


Abstract
DNA replication of phage-plasmid P4 in its host Escherichia coli depends on its replication protein α. In the plasmid state, P4 copy number is controlled by the regulator protein Cnr (copy number regulation). Mutations in α (αcr) that prevent regulation by Cnr cause P4 over-replication and cell death. Using the two-hybrid system in Saccharomyces cerevisiae and a system based on λ immunity in E.coli for in vivo detection of protein–protein interactions, we found that: (i) α protein interacts with Cnr, whereas αcr proteins do not; (ii) both α–α and αcr–αcr interactions occur and the interaction domain is located within the C-terminal of α; (iii) Cnr–Cnr interaction also occurs. Using an in vivo competition assay, we found that Cnr interferes with both α–α and αcr–αcr dimerization. Our data suggest that Cnr and α interact in at least two ways, which may have different functional roles in P4 replication control.
INTRODUCTION
P4 is a natural phasmid that can propagate in Escherichia coli both as a temperate phage and as a plasmid (1–3). The double-stranded P4 DNA circularizes after infection and replication starts from a single site, ori1, proceeding bidirectionally in a θ-type mode (4). DNA replication depends on the product of the P4 α gene, a multifunctional protein organized in distinct domains: the N-terminal region exhibits primase activity, the middle and C-terminal parts display helicase activity and the C-terminal end exhibits DNA binding activity (Fig. 1) (5). Two sites in the P4 genome, ori1 and crr, are essential in cis for replication. Both contain several direct and inverted repeats of a decameric sequence, the type I iterons (6,7), which are bound by the α protein (8). Although essential for replication (6,7), crr is not an origin of replication (4,9). In several iteron-containing plasmids (such as P1, R6K, RK2) (10,11) the replication protein binds to specific sites and DNA looping and/or intermolecular pairing of DNA molecules, mediated by protein–protein interactions, occurs. The formation of the multimeric protein–DNA complexes (handcuffing; 12) inhibits replication initiation and allows plasmid copy number control. However in P4, unlike the above model, crr is positively required in cis for replication and does not appear to be involved in P4 DNA replication control (4,7,13).
Figure 1.

Identification of the α dimerization domain. Schematic representation of the α protein, redrawn from Ziegelin et al. (19). The localization of the domains and the amino acids substitutions of the αcr mutations are indicated. The plasmids carry the α fragments, indicated by the amino acid coordinates and by bars, fused to the N-terminal part of the λ CI repressor. The efficiency of plating of λ, λcY17 (immunity sensitive cI mutant; 24) and λ668 (virulent mutant; 25) on the different strains, relative to the control CSH50, is given (for details see Materials and Methods). Closed bars, fragments conferring immunity; open bars, fragments that do not confer immunity.
Regulation of P4 DNA replication is achieved at different levels. A first level depends on modulation of the expression of phage genes that code for replication functions (2,14–16). However, this regulation is not sufficient to control P4 copy number when P4 propagates as a plasmid. In this case, the P4 Cnr (copy number regulation) protein is essential to modulate the activity of α protein (13,17,18). Deletion of the P4 cnr gene causes P4 DNA over-replication and cell lethality, thus preventing P4 propagation in the plasmid state (13,17); whereas overexpression of Cnr leads to inhibition of P4 DNA replication. However, if the expression of both the Cnr and α proteins is increased, no inhibition of DNA synthesis is observed (17). This suggested that the control of P4 DNA replication depends on the relative concentration of the Cnr and α proteins.
P4 mutants insensitive to the Cnr control carry amino acid substitutions in the C-terminus of α protein (αcr mutations; 18) (Fig. 1). All such mutants are impaired in plasmid propagation. The αcr mutations are in the DNA-binding domain of α, which has been mapped to within a 141-amino acid region, near the C-terminus of the protein (19). Four mutations are clustered (G732V, G732W, L733V and L737V) and a fifth mutation maps at some distance (T675M). This localization suggests that the negative control of Cnr is exerted through a direct interaction with α.
It has been shown in vitro that the Cnr protein increases α affinity for ori1 and crr binding, whereas such an effect could not be observed on αcr mutant proteins (18). It was thus hypothesized that Cnr increases the affinity of the α protein for the origin of replication; however, interaction between the two proteins has not been demonstrated in vivo.
In this work, by making use of the two-hybrid system in yeast and the λ CI dimerization test in E.coli, we investigated in vivo interactions of α and Cnr proteins.
