

Abstract
Expression of oncogenic BCR-ABL in chronic myeloid leukemia (CML) results in increased reactive oxygen species (ROS) that in turn cause increased DNA damage, including DNA double-strand breaks (DSBs). We have previously shown increased error-prone repair of DSBs by nonhomologous end-joining (NHEJ) in CML cells. Recent reports have identified alternative NHEJ pathways that are highly error prone, prompting us to examine the role of the alternative NHEJ pathways in BCR-ABL–positive CML. Importantly, we show that key proteins in the major NHEJ pathway, Artemis and DNA ligase IV, are down-regulated, whereas DNA ligase IIIα, and the protein deleted in Werner syndrome, WRN, are up-regulated. DNA ligase IIIα and WRN form a complex that is recruited to DSBs in CML cells. Furthermore, “knockdown” of either DNA ligase IIIα or WRN leads to increased accumulation of unrepaired DSBs, demonstrating that they contribute to the repair of DSBs. These results indicate that altered DSB repair in CML cells is caused by the increased activity of an alternative NHEJ repair pathway, involving DNA ligase IIIα and WRN. We suggest that, although the repair of ROS-induced DSBs by this pathway contributes to the survival of CML cells, the resultant genomic instability drives disease progression.
Introduction
In the majority of patients with chronic myeloid leukemia (CML), fusion of parts of the BCR and ABL genes generates the BCR-ABL tyrosine kinase.1 This fusion protein interacts with a large number of signaling pathways that allow cells to proliferate in the absence of growth factors and protects them from apoptosis in the absence of external survival factors. In addition, this abnormal signaling leads to defective adherence to the extracellular matrix, and promotes survival and metastasis.2
In addition to, or concomitant with, the roles of BCR-ABL, there is emerging evidence that BCR-ABL expression initiates a cycle of genomic instability that has the potential to create other mutations.3 Specifically, the BCR-ABL kinase induces production of reactive oxygen species (ROS) that causes DNA damage, including double-strand breaks (DSBs) in CML cells.4–6 Thus, cells transformed by BCR-ABL, primary CML cells and cell lines established from CML patients, have increased endogenous DNA damage as measured by various DNA damage assays.7–9 Mutations and large deletions in CML cells result from aberrant repair of DSBs by homologous recombination (HR) and nonhomologous end-joining (NHEJ), the 2 major DSB pathways in mammalian cells.6,9,10 In particular, we have shown that CML cells respond to increasing DNA damage with enhanced DNA repair processes, including an error-prone version of NHEJ that is characterized by a high frequency of large deletions, compared with normal CD34+ hematopoietic progenitor cells.9,10
The major NHEJ pathway in human cells is initiated by binding of the Ku70/86 heterodimer to DSB, followed by the recruitment of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form the active DNA-dependent protein kinase (DNA PK).11–13 In addition to its essential kinase activity, DNA PKcs is the end-bridging factor responsible for synapsis of DNA ends. After protein-mediated end bridging, the DNA ends are subsequently ligated by DNA ligase IV in conjunction with XRCC4.14 The majority of DSBs generated by agents such as ROS and X-radiation produce DSBs that rarely have ligatable 5′P and 3′-OH termini. Therefore, the synapsed DNA ends must be processed before ligation by DNA ligase IV and XRCC4 during NHEJ. Many of these processing events involve a nuclease because repair by NHEJ frequently results in small DNA deletions (up to approximately 20 bp), resulting from resection of the DNA ends back to regions of DNA sequence microhomology.15,16 There appears to be a redundancy in the nucleolytic processing step because the Artemis nuclease that is phosphorylated and activated by DNA PK is required for processing only a subset of “complex” DNA ends.17
Another candidate nuclease is the WRN protein, which is mutated and deleted in the premature aging syndrome, Werner.18,19 WRN interacts with DNA-PKcs and the Ku70/86 heterodimer.20–23 Notably, Li and Comai showed that binding of WRN to Ku86 increases its exonuclease activity.22,23 Although WRN is a nuclease, WS cells lacking WRN expression generate extensive deletions when joining linear plasmids by NHEJ and exhibit chromosomal instability, in particular deletions.24 This, suggests that whereas WRN may participate in the limited nucleolytic processing characteristic of NHEJ, DNA ends may be exposed to degradation by an as yet unidentified nuclease in the absence of WRN.
Recently, other DNA repair proteins have been identified that can substitute for major NHEJ proteins when they are deficient or down-regulated. This “backup” repair appears to be slow, inefficient, and characterized by an increased frequency of errors generated through microhomology-mediated ligation of DNA ends.25–28 In cells with reduced DNA ligase IV activity, DNA ligase IIIα, which usually functions in single-strand break repair (SSB) and base excision repair (BER),29 was shown to participate in DSB repair.25 In cells deficient in the Ku heterodimer, poly (ADP-ribose) polymerase (PARP), an abundant DNA-binding protein that normally initiates the repair of ss breaks, was shown to play a role in repair of DSBs by NHEJ.25,30–32
Although the major NHEJ pathway is considered error prone, the increased frequency and type of repair errors at DSBs in CML suggested altered NHEJ repair activity. Therefore, we have examined the steady state expression levels of NHEJ and candidate backup repair proteins. Notably, 2 proteins from the major NHEJ pathway, Artemis and DNA ligase IV, are down-regulated. In contrast, DNA ligase IIIα, which has been implicated in backup, NHEJ, and WRN are up-regulated and associate in a novel complex in BCR-ABL–positive CML cells. Our data suggest that genomic instability in these cells arises because of the reduced activity of the major NHEJ pathway and the increased activity of an error-prone backup NHEJ pathway involving DNA ligase IIIα and WRN.
Methods
Cell lines and antibodies
Studies of human subjects were performed under University of Maryland institutional review board approval, in accordance with the Declaration of Helsinki, compliance number H25314. K562, MEG01, KU812, and Kasumi4 are BCR-ABL–positive CML cell lines, and U937 is a BCR-ABL–negative cell line (ATCC, Manassas, VA). MO7e, a BCR-ABL–negative human myeloid leukemia, and MO7e BCR-ABL (P210MO7e), stably expressing BCR-ABL, were a kind gift from Dr Van Etten (Tufts University, Boston, MA). NC3, NC10, and NC108, human lymphoblastoid lines established from normal lymphocytes, were a kind gift from Dr Gazdar (University of Texas Southwestern, Dallas, TX). Cell lines were cultured in RPMI (Cellgro, Herndon, VA) with 2 mM l-glutamine (Cellgro), antibiotics, and penicillin-streptomycin (Invitrogen, Carlsbad, CA) supplemented with fetal bovine serum (FBS; ATCC), as indicated by the ATCC. Cells were incubated at 37°C in 5% CO2 in air atmosphere.
Antibodies
The following antibodies were used in the study: Ku86 (Calbiochem, San Diego, CA), mouse monoclonal to human Ku86 at dilution 1:1000; DNA-PKcs (C-19; Santa Cruz Biotechnology, Santa Cruz, CA), goat polyclonal to C-terminal of human DNA-PKcs, at dilution 1:500; Artemis (Abcam, Cambridge, United Kingdom), goat polyclonal to Artemis at dilution 1:1000; DNA ligase IV (Acris Novus Biologicals, Littleton, CO), rabbit polyclonal antibody to human DNA ligase IV, at dilution 1:1000; XRCC4 (Gene Tex, San Antonio, TX), mouse polyclonal to human XRCC4, at dilution 1:1000; DNA ligase IIIα (Gene Tex), mouse monoclonal to human DNA ligase IIIα, at dilution 1:1000; XRCC1 (Gene Tex), rabbit polyclonal antiserum to human XRCC1, at dilution 1:5000; WRN (Abcam), rabbit polyclonal to Werner syndrome helicase WRN, at dilution 1:500; PARP (BD Pharmingen, San Diego, CA), mouse monoclonal to human poly (ADP-ribose) polymerase 1 (PARP1), at dilution 1:1000; anti-myc (Invitrogen, Carlsbad, CA) at dilution 1:1000; actin (Abcam), mouse monoclonal to beta actin, at dilution 1:5000; HRP rabbit anti–goat IgG (Chemicon, Temecula, CA); HRP donkey anti–goat IgG (Promega, Madison, WI); HRP goat antirabbit (sc-2004); HRP goat antimouse (BioRad, Hercules, CA); anti–phospho-histone H2AX (Upstate, Lake Placid, NY); anti–mouse IgG FITC conjugate (Sigma Genosys, The Woodlands, TX); and anti–mouse IgG TRITC conjugate (Sigma Genosys).
