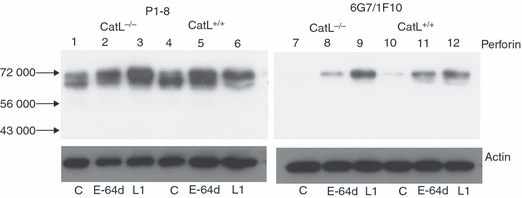
Inhibition of perforin processing by L1 and E-64d in cathepsin L (CatL) -deficient and sufficient natural killer (NK) cells. The NK cells from CatL−/− and CatL+/+ mice were either untreated or treated for 24 hr with either L1 (10 μm) or E-64d (20 μm). Ten micrograms of cell lysate was loaded as indicated, in duplicate. One half of the Western blot was probed with the PI-8 mAb (left hand panel) and the second half was probed with the anti-perforin C-terminus-specific antibody (6G7/IF10) (right hand panel). An anti-actin antibody was used as a loading control. Migration of molecular weight markers is indicated.
As the Western blot analysis indicated that the perforin was processed to some extent, we tested CTLs isolated from CatL-deficient OT-1 (CatL−/−.OT1) mice to evaluate the effect on cytolytic activity. However, we found no significant differences in cytotoxicity compared with CatL+/+ littermate control mice or syngeneic non-littermate B6 mice (Fig. 6a). Nor was there a reduction in the death of target cells exposed to NK cells from the mice (data not shown). When the CatL−/−.OT1 CTLs were pre-incubated with either L1 or E64-d, there was a further reduction in the death of SIINFEKL-pulsed target cells (Fig. 6b) consistent with the effect of the inhibitors on perforin processing (Fig. 5).
Figure 6.
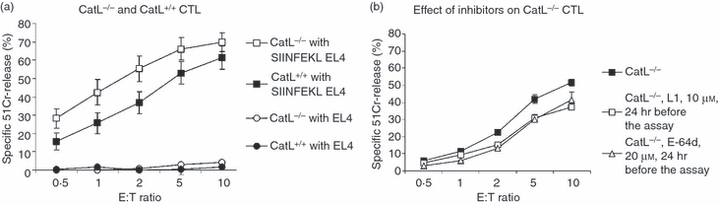
Cytotoxicity mediated by cytotoxic T lymphocytes (CTLs) from cathepsin L (CatL) -deficient and wild-type B6 CTLs. (a) Lysis of 51Cr-labelled and SIINFEKL-pulsed EL4 target cells by CTLs from OT-I transgenic mice. Data have been pooled using CTLs derived from CatL-deficient (CatL−/−) (n = 4) and wild-type (CatL+/+) (n = 4) mice. The total number of experiments performed for each genotype is indicated on the figure. Each individual experiment was performed in triplicate. (b) Effect of Cat inhibitors on lysis of target cells by CTLs from CatL-deficient (CatL−/−) mice (n = 3) Effector cells were pre-incubated with inhibitors, at the concentrations indicated, for 24 hr. Three independent experiments were performed in triplicate and the mean ± SEM was calculated.
Discussion
Although it was published more than a decade ago that perforin is activated by proteolytic cleavage in an acidic compartment,9 the proteinase responsible for this cleavage has not yet been identified. Our current data showed decreased target cell killing by human NK cells and in mouse CTLs pre-incubated with inhibitors E-64d and L1 (Fig. 1b, Fig. 4), indicating that cysteine cathepsins are involved in the process. As we detected impaired perforin processing in human NK cell lines treated with inhibitors E-64d and L1 (Fig. 1a), we reasoned that the decreased target cell killing in our experiments is a consequence of a decreased amount of active perforin. We have shown in human NK cell lines that inhibitors L1 and E-64d in micromolar concentrations completely inhibited cathepsin activity (Fig. 2a) but had no effect on granzyme A, B and H activity (Fig. 2b). We therefore excluded the possibility that the decrease in target cell killing in cells pre-treated with inhibitors was the result of impaired granzyme A, B or H processing. In NK cells several cysteine cathepsins are present: CatB, -H, -L, -S, -X and -W (NCBI GEO: GSE7764). It was already shown that CatB17, Cat C18,19 and CatW32,33 are not essential for CTL target cell killing. The processing of perforin occurs in an acidic compartment (pH 5·5), which is the optimal pH for CatL activity; at neutral pH it is inactivated within minutes.30,34 With antibodies specific for the C-terminal part of perforin we have shown that in vitro CatL preferentially cleaves away the C-terminal part of perforin in a dose-dependent manner (Fig. 3), but can also cleave perforin at other positions. In vivo, this limited proteolysis occurs in cytotoxic granules in which perforin is stored in complex with serglycin and protected from degradation.2