MATERIALS AND METHODS
Microorganisms and media
Manipulation of bacterial as well as yeast strains and of nucleic acids and proteins was carried out using standard methods (20,21). The E.coli K12 strains used were CSH50 [Δ(pro-Lac) F′(proABlacIqZΔM15, traD36)] (22) and 71–18 [Δ(Lac-proAB) F′lacIq, lacZΔM15 pro supE] (20). The Saccharomyces cerevisiae strain was EGY48 (MATα, his3, trp1, ura3-52, leu2::LexAop6-LEU2) (21). Yeast transformation was performed by the lithium acetate procedure (23). Yeast SD medium (23), containing the specific amino acids and/or uracil, supplemented with 2% glucose or 2% galactose/1% raffinose (w/v), was used.
Bacteriophages and plasmids
The bacteriophage strains used were: λ wild-type, λcY17 (24) and λ668 (25). The plasmids are listed in Table 1. The plasmids used in the two-hybrid system are described in detail by Golemis et al. (21). The plasmids used for the λ immunity system are described by Castagnoli et al. (26) and Longo et al. (27).
Table 1. Plasmids.
| Plasmid |
Proteina |
P4 regionb |
Vector |
Relevant phenotype or construction |
Reference |
| pACYC184 | CamR; plac; p15A origin | 31 | |||
| pBR322 | AmpR; plac; ColE1 origin | 32 | |||
| pC132c | CI∼Rop | pBR322 | 26 | ||
| pC168c | CI∼Rop | pACYC184 | 27 | ||
| pC169c | CI*∼Rop | pBR322 | 27 | ||
| pEG202d | LexA | 21 | |||
| pGM283 | Cnr | 6727–7629 | pUC19 | 17 | |
| pGM583 | α | 4595–6969 | pGZ119 | From pGZα by insertion of an EcoRI site at the 5′ of the α gene | |
| pGM584 | Cnr | 6984–7304 | pGZ119 | PCR with 119EcoRI–118SalIe | |
| pGM585 | LexA∼α | 4636–6969 | pEG202 | EcoRI–SalI fragment of pGM583 | |
| pGM587 | LexA∼αcrL733V | 4595–6969 | pGM585 | Substitution of the NotI–SalI fragment derived from pGZαcr4773 | |
| pGM588 | LexA∼αcrG732W | 4595–6969 | pGM585 | As above, derived from pGZα4776A | |
| pGM589 | LexA∼αcrG732V | 4595–6969 | pGM585 | As above, derived from pGZα4775B | |
| pGM590 | LexA∼αcrT675M | 4595–6969 | pGM585 | As above, derived from pGZα4946 | |
| pGM591 | B42∼α | 4595–6969 | pJG4–5 | EcoRI–SalI fragment of pGM583 cloned in the EcoRI–XhoI sites of the vector | |
| pGM592 | B42∼Cnr | 7304–6983 | pJG4–5 | PCR with 119EcoRI–118SalIe | |
| pGM607 | B42∼αcrG732W | 4595–6969 | pJG4–5 | EcoRI–SalI fragment of pGM588 cloned in the EcoRI–XhoI sites of the vector | |
| pGM722f | CI∼α620–777 | 4639–5112 | pC132 | PCR with 401BamHI–402SalIe | |
| pGM723f | CI∼α664–777 | 4639–4980 | pC132 | PCR with 401BamHI–403SalIe | |
| pGM724f | CI∼α707–777 | 4639–4851 | pC132 | PCR with 401BamHI–404SalIe | |
| pGM726f | CI∼Cnr | 6987–7304 | pC132 | PCR with 399BamHI–400SalIe | |
| pGM727f | CI*∼Cnr | 6987–7304 | pC169 | PCR with 399BamHI–400SalIe | |
| pGM729f | CI∼α707–730 | 4780–4851 | pC132 | PCR with 413BamHI–404SalIe | |
| pGM730f | CI∼α707–747 | 4729–4851 | pC132 | PCR with 414BamHI–404SalIe | |
| pGM731f | CI∼α724–777 | 4639–4800 | pC132 | PCR with 401BamHI–411SalIe | |
| pGM732f | CI∼α741–777 | 4639–4749 | pC132 | PCR with 401BamHI–412SalIe | |
| pGM738 | CI∼α664–777 | 4639–4980 | pC168 | Substitution of the SalI–BamHI fragment from pGM723 | |