Western blotting
Nuclear extracts were prepared using CelLytic NuClear Extraction Kit (N-XTRACT, Sigma-Aldrich, St Louis, MO) using a nondetergent protocol. Cells were lysed in 1× hypotonic lysis buffer from a 10× lysis buffer stock, containing 100 mM HEPES, pH 7.9, 15 mM MgCl2, 100 mM KCl in the presence of 0.1 M DTT and protease inhibitor cocktail containing 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF), pepstatin A, bestatin, leupeptin, aprotinin, and trans-epoxysuccinyl-l-leucyl-amido (4-guanidino)-butane (E-64). After centrifugation of the disrupted cells in suspension for 20 minutes at 10 000g to 11 000g, the supernatant (cytoplasmic extract) was transferred to a fresh tube. For nuclear extract, crude nuclear pellets were resuspended in 1× extraction buffer containing 20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% (vol/vol) glycerol. To 147 μL of 1× extraction buffer, 1.5 μL of the prepared 0.1 M DTT solution and 1.5 μL of the protease inhibitor cocktail were added, shaken gently for 30 minutes, and centrifuged for 5 minutes at 20 000g. The supernatant was transferred to a clean, chilled tube, snap-frozen in aliquots with liquid nitrogen, and stored at − 70°C. Nuclear extract (50 μg) was boiled in 2× Laemmli sample buffer (BioRad) for 10 minutes. The proteins were separated on either 7.5% or 4% to 15% SDS–polyacrylamide gel electrophoresis (PAGE) and transferred on PVDF membrane. After blocking, membranes were probed with first antibody and secondary antibody as indicated above. Enhanced chemiluminescence (ECL; Amersham Biosciences, Arlington Heights, IL) was used for detection of the proteins.
Immunoprecipitation
Protein A agarose (Upstate) was washed twice with PBS and restored to a 50% slurry with PBS. The nuclear lysate (1 mg/mL), using CelLytic NuClear Extraction Kit (N-XTRACT), was precleared adding 100 μL protein A bead slurry per 1 mg cell lysate and incubating at 4°C for 10 minutes. Protein A beads were removed by centrifugation at 16 000g at 4°C for 5 seconds. The supernatant was transferred to a fresh centrifuge tube. WRN or DNA ligase IIIα antibodies were added to cell lysate and gently mixed overnight at 4°C on a rocker. The immunocomplex was captured by adding 100 μL protein A beads slurry and gently rocking for 2 hours at 4°C. The agarose beads were collected by pulse centrifugation for 5 seconds at 16 000g. The supernatant fraction was discarded and the beads were washed 3 times with 1 mL ice-cold PBS. The agarose beads were resuspended in 60 μL 2× Laemmli sample buffer, mixed gently, and boiled for 5 minutes to dissociate the immunocomplex from the beads. The beads were collected by centrifugation and SDS-PAGE was performed with the supernatant fraction.
K562 and NC10 DRneo transfectants
K562 and NC10 cells were stably transfected with DRneo plasmid in the presence of 350 μg/mL hygromycin B (Cellgro). Stable K562 and NC10 cells containing DRneo plasmid were engineered with an inducible DSB, ISce-1.33 DSBs were induced in stable transfectants by transient transfection of the unique ISce-1 restriction enzyme construct (1 μg/106 cells) using Amaxa Nucleofector Kit V (Gaithersburg, MD). After induction of the ISce-1 DSB, fluorescence in situ hybridization (FISH) coimmunostaining was performed as previously described34 to colocalize γH2AX, Ku86, DNA ligase IV, DNA ligase IIIα, and WRN proteins to the ISce-1 site of DRneo. Briefly, DRneo was labeled with biotin and detected with FITC-conjugated avidin (green signal). Thereafter, cells were immunostained for these proteins and detected indirectly with TRITC-conjugated IgG.
Fluorescence in situ hybridization
FISH was performed as previously described.34 Probes were prepared by nick translation using Bio-11-dUTP (Enzo Diagnostics, New York, NY) and digoxygenin-11-dUTP (Boehringer Mannheim, Mannheim, Germany) or using directly labeled nucleotides (Invitrogen or Vysis, Downers Grove, IL). The hybridization mixture contained the labeled DRneo probe (5.0-10 μg/mL), competitor DNA (unlabeled, sonicated, total human genomic DNA, 0.05-0.1 mg/mL), and the Cot1 fraction of DNA (0.05-0.1 mg/mL). Probes were hybridized to cytospin preparations of cells overnight at 37°C in a humidity chamber, and subsequently slides were washed according to standard protocols. Slides were preincubated in blocking buffer (4× SSC containing 2% bovine serum albumin [BSA] and 0.2% Triton X-100) for 30 minutes, and then incubated for up to 1 hour with FITC-conjugated antibody. After washing and blocking, slides were incubated for 1 hour with primary antibody diluted in blocking buffer at 37°C in a humidity chamber. Slides were then washed 3 times for 4 minutes each in 4× SSC containing 2% Tween 20 and incubated with secondary antibody conjugated to TRITC fluorochrome. Secondary antibodies (Sigma Genosys) were used at 1:100 dilution. Interphase nuclei were counterstained with 4,6 diamino-2-phenylindole-dihydrochloride (DAPI).
Immunocytochemistry
Cells were harvested and washed in PBS and centrifuged at 800g for 5 minutes. Cells were cytospun and fixed in 2% paraformaldehyde (P-6148; Sigma Genosys) for 10 minutes at room temperature and washed 3 times for 3 minutes each. Cells were permeabilized in a Triton X-100 solution for 5 minutes at room temperature. The slides were blocked in a humidified chamber at 37°C for 1 hour with a solution of 10% FBS in PBS. After washing, slides were incubated for 1 hour with anti–phospho-histone H2AX antibody (1:100; Upstate) at 37°C in humidified chamber. Secondary antibody was anti–mouse IgG FITC conjugate (Sigma Genosys) at the dilution 1:100. After washing and drying the slide, DAPI (Vector Laboratories, Burlingame, CA) was used for counterstaining.
Imaging microscopic analysis
The slides were examined using Nikon fluorescent microscope Eclipse 80i (Melville, NY) with DAPI/FITC/TRITC triple-pass filters. The magnification of the objective lenses were 60×/1.40 NA oil objective. Images were captured using a CCD (charge-coupled device) camera and software (Smart capture VP; Nikon); and data were analyzed.
siRNA
siRNA oligonucleotides were purchased from Dharmacon RNA Technologies (Chicago, IL). siRNA oligonucleotides used for WRN were 5′- AAGGCAUGUGUUCGGAAGAUU-3′ and 3′-UUUUCCGUACACAAGCCUUCU-5′. For DNA ligase IIIα, a target plus SMART pool from Dharmacon RNA Technologies were used. These oligonucleotides were transiently transfected into K562 and NC10 cell lines using Cell Line Nucleofector Kit V (VCA-1003) in Nucleofector II Amaxa biosystems.