The use of primary CD8+ CTL from transgenic mice that express the OT-1 T-cell receptor, allowed us to examine the effect of inhibitors in a peptide- and major histocompatibility complex-restricted cellular context. The treatment of mouse CD8+ CTLs with L1 and E-64d inhibitors almost completely prevented target cell killing (Fig. 4). To verify the importance of CatL, activated CD8+ CTLs and NKs were generated from CatL-deficient mice, which allowed us to study the contribution of a single proteinase in perforin processing and consequently target cell killing. The CatL is essential for major histocompatibility complex II invariant chain degradation in thymus and in Cat L-deficient mice the number of CD4+ T cells in the thymus and periphery is reduced by 60–80%, whereas the number of CD8+ T cells is increased.35 Western blot analysis revealed a larger amount of unprocessed perforin in CatL−/− NKs than in CatL+/+ NKs, clearly indicating that CatL played a role in processing perforin. However, the results also indicated that additional proteases are also involved in the processing and hence compensate for the loss of CatL (Fig. 5). Furthermore, treatment with the inhibitors led to a marked increase in the amount of unprocessed perforin detected in both the wild-type and the CatL-deficient NKs. This indicated that in CatL-deficient NKs additional cathepsins were active and processing perforin (Fig. 5).
We did not observe any significant difference in target cell killing by CatL+/+ and CatL−/− CD8+ CTLs (Fig. 6a). Similarly, both CatL+/+ NK cells and CatL−/− NK cells were equally efficient in causing target cell lysis (data not shown). Hence, although there was an apparent decrease in the level of processed perforin detected, sufficient active perforin was remaining in the CatL−/− effector cells to cause comparable lysis of the target cells. This was not completely surprising because effector cell granules contain large stores of processed perforin, of which only a proportion is released as each target cell is ‘hit’. This mechanism enables the killer cell to kill multiple targets in succession.36,37 Although we observed a reduction in total levels of processed perforin with CatL inhibition or deficiency, still sufficient perforin remained to result in residual target cell killing.
These findings are probably also consistent with the observations that like other cathepsin-deficient mice (CatB, -C or -S), CatL-deficient mice do not display the same immune-deficient phenotype as the perforin-deficient mice.19,38,39 However, it was recently reported that asparaginyl endopeptidase (AEP)-deficient mice developed a classical syndrome resembling HLH and had reduced natural killer cell activity.40 The AEP is insensitive to E-64d or leupeptin, and therefore cannot cleave perforin directly but it cleaves the CatB, -H and -L single-chain forms into the two-chain form.41 Therefore we propose that the perforin could be cleaved by several proteases that are processed by AEP. When only CatL is deficient, redundancy in the system exists and other cathepsins cleave and activate perforin. Loss of a protease such as AEP, which has the capability to process a number of different cathepsins, could, however, have a more profound effect, similar to perforin deficiency.
In conclusion, we can say that collectively cysteine cathepsins contribute to perforin processing and consequently target cell killing by NK cells and CTLs. Although CatL contributes to perforin processing, in its absence the activity of other proteases in cytotoxic granules is sufficient and the effector functions of NK cells and CTLs are not compromised.
Acknowledgments
The authors thank Eric Vivier and Phillip Bird for NK-92 and K562 cells; Mark Smyth for the YT 5 cell line, Garnett Suck for KHYG1, Frank Carbone for EL-4, Marko Mihelic for active recombinant mouse Cat L, James Powers for granzyme B substrate, and Jose Villadangos for making the CatL−/− mice available for Australian investigators. This work was supported by the Research Agency of the Republic of Slovenia, grant P-0140 (to V.T. and B.T.) and Š.K. was supported by Slovenian human resources development and scholarship foundation, Slovenia. J.A.T. and V.R.S. are supported by a programme grant from the National Health and Medical Research Council (NHMRC; 454569) of Australia. J.A.T. received a senior fellowship (288999) from NMHRC. S.H. was supported by Deutsche Forschungsgemeinschaft (Ho 4007/1-1).
Glossary
Abbreviations:
- AEP
asparaginyl endopeptidase
- BLT
N-α-carbobenzyloxy-l-lysine thiobenzyl ester
- CTL
cytotoxic T lymphocyte
- CatL
cathepsin L
- FCS
fetal calf serum
- HLH
haemophagocytic lymphohistiocytosis
- mAb
monoclonal antibody
- NK
natural killer
- SDS–PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
Disclosure
The authors have no potential conflicts of interest.