| pGM740 | CI∼α741–777 | 4639–4749 | pC168 | Substitution of the SalI–BamHI fragment from pGM732 | |
| pGM773f | CI∼αcr620–777G732W | 4639–5112 | pC132 | PCR on pGZα4776A DNA with 401BamHI–402SalIe | |
| pGM774f | CI∼αcr620–777G732V | 4639–5112 | pC132 | PCR on pGZα4775B DNA with 401BamHI–402SalIe | |
| pGM775f | CI∼αcr620–777T675M | 4639–5112 | pC132 | PCR on pGZα4946 DNA with 401BamHI–402SalIe | |
| pGM776 | CI∼α620–777 | 4639–5112 | pC168 | PCR with 401BamHI–402SalIe | |
| pGM778 | CI∼αcr620–777G732W | 4639–5112 | pC168 | PCR on pGZα4776A DNA with 401BamHI–402SalIe | |
| pGM779 | CI∼αcr620–777G732V | 4639–5112 | pC168 | PCR on pGZα4775B DNA with 401BamHI–402SalIe | |
| pGM780 | CI∼αcr620–777T675M | 4639–5112 | pC168 | PCR on pGZα4946 DNA with 401BamHI–402SalIe | |
| pGM794 | CI*∼Cnr | 6987–7304 | pC168 | Substitution of the BamHI–EcoRI fragment from pGM727 | |
| pGZ119 | CamR; ptac; V origin | 33 | |||
| pGZαg | α | 4595–7041 | pGZ119 | 18 | |
| pGZα4773g | αcrL733V | 4595–7041 | pGZ119 | 18 | |
| pGZα4776Ag | αcrG732W | 4595–7041 | pGZ119 | 18 | |
| pGZα4775Bg | αcrG732V | 4595–7041 | pGZ119 | 18 | |
| pGZα4946g | αcrT675M | 4595–7041 | pGZ119 | 18 | |
| pJG4–5d | B42 | 21 | |||
| pJK101d | 21 | ||||
| pMS119HE | AmpR; ptac; V origin; | 34 | |||
| pSH17–4d | LexA∼Gal4 | 21 | |||
| pSH18–34d | 21 | ||||
| pST106 | Cnr | 7307–6976 | pMS119HE | 17 | |
| pUC19 | AmpR; plac; ColE1 origin | 35 |
aThe protein expressed by the plasmid is indicated. CI*, mutant λ CI repressor protein unable to bind DNA (27); ∼, fusion protein.
bCoordinates of the cloned P4 region are from the complete P4 sequence (36; GenBank accession no. X51522).
cKindly provided by F. Gigliani.
dKindly provided by R. Brent.
eThe P4 fragment obtained by PCR amplification with the pair of oligonucleotides indicated was digested with the appropriate enzymes and cloned in the corresponding sites of the vector. Unless otherwise stated, the PCR has been performed on P4 DNA. The restriction site is in italic. The sequence complementary to P4 is underlined. 118SalI(ACGCGTCGACTCAGTGCAGTACCGGCGC); 119EcoRI(ACGAATTCATGAAAACACCCTTACCGCC); 399BamHI(ATTGGATCCGCTAGTGCAGTACCGGCGCTTTTATGTG); 400SalI(GATCGTCGACTATGAAAACACCCTTACCGCCCG);
401BamHI(GAAGGATCCGCTAGGCTGTTGTAGGGTCGTCAC); 402SalI(GATTGTCGACTCCGCAGGAGCGCGAC);
403SalI(GATTGTCGACTGAGGCACTGAACATCAAACGG); 404SalI(CTCGGTCGACTCTCTATCACGCCTATCTGGCC);
411SalI(CTCGGTCGACTCTCAGTCTGAAAATGTTCGGG); 412SalI(GTGGGTCGACTGGACTGAATTACGAGAAACGC);
413BamHI(GAAGGATCCGCTACCCGAACATTTTCAGACTGAG); 414BamHI(GAAGGATCCGCTAGCGTTTCTCGTAATTCAGTCC). The sequence of the cloned fragments was confirmed by sequencing.
fIn such constructs, a UAG stop codon separates the α gene from a downstream in frame lacZ gene. Thus, after transformation of strain 71.18, which carries a tRNA amber suppressor, the colonies had a blue color in the presence of X-Gal.
gKindly provided by R. Calendar.
Identification of protein–protein interactions
Two different systems were used for the identification of protein–protein interactions. The two-hybrid system in S.cerevisiae was performed as described previously (21). Experimental details are reported in the legend to Table 2.