For transfection, 10 μL of 20 μM siRNA was used for 106 cells. At the time optimal for silencing of these genes (72 hours after transfection), cells were harvested either to proceed Western blotting or DSB in vivo plasmid misrepair assay. Mock transfection and siRNA nontarget were used as controls.
In vivo plasmid misrepair assay
EcoR1–linearized pUC18 plasmids (Fermenta, Hanover, MDs) were transfected in siRNA nontarget, siRNA-silenced cells and Artemis-overexpressed cells using Amaxa Nucleofector Kit V. Plasmid DNA was extracted and used to transform E coli strain DH5α (Invitrogen), and the transformation mixture was plated onto agar plates containing X-gal and IPTG. Colonies were analyzed by counting the total number of white (misrepaired colonies) and blue (properly repaired colonies). Misrepair frequency were calculated by the number of white colonies as a percentage of total blue and white colonies. White colonies were characterized by polymerase chain reaction (PCR) amplification using primers 5′-CGGCATCAGAGCAGATTGTA-3′ and 5′-TGGATAACCGTATTACCGCC-3′ (Sigma Genosys) running 35 cycles at 60°C and detected on 1% agarose gel. The same primers were used for sequencing of repaired plasmids. DNA sequences at the deletion breakpoints were analyzed for regions of microhomology by comparison with wild-type pUC18 DNA sequences (BLAST search35). Microhomologous DNA sequences are defined as small regions of nucleotide sequence identity between the broken DNA ends ranging from 1 to several base pairs in length, according to Paull and Gellert.36
Results
Reduced steady state levels of Artemis and DNA ligase IV in BCR-ABL–positive CML cell lines
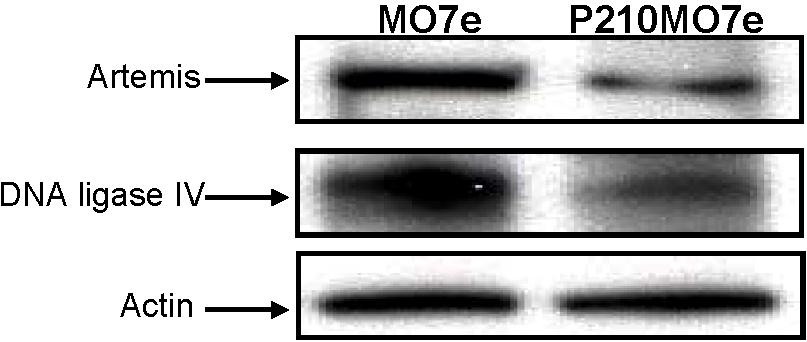
BCR-ABL–positive CML cells produce increased ROS4 (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article) that result in an increase in DSBs.3,5,6 Notably, the repair of DSBs is clearly abnormal in BCR-ABL–positive CML cells, although the mechanism by which this altered repair occurs is unknown.3,5,6 To examine the steady state levels of the key NHEJ proteins, including Ku70/86, DNA-PKcs, DNA ligase IV/XRCC4, and Artemis, Western blotting analysis was performed in BCR-ABL–positive CML cells and compared with controls. We find that the CML cell lines (K562, MEG01, KU812, and Kasumi4) have significantly reduced steady state levels (4- to 7-fold) of Artemis (Figure 1Ai), compared with 3 Epstein-Barr virus (EBV)–transformed B-cell lines (NC3, NC10, and NC108), established from 3 different healthy individuals, and a BCR-ABL–negative leukemia cell line MO7e (Figure 1Aii). In addition, DNA ligase IV levels were 2- to 3-fold lower in CML cell lines, compared with appropriate controls (Figure 1B). Importantly, BCR-ABL–positive P210MO7e cells, compared with isogenic MO7e controls, confirm that the expression of BCR-ABL is associated with decreased levels of DNA ligase IV and Artemis (Figure S2). Treatment of CML cells with the proteosome inhibitor MG132 had no effect on the levels of Artemis and DNA ligase IV, indicating that the reduced levels of these proteins in CML cells is not due to proteosomal degradation (data not shown).
Figure 1.

BCR-ABL–positive CML cell lines show down-regulation of major NHEJ proteins, Artemis, and DNA ligase IV, and up-regulation of alternative NHEJ proteins, DNA ligase IIIα, and WRN. (Ai) Western blotting using nuclear extracts from 2 BCR-ABL–positive CML cell lines (K562 and MEG01), and 3 EBV-transformed B-cell lines, established from healthy individuals (NC3, NC10, and NC108), and (Aii) 1 BCR-ABL–negative CML cell line (MO7e). Artemis shows down-regulation (4- to 7-fold) in BCR-ABL–positive CML cell lines. Either actin or Ku86 was used as loading control. (B) Western blotting using nuclear extracts from K562 shows 2- to 3-fold decrease in DNA ligase IV expression, compared with EBV-transformed B-cell lines (NC3 and NC10). Ku86 was used as loading control. (C) Western blotting using nuclear extracts from 2 BCR-ABL–positive CML cell lines (K562 and MEG01) and 3-EBV transformed B-cell lines (NC3, NC10, and NC108). CML cell lines show up-regulation of DNA ligase IIIα (3- to 6-fold). PARP1 expression is unchanged. Werner syndrome protein, WRN, shows up-regulation (4- to 7-fold) in CML cell lines. Actin was used as loading control. (D) BCR-ABL–negative MO7e shows down-regulation of DNA ligase IIIα and WRN compared with BCR-ABL–positive P210MO7e and K562. Western blotting using nuclear extracts from 2 BCR-ABL–positive cell lines (K562 and P210MO7e) and BCR-ABL–negative cell lines (U937 and MO7e). Actin was used as loading control. (E) Primary CML bone marrow mononuclear cells show down-regulation of DNA ligase IIIα and WRN. Western blotting using nuclear extracts from CML cell line (K562) and 2 CML patients (CML1, CML2) at diagnosis. Patient CML1 shows decreased DNA ligase IIIα and WRN expression with decreased levels of BCR-ABL positivity by FISH. Patient CML2 shows increased expression of DNA ligase IIIα and WRN, with 100% BCR-ABL positivity. BCR-ABL–positive CML cell line K562 was used as a positive control. Ku86 and actin were used as loading controls.
Up-regulation of DNA ligase IIIα and WRN in BCR-ABL–positive CML cell lines
Recent studies have identified an alternative or backup NHEJ repair, involving DNA repair proteins, such as, DNA ligase IIIα and PARP1, that is characterized by increased repair errors.25,32,37 To determine whether this backup NHEJ may contribute to altered DSB repair in CML,38 we initially examined the steady state levels of candidate backup repair proteins, such as DNA ligase IIIα, XRCC1, and PARP1, in BCR-ABL–positive CML cell lines (K562, MEG01, KU812, and Kasumi4). The steady state levels of DNA ligase IIIα were significantly elevated (3- to 6-fold) in BCR-ABL–positive cell lines, compared with control EBV-transformed B-cell lines (NC3, NC10, and NC108) (Figure 1C). DNA ligase IIIα was also elevated in BCR-ABL–positive CML, compared with BCR-ABL–negative leukemia cell lines, U937 and MO7e (Figure 1D). Furthermore, the steady state level of DNA ligase IIIα is increased in MO7e cells stably expressing BCR-ABL (P210MO7e), compared with isogenic MO7e cells (Figure 1D). In contrast, there were no significant changes in the levels of the single-strand break repair proteins, PARP1 and XRCC1, the partner protein of DNA ligase IIIα (Figure 1C).