References
- 1.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–56. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 2.Metkar SS, Wang B, Aguilar-Santelises M, Raja SM, Uhlin-Hansen L, Podack E, Trapani JA, Froelich CJ. Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity. 2002;16:417–28. doi: 10.1016/s1074-7613(02)00286-8. [DOI] [PubMed] [Google Scholar]
- 3.Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15:751–61. doi: 10.1016/s1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 4.Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, Tschopp J, Slot JW, Geuze HJ. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med. 1991;173:1099–109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth MJ, Kelly JM, Sutton VR, Davis JE, Browne KA, Sayers TJ, Trapani JA. Unlocking the secrets of cytotoxic granule proteins. J Leukoc Biol. 2001;70:18–29. [PubMed] [Google Scholar]
- 6.Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol. 2002;3:122–31. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 7.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–52. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 8.Rosado CJ, Buckle AM, Law RH, et al. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317:1548–51. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 9.Uellner R, Zvelebil MJ, Hopkins J, et al. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J. 1997;16:7287–96. doi: 10.1093/emboj/16.24.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masson D, Peters PJ, Geuze HJ, Borst J, Tschopp J. Interaction of chondroitin sulfate with perforin and granzymes of cytolytic T-cells is dependent on pH. Biochemistry. 1990;29:11229–35. doi: 10.1021/bi00503a011. [DOI] [PubMed] [Google Scholar]
- 11.Fischer A, Latour S, de Saint Basile G. Genetic defects affecting lymphocyte cytotoxicity. Curr Opin Immunol. 2007;19:348–53. doi: 10.1016/j.coi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–9. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 13.Kagi D, Ledermann B, Burki K, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–7. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 14.Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–60. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binder D, van den Broek MF, Kagi D, Bluethmann H, Fehr J, Hengartner H, Zinkernagel RM. Aplastic anemia rescued by exhaustion of cytokine-secreting CD8+ T cells in persistent infection with lymphocytic choriomeningitis virus. J Exp Med. 1998;187:1903–20. doi: 10.1084/jem.187.11.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA. Surface cathepsin B protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med. 2002;196:493–503. doi: 10.1084/jem.20011836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baran K, Ciccone A, Peters C, Yagita H, Bird PI, Villadangos JA, Trapani JA. Cytotoxic T lymphocytes from cathepsin B-deficient mice survive normally in vitro and in vivo after encountering and killing target cells. J Biol Chem. 2006;281:30485–91. doi: 10.1074/jbc.M602007200. [DOI] [PubMed] [Google Scholar]
- 18.Meade JL, Wilson EB, Holmes TD, et al. Proteolytic activation of the cytotoxic phenotype during human NK cell development. J Immunol. 2009;183:803–13. doi: 10.4049/jimmunol.0713829. [DOI] [PubMed] [Google Scholar]
- 19.Sutton VR, Waterhouse NJ, Browne KA, et al. Residual active granzyme B in cathepsin C-null lymphocytes is sufficient for perforin-dependent target cell apoptosis. J Cell Biol. 2007;176:425–33. doi: 10.1083/jcb.200609077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawasaki A, Shinkai Y, Kuwana Y, et al. Perforin, a pore-forming protein detectable by monoclonal antibodies, is a functional marker for killer cells. Int Immunol. 1990;2:677–84. doi: 10.1093/intimm/2.7.677. [DOI] [PubMed] [Google Scholar]
- 21.Voskoboinik I, Thia MC, De Bono A, et al. The functional basis for hemophagocytic lymphohistiocytosis in a patient with co-inherited missense mutations in the perforin (PFN1) gene. J Exp Med. 2004;200:811–6. doi: 10.1084/jem.20040776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mihelic M, Dobersek A, Guncar G, Turk D. Inhibitory fragment from the p41 form of invariant chain can regulate activity of cysteine cathepsins in antigen presentation. J Biol Chem. 2008;283:14453–60. doi: 10.1074/jbc.M801283200. [DOI] [PubMed] [Google Scholar]