Table 2. Interaction between α and Cnr proteins detected by the yeast two-hybrid system.
| Plasmidsa | Protein fused tob | Activation of lexAop6-LEU2d | Activation of lexAop8-lacZe | |
| |
LexA DNA-binding domainc |
B42 activation domain |
(Eop) |
(U β-galactosidase) |
| pEG202 pJG4–5 | – | – | 1 × 10–3 | 3.16 ± 0.68 |
| pEG202 pGM592 | – | Cnr | <7 × 10–3 | 2.81 ± 0.97 |
| pGM585 pJG4–5 | α | – | <5 × 10–3 | 1.02 ± 1.12 |
| pGM585 pGM592 | α | Cnr | 0.62 | 11.42 ± 1.20 |
| pGM587 pJG4–5 | αcrL733V | – | <4 × 10–3 | 2.10 ± 0.62 |
| pGM587 pGM592 | αcrL733V | Cnr | <8 × 10–3 | 1.74 ± 1.45 |
| pGM588 pJG4–5 | αcrG732W | – | <3 × 10–3 | 0.82 ± 0.40 |
| pGM588 pGM592 | αcrG732W | Cnr | <6 × 10–3 | 1.02 ± 0.43 |
| pGM589 pJG4–5 | αcrG732V | – | <4 × 10–3 | 1.52 ± 1.10 |
| pGM589 pGM592 | αcrG732V | Cnr | <7 × 10–3 | 2.24 ± 0.30 |
| pGM590 pJG4–5 | αcrT675M | – | <4 × 10–3 | 2.03 ± 0.46 |
| pGM590 pGM592 | αcrT675M | Cnr | 0.14 | 3.80 ± 1.73 |
aThe plasmids are carried by the S.cerevisiae strain EGY48 (pSH18–34).
bThe proteins fused to either the DNA-binding domain or the transcription activation domain are indicated. The fusion proteins were expressed in S.cerevisiae EGY48 (pSH18–34) in a galactose/raffinose medium lacking uracil, hystidine and tryptophan. Three independent transformants were tested for each strain.
cActivation and repression assays (21) confirmed that the fusion protein by itself did not activate the reporter genes and that it is localized in the nucleus (data not shown).
dExpression of the lexAop6-LEU2 reporter gene was tested by measuring the efficiency of plating (Eop) in a galactose/raffinose medium in the presence or absence of leucine.
eExpression of the lexAop8-lacZ reporter gene was tested by measuring the β-galactosidase specific activity. The activities are calculated as nanomoles of O-nitrophenyl galactoside hydrolyzed per minute per milligram of protein (U β-galactosidase; 37). The values are the mean of assays on three independent transformants each assayed twice.
The λ immunity system is described by Castagnoli et al. (26) and the competition essay by Longo et al. (27).
Efficiency of plating of λ
Overnight cultures of CSH50, carrying the different plasmids, were grown at 37°C with aeration in TB broth, supplemented with 0.2% maltose and 0.01 M MgSO4 and ampicillin (50 µg/ml) or chloramphenicol (30 µg/ml), as required. In the case of low copy number plasmids, 1 mM isopropyl-β-d-thiogalactoside (IPTG) was added to the medium. Cultures of both high and low copy number plasmids (0.3 ml) were plated with soft agar containing 1 mM IPTG. Drops (10 µl) of a suspension of the λ wild-type, the immunity sensitive λcY17 and the virulent λ668 mutant were streaked on the plates. The efficiency of plating relative to the control strain CSH50 was evaluated. CSH50/pC132 and CSH50/pC168, expressing the λ CI repressor fused to the Rop protein, were used as positive controls (26).
RESULTS
Detection of α–Cnr interaction by the two-hybrid system in yeast
The possibility that the Cnr protein interacts with the α protein was suggested by the existence of mutations in the P4 α gene that abolish the negative control exerted by Cnr on P4 replication (αcr mutations; 18) (Fig. 1). To demonstrate Cnr–α interaction in vivo, we used the two-hybrid system in yeast (21): the wild-type α gene and four αcr mutant genes (αT675M, αG732V, αG732W and αL733V) were fused to the LexA DNA-binding domain in pEG202 (pGM585, pGM590, pGM589, pGM588 and pGM587, respectively), whereas cnr was fused to the B42 activation domain in pJG4–5 (pGM592). After transformation of the S.cerevisiae strain EGY48 (pSH18–34), α–Cnr interaction was revealed by the expression of two different reporter genes: the chromosomal LEU2 gene and the plasmid lacZ gene, both under control of LexA operators. The results, reported in Table 2, showed that concomitant expression in EGY48 (pSH18–34) of Lex∼α wild-type and B42∼Cnr activated both the LEU2 and the lacZ reporter genes, indicating that the two proteins interact with each other.