Next, we examined expression levels of WRN protein, a RecQ helicase with 3′-5′ exonuclease activity that has been implicated in NHEJ repair.39–42 Intriguingly, the steady state levels of WRN protein were also elevated (4- to 7-fold) in CML cell lines (K562, MEG01, KU812, Kasumi4) and P210MO7e, compared with control lymphoblastoid cell lines (NC3, NC10, and NC108) and the BCR-ABL–negative leukemia cell lines, U937 and MO7e (Figure 1C,D).
To provide additional evidence for the relationship between DNA ligase IIIα and WRN and expression of the BCR-ABL, we examined the levels of these proteins in bone marrow mononuclear samples (BM MNCs) from patients (n = 4) at diagnosis, with different levels of BCR-ABL (Table S1). Nuclear extracts from patient cells demonstrate that the steady state levels of WRN and DNA ligase IIIα are reduced in patient sample (CML1) where BCR-ABL levels (60%) are significantly decreased, as assessed by fluorescence in situ hybridization (FISH) (Figure 1E; Table S1). In contrast, the level of the Ku86 subunit does not change significantly (Figure 1E). These data, together with those in Figure 1D, suggest that the increased steady state levels of DNA ligase IIIα and WRN may either be a direct effect of BCR-ABL or a consequence of the increased levels of ROS and DNA damage caused by BCR-ABL expression.
WRN and DNA ligase IIIα colocalize with DRneo at ISce-1–induced DSBs
To investigate the role of WRN and DNA ligase IIIα in DSB repair in BCR-ABL–positive CML, K562 and control NC10 cells were stably transfected with the DRneo plasmid that contains the ISce-1 restriction site that is not present in the mammalian genome (K562DRneo and NC10DRneo, Figure 2A).43 Subsequent transfection of these cells with expression constructs for ISce-1 endonuclease specifically induces a DSB into the genome of these cells.43 A combined fluorescence in situ hybridization (FISH) immunostaining technique was used to determine whether DNA ligase IIIα and WRN localize to the ISce-1–induced DSBs.34,44 The efficiency of DSB induction, after transfection of the ISce-1 endonuclease, was determined by the nuclear colocalization of γH2AX, a key marker for DSBs, and the DRneo signal. Two hundred nuclei were analyzed from each of at least 3 experiments. Approximately 12% of K562DRneo (Figures 2B, S3B) and 8% of NC10DRneo (Figure S3A,B) cells had induced DSBs, as shown by colocalization of γ-H2AX and DRneo. In CML (Figures 2B, S3B), 8% of cells show colocalization of Ku86 with DRneo versus 6% in NC10 cells, following ISce-1 expression. In contrast, the colocalization of DNA ligase IV with DRneo (Figures 2B, S3B) was significantly lower in CML cells (1%) compared with NC10 cells (4%). More strikingly, although we had difficulty detecting colocalization of either DNA ligase IIIα or WRN with DRneo in NC10 cells (Figure S3A-B), DNA ligase IIIα and WRN (Figures 2B, S3B) did colocalize with DRneo in K562 cells at a frequency of approximately 2%. Because the antibodies have different affinities for their target protein, it is not possible to compare the colocalization frequencies obtained with different antibodies. Nonetheless, this approach does reveal quantitative differences in the colocalization of a specific protein in different cell lines and, importantly, showed that DNA ligase IIIα and WRN are specifically recruited to DSBs in CML cells, providing evidence that these proteins are participants in the altered DSB repair in these cells.
Figure 2.

PCR and FISH localization of the DRneo probe in K562DRneo and NC10DRneo cells. (Ai) Detection of integrated DRneo in K562 and NC10 cell clones stably transfected with DRneo (K562DRneo and NC10DRneo, respectively) by PCR. A vertical black line has been inserted to indicate a repositioned gel lane. (Aii) The DRneo construct labeled with spectrum red was used for FISH analysis of metaphase spreads prepared from K562DRneo cells. The DRneo was integrated at a telomeric position on the chromosomes as shown in DAPI-stained (left, red arrow) and G-banded (middle, red arrow) images. Two-color FISH was used to localize DRneo to chromosome 4 in K562DRneo cells (right image). Chromosomes from 2 different metaphase cells are shown with yellow signal indicating chromosome 4 alpha satellite probe, localized to the centromere (black arrow) and red signal localizing the DRneo probe to the telomeric region of the chromosome (red arrow). (B) Colocalization of DRneo and γH2AX, Ku86, DNA ligase IV, DNA ligase IIIα, and WRN in K562DRneo cells by FISH. Images show colocalization of these proteins (TRITC, red signal) with DRneo (FITC, green signal) in K562DRneo cells transfected with ISce-1. Nuclei are stained with DAPI (blue). Right-hand panels are merged images of FITC and TRITC showing colocalization.
DNA ligase IIIα immunoprecipitates with WRN
To determine whether DNA ligase IIIα and WRN proteins associate in a complex, we performed immunoprecipitation experiments with WRN antibody. Notably, significantly higher amounts of DNA ligase IIIα were specifically coimmunoprecipitated from CML extracts compared with extracts from comparable control cell lines (Figure 3A). Similar results were obtained in reciprocal experiments using the DNA ligase IIIα antibody (Figure 3B). These data provide evidence for a novel complex containing WRN and DNA ligase IIIα that is present at higher levels in CML cells, compared with control cell lines. This presumably reflects the increased levels of WRN and DNA ligase IIIα in CML cells.
Figure 3.

DNA ligase IIIα immunoprecipitates with WRN. Nuclear extracts from CML cell line K562 and EBV-transformed B-cell line NC10 were used to immunoprecipitate (A) DNA ligase IIIα, using WRN antibody, followed by Western blotting for DNA ligase IIIα (top panel) and WRN (bottom panel). (B) Reciprocal immunoprecipitation of WRN, using DNA ligase IIIα antibody, followed by Western blotting for WRN (top panel) and DNA ligase IIIα (bottom panel). Western blotting was also performed for negative control nonspecific antibodies.
siRNA down-regulation of WRN and DNA ligase IIIα results in increased DSBs
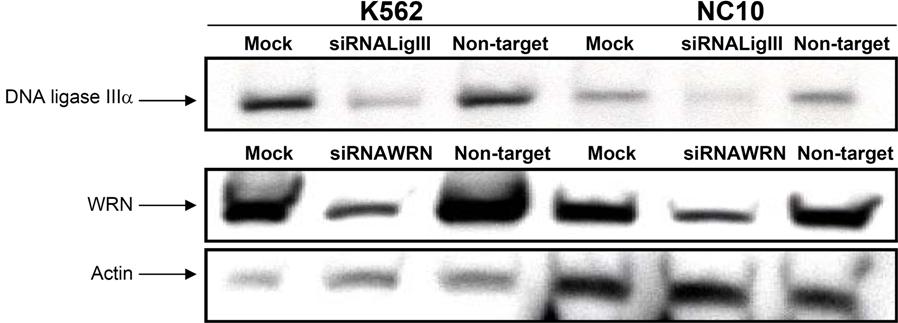
To demonstrate that WRN and DNA ligase IIIα play a role in repairing DSBs in BCR-ABL–positive CML, we examined whether their down-regulation results in an increased level of unrepaired DSBs. After siRNA knockdown of either WRN (approximately 90% reduction at 72 hours) or DNA ligase IIIα (approximately 75% reduction at 72 hours) in CML cells (Figure 4A), there was a significant increase (P < .01) in the number of γH2AX foci (Figure 4B,C; Table S2). Together, these results suggest that both WRN and DNA ligase IIIα play a role in repair of endogenously generated DSBs in CML cells.
Figure 4.