- 23.Reinheckel T, Hagemann S, Dollwet-Mack S, et al. The lysosomal cysteine protease cathepsin L regulates keratinocyte proliferation by control of growth factor recycling. J Cell Sci. 2005;118:3387–95. doi: 10.1242/jcs.02469. [DOI] [PubMed] [Google Scholar]
- 24.Yodoi J, Teshigawara K, Nikaido T, et al. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells) J Immunol. 1985;134:1623–30. [PubMed] [Google Scholar]
- 25.Suck G, Branch DR, Smyth MJ, Miller RG, Vergidis J, Fahim S, Keating A. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol. 2005;33:1160–71. doi: 10.1016/j.exphem.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 26.Urbich C, Heeschen C, Aicher A, et al. Cathepsin L is required for endothelial progenitor cell-induced neovascularization. Nat Med. 2005;11:206–13. doi: 10.1038/nm1182. [DOI] [PubMed] [Google Scholar]
- 27.Ravanko K, Jarvinen K, Helin J, Kalkkinen N, Holtta E. Cysteine cathepsins are central contributors of invasion by cultured adenosylmethionine decarboxylase-transformed rodent fibroblasts. Cancer Res. 2004;64:8831–8. doi: 10.1158/0008-5472.CAN-03-2993. [DOI] [PubMed] [Google Scholar]
- 28.Sutton VR, Vaux DL, Trapani JA. Bcl-2 prevents apoptosis induced by perforin and granzyme B, but not that mediated by whole cytotoxic lymphocytes. J Immunol. 1997;158:5783–90. [PubMed] [Google Scholar]
- 29.Edwards KM, Kam CM, Powers JC, Trapani JA. The human cytotoxic T cell granule serine protease granzyme H has chymotrypsin-like (chymase) activity and is taken up into cytoplasmic vesicles reminiscent of granzyme B-containing endosomes. J Biol Chem. 1999;274:30468–73. doi: 10.1074/jbc.274.43.30468. [DOI] [PubMed] [Google Scholar]
- 30.Barrett AJ, Kirschke H. Cathepsin B, cathepsin H and cathepsin L. Methods Enzymol. 1981;80:535–61. doi: 10.1016/s0076-6879(81)80043-2. [DOI] [PubMed] [Google Scholar]
- 31.Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, et al. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 2003;10:881–8. doi: 10.1038/sj.cdd.4401247. [DOI] [PubMed] [Google Scholar]
- 32.Ondr JK, Pham CT. Characterization of murine cathepsin W and its role in cell-mediated cytotoxicity. J Biol Chem. 2004;279:27525–33. doi: 10.1074/jbc.M400304200. [DOI] [PubMed] [Google Scholar]
- 33.Stoeckle C, Gouttefangeas C, Hammer M, Weber E, Melms A, Tolosa E. Cathepsin W expressed exclusively in CD8+ T cells and NK cells, is secreted during target cell killing but is not essential for cytotoxicity in human CTLs. Exp Hematol. 2009;37:266–75. doi: 10.1016/j.exphem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 34.Turk B, Bieth JG, Bjork I, et al. Regulation of the activity of lysosomal cysteine proteinases by pH-induced inactivation and/or endogenous protein inhibitors, cystatins. Biol Chem Hoppe Seyler. 1995;376:225–30. doi: 10.1515/bchm3.1995.376.4.225. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa T, Roth W, Wong P, et al. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science. 1998;280:450–3. doi: 10.1126/science.280.5362.450. [DOI] [PubMed] [Google Scholar]
- 36.Trapani JA. Dual mechanisms of apoptosis induction by cytotoxic lymphocytes. Int Rev Cytol. 1998;182:111–92. doi: 10.1016/s0074-7696(08)62169-5. [DOI] [PubMed] [Google Scholar]
- 37.Bhat R, Watzl C. Serial killing of tumor cells by human natural killer cells – enhancement by therapeutic antibodies. PLoS ONE. 2007;2:e326. doi: 10.1371/journal.pone.0000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem. 2001;382:735–41. doi: 10.1515/BC.2001.089. [DOI] [PubMed] [Google Scholar]
- 39.Shi GP, Villadangos JA, Dranoff G, et al. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 1999;10:197–206. doi: 10.1016/s1074-7613(00)80020-5. [DOI] [PubMed] [Google Scholar]
- 40.Chan CB, Abe M, Hashimoto N, et al. Mice lacking asparaginyl endopeptidase develop disorders resembling hemophagocytic syndrome. Proc Natl Acad Sci U S A. 2009;106:468–73. doi: 10.1073/pnas.0809824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shirahama-Noda K, Yamamoto A, Sugihara K, Hashimoto N, Asano M, Nishimura M, Hara-Nishimura I. Biosynthetic processing of cathepsins and lysosomal degradation are abolished in asparaginyl endopeptidase-deficient mice. J Biol Chem. 2003;278:33194–9. doi: 10.1074/jbc.M302742200. [DOI] [PubMed] [Google Scholar]