siRNA down-regulation of WRN and DNA ligase IIIα in CML cell line. (A) Nuclear extracts were prepared at 4 different time points (0h, 24h, 48h, and 72h) after siRNA treatment of cells. Western blotting was performed using WRN antibody (90% knockdown; top panel) and DNA ligase IIIα antibody (75% knockdown; middle panel). siRNA using nontarget oligonucleotides were used as controls. Ku86 was used as loading control (bottom panel). (B) Bar graph showing the percentage of K562 cells with more than 5 γH2AX foci examined in siRNA nontarget, siRNA knockdown of WRN and DNA ligase IIIα cells with or without irradiation (IR; 2.5 Gy). Black bars indicate no IR and gray bars are with IR treatment. Two hundred nuclei were examined from each of 3 different experiments. Values significantly different are marked with an asterisk (P < .01 by Student t test). Error bars reflect the standard error of the mean. (C) Images of 3 different cells showing γH2AX foci (FITC, green signal) in K562 following siRNA nontarget (left panels), siRNA down-regulation of WRN (middle panels), and DNA ligase IIIα (right panels), with and without irradiation.
Next, we examined the role of WRN and DNA ligase IIIα in the repair of DSBs generated by exogenous agents in CML cells. The number of γH2AX foci was determined in K562 cells 6 hours after exposure to 2.5 Gy γ-radiation. There was a marked defect in the repair of DSBs induced by ionizing radiation in cells with reduced levels of DNA ligase IIIα (P < .01; Figure 4B,C; Table S2). In contrast, ionizing radiation in cells with low levels of WRN did not result in a significant increase in the number of γH2AX foci (Figure 4B,C; Table S2), compared with nontarget controls, but the foci were much larger and were similar to those remaining in DNA ligase IIIα (knockdown cells).
Finally, to determine whether backup NHEJ plays a role in control lymphoblastoid cells, established from healthy individuals, we examined the frequency of DSB after siRNA knockdown of DNA ligase IIIα or WRN in NC10 cells (Figure S4). As expected, NC10 cells had a lower number of spontaneous DSBs, as measured by γH2AX foci (1.5%; Table S3), compared with K562 cells (5%; Table S2). In response to 2.5 Gy irradiation, the frequency of γH2AX foci in NC10 cells increased to 5% (Table S3), whereas 32% of K562 cells had more than 5 γH2AX foci (Table S2). Knockdown of DNA ligase IIIα or WRN in NC10 control cells increased the frequency of spontaneous DSBs to 3% and, radiation-induced DSBs to 8% (Table S3). In contrast, knockdown of DNA ligase IIIα or WRN in K562 cells resulted in a much higher frequency of γH2AX foci in unirradiated cells (17%-18%) and in irradiated cells (38%-56%; Table S2).
siRNA down-regulation of WRN and DNA ligase IIIα decreases the end-joining efficiency in CML cells
To more directly define the role of WRN and DNA ligase IIIα in the repair of DSBs in CML, an in vivo plasmid end-joining assay was performed. After knockdown of either WRN or DNA ligase IIIα in K562 cells, a pUC18 plasmid containing a DSB within LacZα was transfected into K562 cells. Circularized (repaired) plasmid DNA was extracted from the K562 cells and used to transform E coli, as previously reported.10 Reducing the levels of either DNA ligase IIIα (Figure 5A; Table S4) or WRN (Figure 6A; Table S4) results in a 2- to 3-fold reduction in the number of colonies, indicating a decrease in end-joining efficiency in the BCR-ABL–positive CML cells (P < .01). Analysis of the colonies from the plasmid reactivation assays for their frequency of repair errors, that is, white (incorrectly repaired) versus total colonies (blue [correctly repaired] + white)10, revealed that knockdown of DNA ligase IIIα results in a significant increase (P < .01) in the frequency of misrepaired colonies (Figure 5B; Table S4). There was no significant change in the percentage of misrepair events that resulted in large deletions, defined as more than 20 bp (Figure 5C,D; Table S4). In contrast, knockdown of WRN did not increase the frequency of misrepaired colonies (Figure 6B; Table S4), but there was a significant (P < .01) increase (approximately 3-fold) in the percentage of colonies with large deletions (Figure 6C,D; Table S4).
Figure 5.

Down-regulation of DNA ligase IIIα decreases the end-joining efficiency and repair using DNA sequence microhomologies, but increases the misrepair frequency and has no effect on the percentage of large deletions. An in vivo LacZα plasmid reactivation assay was used to measure end joining in siRNA DNA ligase IIIα down-regulated cells and compared with that using siRNA controls. (A) Relative percentage of colonies, indicating the efficiency of end joining. (B) The percentage of misrepair, that is, the number of white colonies as a percentage of total colonies (blue + white). (C) Graph of percentage of large deletions, defined as more than 20 bp. (D) Agarose gel showing PCR products of repaired colonies in siRNA nontarget controls and siRNA knockdown of DNA ligase IIIα. (E) The percentage of plasmids repaired using DNA sequence microhomologies of 1 to 6 bp. Fifteen plasmids were sequenced. Values significantly different are marked with an asterisk (Student t test; P < .01 for panels A and B and P < .001 for panel E). Error bars reflect the standard error of the mean.
Figure 6.

Down-regulation of WRN decreases the efficiency of end joining and repair using DNA sequence microhomologies but increases large deletions in misrepaired plasmids. An in vivo LacZα plasmid reactivation assay was used to measure end joining in siRNA down-regulated WRN K562 cells compared with that using siRNA controls. (A) Relative percentage of colonies indicating the efficiency of end joining. (B) The percentage of misrepair, that is, the percentage of the white colonies as a percentage of total colonies (blue + white). (C) The percentage of large deletions, defined as more than 20 bp. (D) Agarose gel showing PCR products of repaired colonies. (E) The percentage of plasmids repaired using DNA sequence microhomologies of 1 to 6 bp. Fifteen plasmids were sequenced. Values significantly different are marked with an asterisk (Student t test; P < .01 for panel A and P < .001 for panels C and E). Error bars reflect the standard error of the mean.
To determine whether knockdown of either DNA ligase IIIα or WRN leads to differences in repair using DNA sequence microhomologies, we sequenced the breakpoint junctions of 15 repaired plasmids from each of the LacZα reactivation experiments. The majority (80%) of plasmids in K562 cells are repaired using DNA microhomologies of 1 to 6 bp. In contrast, plasmids from cells with reduced levels of either DNA ligase IIIα (P < .001; Figure 5E) or WRN (P < .001; Figure 6E) had a reduction in the overall percentage of microhomologies and these constituted 1 to 3 bp in length (DNA ligase IIIα, 25%; WRN, 40%) at the breakpoint junctions in repaired plasmids (Table 1 and 2; Table S4). We next determined that siRNA knockdown of either WRN or DNA ligase IIIα in CML cells results in a small but reproducible decrease in viability as measured by trypan blue exclusion (Figure S5).
Table 1.
Junction sequences and microhomology for NC10 and K562 clones
| Bacterial clone | Sequence at the junction | Deleted region | Bp deleted, no. | *Microhomology sequences |
|---|---|---|---|---|
| NC10 | ||||
| siRNA Control | ||||
| No. 1 | CGAGCTCGAA/AATCATGGTC | 452-458 | 5 | AA |
| No. 2 | CGAGCTCGAA/TCGTAATCAT | 452-454 | 1 | none |
| No. 3 | AAACGACGGC/AATTCGTAAT | 391-451 | 59 | none |
| No. 4 | AACGACGGCC/ACACAACATA | 392-510 | 117 | none |
| No. 5 | CGACGTTGTA/CCACACAACA | 381-508 | 126 | none |
| siRNA WRN | ||||
| No. 1 | CCCGGGTACC/TGGTCATAGC | 443-463 | 19 | none |
| No. 2 | CTGCGCAACT/TGCCCGCTTT | 264-596 | 331 | T |
| No. 3 | TGCGTAAGGA/AAAGCCTGGG | 219-541 | 321 | A |
| No. 4 | ACCGAGCTCG/AATCATGGTC | 450-458 | 7 | none |
| No. 5 | CTCTAGAGGA/CTTTCCAGTC | 431-602 | 170 | none |
| siRNA DNA ligase III α | ||||
| No. 1 | ACCGAGCTCG/AATTCGTAAT | 450-455 | 4 | none |
| No. 2 | CCGAGCTCGA/TTCCTGTGTG | 451-476 | 24 | none |
| No. 3 | CCGAGCTCGA/CGAGCCGGAA | 451-520 | 68 | CGA |
| No. 4 | GCTCGAATTC/AATCATGGTC | 455-458 | 2 | none |
| No. 5 | TACCGAGCTC/GGTCATAGCT | 449-464 | 14 | none |
| K562 | ||||
| siRNA Control | ||||
| No. 1 | CGACGGCCAG/AGCCTGGGGT | 394-543 | 148 | AG |
| No. 2 | CATGCCTGCA/CAATTCCACAC | 416-503 | 86 | CA |
| No. 3 | GCGCAA(G)TGT/AAGTGTAAAG | 266-535 | 268 | AAGTGT(C at position 263 is mutated to G) |
| No. 4 | GTGCTGCAAG/TGC(A)AGCTGC | 335-625 | 289 | TGCAAG(C at position 628 is mutated to A) |
| No. 5 | CAAGCTTGCA/AGCCTGGGGT | 408-543 | 134 | A |
| siRNA WRN | ||||
| No. 1 | GACTCTAGAG/GTTGCGCTCA | 429-585 | 155 | G |
| No. 2 | CAGGCTGCGC/GAGTGAGCT | 259-560 | 300 | none |
| No. 3 | AAAGGGGGAT/CTAACTCACA | 324-567 | 242 | none |
| No. 4 | GGGGATGTGC/GAAGCATAA | 328-527 | 198 | none |
| No. 5 | CCCGGGTACC/CCCGCTTTCC | 443-598 | 154 | CC |
| siRNA DNA ligase III α | ||||
| No. 1 | GTAAAACGACGGC/TGTAAAG | 391-538 | 146 | none |
| No. 2 | ATGCCTGCAG/CGAGCCGGAA | 416-520 | 103 | none |
| No. 3 | ACCGAGCTCG/(C)GTCATAGCT | 450-464 | 13 | CG(G at position 464 is mutated to C) |
| No. 4 | GCTCGAATTC/TTCCTGTGTG | 455-476 | 20 | TCC |
| No. 5 | CGAGCTCGA/TTGTAATCAT | 452 | 1 | none |
| Over-expression of Artemis | ||||
| No. 1 | TACGCCAGCT/TCGTAATCAT | 310-454 | 143 | T |
| No. 2 | GGCCAGTGCC/TATCCGCTCA | 398-493 | 94 | none |
| No. 3 | GTCGACTCTA/TATCCGCTCA | 426-493 | 66 | TA |
| No. 4 | GAGCTCGAAT/TCCTGTGTGA | 453-477 | 23 | T |
| No. 5 | CGAGCTCGAA/TGGTCATAGC | 452-463 | 10 | none |
Underlined bolded sequences indicate microhomologies at the breakpoint junctions.
Microhomology is defined as one or more identical DNA sequences at the break point junctions. The mutated nucleotides are depicted in parentheses.
Table 2.
Puc18 sequence
| Puc18 sequence | |
|---|---|
| 1 | tcgcgcgttt cggtgatgac ggtgaaaacc tctgacacat gcagctcccg gagacggtca |
| 61 | cagcttgtct gtaagcggat gccgggagca gacaagcccg tcagggcgcg tcagcgggtg |
| 121 | ttggcgggtg tcggggctgg cttaactatg cggcatcaga gcagattgta ctgagagtgc |
| 181 | accatatgcg gtgtgaaata ccgcacagat gcgtaaggag aaaataccgc atcaggcgcc |
| 241 | attcgccatt caggctgcgc aactgttggg aagggcgatc ggtgcgggcc tcttcgctat |
| 301 | tacgccagct ggcgaaaggg ggatgtgctg caaggcgatt aagttgggta acgccagggt |
| 361 | tttcccagtc acgacgttgt aaaacgacgg ccagtgccaa gcttgcatgc ctgcaggtcg |
| 421 | actctagagg atccccgggt accgagctcg aattcgtaat catggtcata gctgtttcct |
| 481 | gtgtgaaatt gttatccgct cacaattcca cacaacatac gagccggaag cataaagtgt |
| 541 | aaagcctggg gtgcctaatg agtgagctaa ctcacattaa ttgcgttgcg ctcactgccc |
| 601 | gctttccagt cgggaaacct gtcgtgccag ctgcattaat gaatcggcca acgcgcgggg |
| 661 | agaggcggtt tgcgtattgg gcgctcttcc gcttcctcgc tcactgactc gctgcgctcg |
| 721 | gtcgttcggc tgcggcgagc ggtatcagct cactcaaagg cggtaatacg gttatccaca |
Forward primer: cggcatcaga gcagattgta; reverse primer: gg cggtaatacg gttatcca; EcoR1 Site: g aattcg.
Finally, to determine the contribution of the backup NHEJ pathway in control lymphoblastoid cells, we examined DSB repair after siRNA knockdown of DNA ligase IIIα or WRN in NC10 cells (Figure S4). As expected, untreated NC10 cells have a very low frequency of misrepair (2.2%; Table S5), compared with K562 cells (18%; Table S4). Thus, a large number of colonies were plated to have a sufficient number of misrepaired colonies for sequencing of the deletion breakpoints generated in NC10 cells This revealed that a small percentage of DSBs are repaired using DNA sequence microhomology (12%; Table S5; Table 1), compared with K562 cells (80%; Table S4; Table 1). Notably, knockdown of DNA ligase IIIα or WRN did not result in a change in microhomologous sequences at the deletion breakpoints in NC10 cells (12%; Table S5; Table 1). Whereas in K562 cells, there was a decrease in microhomologies (approximately 44%; Table S4; Table 1). Based on these results, we conclude that backup NHEJ does not contribute significantly to DSB repair in cells with no abnormalities in the major NHEJ pathway.
Artemis overexpression leads to a decrease in large deletions
To determine whether the reduced levels of Artemis contribute to the aberrant DSB repair in CML, Artemis was overexpressed in K562 cells (Figure 7A). Although Artemis overexpression did not significantly alter the end-joining efficiency (Figure 7B), there was a considerable decrease in the size of DNA deletions at misrepaired DSBs (P < .001; Figure 7C,D). In addition, analysis of the breakpoint junctions from 15 repaired plasmids in K562 cells overexpressing Artemis showed a decrease in repair events involving DNA sequence microhomologies down to 33%, compared with empty vector controls (80%) (P < .001; Figure 7E; Table 1). Thus, down-regulation of Artemis in CML results in abnormal processing of DSBs.
Figure 7.

Artemis overexpression in K562 leads to a decrease in large deletions. (A) Western blotting in K562 cells transfected with the myc-tagged Artemis cDNA construct (pcDNA huASC1D) showing Artemis overexpression. Ku86 was used as loading control. An in vivo LacZα plasmid reactivation assay was used to measure end joining in Artemis overexpressed cells, compared with empty vector–transfected controls. (B) Relative percentage of colonies, indicating the efficiency of end joining. (C) Graph of percentage of large deletions, defined as more than 20 bp. (D) Agarose gel showing PCR products of repaired colonies in empty vector controls and Artemis overexpressed K562 cells. (E) The percentage of plasmids repaired using DNA sequence microhomologies of 1 to 6 bp. Fifteen plasmids were sequenced. Values significantly different are marked with an asterisk (Student t test; P < .001 for panels C and E). Error bars reflect the standard error of the mean.
Discussion
BCR-ABL has been shown to induce a cycle of events whereby increased ROS production leads to DSBs that are repaired by an error-prone mechanism. Here we have identified a novel protein complex containing DNA ligase IIIα and WRN that is present in elevated levels and is involved in the repair of DSBs in CML cells. This complex participates in an alternative end-joining pathway that compensates for the decreased activity of the major NHEJ pathway resulting from reduced levels of Artemis and DNA ligase IV, in CML. We suggest that alternative end-joining mediated by DNA ligase IIIα and WRN contributes to disease progression by enabling cells to repair ROS-induced DSBs, albeit by a more error-prone pathway, ensuring the survival of CML cells at the cost of increased genomic instability.
Ku binding to DSBs is essential to initiate repair via the main NHEJ pathway.11 Recent studies show that DNA-PKcs and DNA ligase IV are recruited independently to Ku-bound DSBs.45 In addition to catalyzing the last step of the major NHEJ repair pathway,14 studies in yeast show that the DNA ligase IV homologue, Dnl4, acts early in NHEJ, stabilizing Ku binding and that, in the absence of Dnl4, there is increased end resection.46 In BCR-ABL–positive CML cells, we have shown that the steady state levels of DNA ligase IV are significantly reduced. This suggests that Ku binding may be destabilized in CML cells, resulting in increased end resection. In addition, because DNA ligase IV performs the final ligation of DNA ends, the reduced levels of DNA ligase IV are likely to cause a delay in the joining of DNA ends synapsed by DNA PK. Such a delay may lead to the loss of a few additional nucleotides from the ends.
Here, we show that the steady state levels of DNA ligase IIIα are elevated in CML cells and recruited to DSBs. This work provides further evidence that DNA ligase IIIα, which normally plays a role in the repair of DNA single strand breaks and BER,29,47 participates in a backup pathway for NHEJ when DNA ligase IV levels are reduced.25,37 Notably, knockdown of DNA ligase IIIα in CML cells results in an increased frequency of misrepair but has no effect on the size of deletions. Based on these results, we propose that DNA ligase IIIα compensates for the reduced levels of DNA ligase IV by acting at DNA ends synapsed by DNA-PK. In addition, our data suggest that another ligase, possibly DNA ligase I, completes the repair events generating the deletions.
In addition to DNA ligase IV, the level of another key factor in the major NHEJ pathway, Artemis, is also markedly reduced in CML cells. Although the Artemis nuclease interacts with and is activated by DNA PKcs, it is thought to be required only for processing a subset of DSBs produced by such agents as ionizing radiation and ROS, defined as complex DSBs.17 The reduced levels of Artemis do not appear to be caused by proteosomal degradation and cannot be explained by changes in the level of DNA PKcs because, we find that, in contrast to the report of Deutsch et al,48 DNA PKcs is not reduced in CML cells. Notably, overexpression of Artemis in CML cell lines results in a reduction in the size of deletions. These results suggest that Artemis, in addition to participating in end processing, may play a role in stabilizing complexes containing DNA-PK that are assembled on DNA ends.
We find that the steady state levels of another DNA repair protein, WRN, are also elevated in BCR-ABL–positive CML cells. Although WRN is not considered to be one of the core NHEJ factors, it does bind to Ku, thereby increasing its exonuclease activity.20–23 However, the biologic relevance of the stimulation of WRN nuclease activity by Ku is not obvious because cells lacking WRN protein are not hypersensitive to ionizing radiation like other NHEJ-deficient cell lines and exhibit an increased incidence of deletions.24 In addition, WRN is a substrate of the c-Abl kinase and is constitutively phosphorylated by BCR-ABL in CML cells, resulting in inhibition of exonuclease and helicase activities.49 Here, we have shown that knockdown of WRN in CML cells leads to an increase in the size of deletions. Thus, it appears that WRN acts to protect DNA ends from nucleolytic resection. Presumably, the elevated levels of WRN in the CML cells reflect an attempt to prevent deletions at the ROS-induced DSBs. The mechanism by which WRN suppresses deletions is not known. It is possible that WRN stabilizes complexes of NHEJ proteins at DNA ends. Alternatively, WRN may negatively regulate the activity of nuclease(s) that act at DSBs, thereby limiting resection.
One of the standard methods for discriminating between the end-joining pathways used to repair DNA ends is to examine the sequences at the joins; alternative NHEJ repair appears to use microhomology-mediated repair at DNA ends.25–28 CML cells repair approximately 80% of DNA ends using microhomologies. Knockdown of DNA ligase IIIα and WRN and overexpression of Artemis, proteins specifically altered in CML, led to a significant reduction in microhomology sequences at repair sites. Furthermore, the size of microhomology sequences appears to decrease with these experimental maneuvers. These data provide additional evidence for an alternative end-joining pathway that is operative in CML. In contrast, control lymphoblastoid cells established from healthy individuals infrequently repair DSBs using DNA sequence microhomologies. This is in line with the data of Yan et al,28 suggesting that the contribution and/or activity of the alternative NHEJ pathway is low in wild-type cells.
Abnormal repair of ROS-induced DSBs is a characteristic feature of BCR-ABL–positive CML. Here, we have shown that both down-regulation of the major NHEJ pathway and up-regulation of an error-prone alternative NHEJ pathway contribute to the altered DNA repair and genomic instability in CML cells. Elevated levels of DNA ligase IIIα and WRN compensate for DNA ligase IV deficiency. Because these proteins associate and colocalize with DSBs in CML cells, it seems likely that they functionally interact with DNA PK complexes formed at DNA ends. We suggest that the reduced levels of Artemis and/or DNA ligase IV result in decreased stability of DNA PK complexes at DNA ends that in turn leads to increased end resection. The identity of the protein factors involved in the repair pathway acting on the resected ends and generating the deletions is not known. Nonetheless, it is evident that there are major differences in the repair of DSBs in BCR-ABL–positive CML cells. Because resistance to imatinib is being reported with greater frequency,50 this altered repair, in particular the alternative NHEJ pathway, constitutes an attractive target for the development of novel therapeutic strategies for CML. Thus, further characterization of the abnormal DNA repair in BCR-ABL–positive CML will not only provide insights into the role of genomic instability in disease progression, but may also lead to the development of novel therapeutic strategies in patients resistant to tyrosine kinase inhibitors.
Acknowledgments
We thank Professor Stephen Baylin (Johns Hopkins University [JHU], Baltimore, MD) for insightful comments and careful reading of our paper. We thank Matt Whitehurst for technical support. We thank Drs Rapoport (University of Maryland) and Douglas Smith (JHU) for CML patient samples (IRB H25314).
F.V.R. and A.S. are funded by the Leukemia Lymphoma Society, New York, NY and the Department of Defense, Washington, DC. A.E.T. is funded by National Institutes of Health (NIH, Bethesda, MD) grants (ES 012512).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.S. designed and performed the experiments, analyzed the data, and participated in writing the paper; F.V.R. designed the experiments, analyzed the data, and wrote the paper; A.E.T. provided reagents and critically read the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Feyruz Rassool, University of Maryland School of Medicine, 655 W Baltimore St, BRB 7-023, Baltimore, MD 21201; e-mail: frassool@som.umaryland.edu.
References
- 1.Druker BJ, Sawyers CL, Capdeville R, Ford JM, Baccarani M, Goldman JM. Chronic myelogenous leukemia. Hematology (Am Soc Hematol Educ Program) 2001:87–112. doi: 10.1182/asheducation-2001.1.87. [DOI] [PubMed] [Google Scholar]
- 2.Sawyers CL. Signal transduction pathways involved in BCR-ABL transformation. Baillieres Clin Haematol. 1997;10:223–231. doi: 10.1016/s0950-3536(97)80004-2. [DOI] [PubMed] [Google Scholar]
- 3.Rassool FV. Genetic rearrangements beget genomic instability. Blood. 2004;104:3424–3425. [Google Scholar]
- 4.Sattler M, Verma S, Shrikhande G, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–24278. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 5.Skorski T. BCR/ABL regulates response to DNA damage: the role in resistance to genotoxic treatment and in genomic instability. Oncogene. 2002;21:8591–8604. doi: 10.1038/sj.onc.1206087. [DOI] [PubMed] [Google Scholar]
- 6.Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 7.Majsterek I, Blasiak J, Mlynarski W, Hoser G, Skorski T. Does the bcr/abl-mediated increase in the efficacy of DNA repair play a role in the drug resistance of cancer cells? Cell Biol Int. 2002;26:363–370. doi: 10.1006/cbir.2002.0865. [DOI] [PubMed] [Google Scholar]
- 8.Slupianek A, Hoser G, Majsterek I, et al. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol Cell Biol. 2002;22:4189–4201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brady N, Gaymes TJ, Cheung M, Mufti GJ, Rassool FV. Increased error-prone NHEJ activity in myeloid leukemias is associated with DNA damage at sites that recruit key nonhomologous end-joining proteins. Cancer Res. 2003;63:1798–1805. [PubMed] [Google Scholar]
- 10.Gaymes TJ, Mufti GJ, Rassool FV. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002;62:2791–2797. [PubMed] [Google Scholar]
- 11.Falzon M, Fewell JW, Kuff EL. EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single- to double-strand transitions in DNA. J Biol Chem. 1993;268:10546–10552. [PubMed] [Google Scholar]
- 12.Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 13.Mimori T, Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- 14.Grawunder U, Wilm M, Wu X, et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–495. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- 15.Roth DB, Porter TN, Wilson JH. Mechanisms of nonhomologous recombination in mammalian cells. Mol Cell Biol. 1985;5:2599–2607. doi: 10.1128/mcb.5.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth DB, Wilson JH. Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol Cell Biol. 1986;6:4295–4304. doi: 10.1128/mcb.6.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeggo PA, Lobrich M. Artemis links ATM to double strand break rejoining. Cell Cycle. 2005;4:359–362. doi: 10.4161/cc.4.3.1527. [DOI] [PubMed] [Google Scholar]
- 18.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 19.Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature ageing. Biochem J. 2003;374(pt 3):577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orren DK, Machwe A, Karmakar P, Piotrowski J, Cooper MP, Bohr VA. A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 2001;29:1926–1934. doi: 10.1093/nar/29.9.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912. [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, Comai L. Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J Biol Chem. 2000;275:28349–28352. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- 23.Li B, Comai L. Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-Ku70/80 complex. J Biol Chem. 2001;276:9896–9902. doi: 10.1074/jbc.M008575200. [DOI] [PubMed] [Google Scholar]
- 24.Oshima J, Huang S, Pae C, Campisi J, Schiestl RH. Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 2002;62:547–551. [PubMed] [Google Scholar]
- 25.Wang H, Rosidi B, Perrault R, et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 26.Corneo B, Wendland RL, Deriano L, et al. Rag mutations reveal robust alternative end joining. Nature. 2007;449:483–486. doi: 10.1038/nature06168. [DOI] [PubMed] [Google Scholar]
- 27.Klassen R, Krampe S, Meinhardt F. Homologous recombination and the y Nospacekuqq70/80 complex exert opposite roles in resistance against the killer toxin from Pichia acaciae. DNA Repair (Amst) 2007;6:1864–1875. doi: 10.1016/j.dnarep.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Yan CT, Boboila C, Souza EK, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 29.Gottlich B, Reichenberger S, Feldmann E, Pfeiffer P. Rejoining of DNA double-strand breaks in vitro by single-strand annealing. Eur J Biochem. 1998;258:387–395. doi: 10.1046/j.1432-1327.1998.2580387.x. [DOI] [PubMed] [Google Scholar]
- 30.Riballo E, Critchlow SE, Teo SH, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- 31.Difilippantonio MJ, Zhu J, Chen HT, et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang M, Wu W, Wu W, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang F, Han M, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci U S A. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rassool FV, Le Beau MM, Shen ML, et al. Direct cloning of DNA sequences from the common fragile site region at chromosome band 3p14.2. Genomics. 1996;35:109–117. doi: 10.1006/geno.1996.0329. [DOI] [PubMed] [Google Scholar]
- 35.National Center for Biotechnology Information. Basic Local Alignment Search Tool (BLAST). [Accessed 2006–2007]. http://www.ncbi.nlm.nih.gov/blast/Blast.cgi.
- 36.Paull TT, Gellert M. A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc Natl Acad Sci U S A. 2000;97:6409–6414. doi: 10.1073/pnas.110144297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, Perrault AR, Takeda Y, Qin W, Iliakis G. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2003;31:5377–5388. doi: 10.1093/nar/gkg728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaymes TJ, North PS, Brady N, Hickson ID, Mufti GJ, Rassool FV. Increased error-prone non homologous DNA end-joining: a proposed mechanism of chromosomal instability in Bloom's syndrome. Oncogene. 2002;21:2525–2533. doi: 10.1038/sj.onc.1205331. [DOI] [PubMed] [Google Scholar]
- 39.Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry JJ, Yannone SM, Holden LG, et al. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 41.Li B, Comai L. Displacement of DNA-PKcs from DNA ends by the Werner syndrome protein. Nucleic Acids Res. 2002;30:3653–3661. doi: 10.1093/nar/gkf488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karmakar P, Snowden CM, Ramsden DA, Bohr VA. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 2002;30:3583–3591. doi: 10.1093/nar/gkf482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson C, Jasin M. Coupled homologous and nonhomologous repair of a double-strand break preserves genomic integrity in mammalian cells. Mol Cell Biol. 2000;20:9068–9075. doi: 10.1128/mcb.20.23.9068-9075.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rassool FV, North PS, Mufti GJ, Hickson ID. Constitutive DNA damage is linked to DNA replication abnormalities in Bloom's syndrome cells. Oncogene. 2003;22:8749–8757. doi: 10.1038/sj.onc.1206970. [DOI] [PubMed] [Google Scholar]
- 45.Mari PO, Florea BI, Persengiev SP, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci U S A. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Hefferin ML, Chen L, et al. Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat Struct Mol Biol. 2007;14:639–646. doi: 10.1038/nsmb1261. [DOI] [PubMed] [Google Scholar]
- 47.Tomkinson AE, Vijayakumar S, Pascal JM, Ellenberger T. DNA ligases: structure, reaction mechanism, and function. Chem Rev. 2006;106:687–699. doi: 10.1021/cr040498d. [DOI] [PubMed] [Google Scholar]
- 48.Deutsch E, Dugray A, AbdulKarim B, et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood. 2001;97:2084–2090. doi: 10.1182/blood.v97.7.2084. [DOI] [PubMed] [Google Scholar]
- 49.Cheng WH, von Kobbe C, Opresko PL, et al. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol Cell Biol. 2003;23:6385–6395. doi: 10.1128/MCB.23.18.6385-6395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kantarjian H, O'Brien S, Talpaz M, et al. Outcome of patients with Philadelphia chromosome-positive chronic myelogenous leukemia post-imatinib mesylate failure. Cancer. 2007;109:1556–1560. doi: 10.1002/cncr.22569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}