When the LexA∼αcr mutant hybrid proteins αcrG732V, αcrG732W or αcrL733V and B42∼Cnr were expressed in the same strain, activation of the reporter genes was not observed. Thus, Cnr does not interact with these αcr proteins. A low level of activation of the chromosomal LEU2 gene, but not of the plasmid lacZ reporter gene, was detected with the LexA∼αT675M protein and might indicate leakiness of the mutant.
Detection of α–α interactions by the two-hybrid system in yeast
Using the two-hybrid system in yeast, we also investigated whether α proteins were able to self-interact. The wild-type α protein was fused both to the LexA DNA-binding domain and to the B42 transactivation domain (pGM591). The results, reported in Table 3, showed that only co-expression of LexA∼α and B42∼α led to activation of LEU2 and lacZ reporter genes. Thus, α proteins interact with each other.
Table 3. Interaction between α proteins detected by the yeast two-hybrid system.
| Plasmidsa | Protein fused tob | Activation of | Activation of lexAop8-lacZ | ||
| |
LexA DNA-binding domainc |
B42 activation
domain |
lexAop6-LEU2d (Eop) |
Activitye (U β-galactosidase) |
Colorf |
| pEG202 pJG4–5 | – | – | <1 × 10–3 | 3.16 ± 0.68 | White |
| pEG202 pGM591 | – | α | <3 × 10–3 | 3.46 ± 0.10 | White |
| pGM585 pJG4–5 | α | – | <5 × 10–3 | 1.02 ± 1.12 | White |
| pGM585 pGM591 | α | α | 1 | 350.30 ± 29.20 | Dark blue |
| pGM588 pGM607 | αcrG732W | αcrG732W | 1 | Not tested | Dark blue |
| pGM585 pGM607 | α | αcrG732W | 1 | Not tested | Dark blue |
| pGM588 pGM591 | αcrG732W | α | 1 | Not tested | Dark blue |
a–eSee Table 2.
fColor of colonies grown in glucose medium in the presence of tryptophan, leucine and X-Gal.
We also tested the ability of a mutant αcrG732W protein to dimerize. Both αcr–αcr homo- and αcr–α heterodimerization was observed (Table 3), indicating that the presence of the αcrG732W mutation did not impair α–α interaction.
Identification of the α interaction domain
The α protein is 777 amino acids long and has a modular organization (5). We first attempted to identify its interaction domain by the two-hybrid system in yeast. However, fusion of portions of the α proteins with LexA produced false positives. Thus, we used an in vivo assay for detection of protein–protein interactions in E.coli, based on fusions with the λ CI repressor. In this system, the N-terminal part of the λ CI repressor, which contains the DNA-binding domain, is fused to the protein to be tested. A truncated CI protein, which lacks the C-terminal dimerization domain, is inactive. However, if the fused polypeptide can dimerize, CI functionality is restored and the hybrid protein confers immunity to λ infection (24,26,28).
CI fusion with the whole α protein did not express λ immunity (data not shown); this might be due to steric hindrance of the fusion protein. Thus, fragments of decreasing length of the C-terminal part of the α gene were cloned, creating fusions with the N-terminal part of CI; the different α regions used are indicated in Figure 1. Strain CSH50 was transformed with the plasmids and the resistance to λ infection tested. Four constructs displayed λ immunity and they all cover the C-terminal part of the α gene. The smallest fragment contains the α portion from amino acid 707 to 747 (pGM730). Neither the 724–777 nor the 707–730 α regions expressed immunity (pGM731 and pGM729). Thus, the results of the λ immunity analysis indicated that α protein residues 707–747 are critical for dimerization in vivo.
Most αcr mutations map in this region (Fig. 1). Thus, we tested whether CI∼αcr fusion proteins could dimerize. The DNA regions encoding the 620–777 α amino acids from αcrG732W, G732V and T675M fused to CI conferred λ immunity (Table 4), indicating that the αcr mutations did not affect dimerization ability.
Table 4. Interaction of αcr–αcr and Cnr–Cnr proteins.
| Plasmida | Fusion proteinb | Efficiency of plating ofc | ||
| λ | λcY17 | λ668 | ||
| – |
– |
1 |
1 |
1 |
| pC132 | CI∼Ropd | <10–5 | <10–5 | 1 |
| pGM722 | CI∼α620–777 | <10–5 | <10–5 | 1 |
| pGM773 | CI∼αcr620–777G732W | <10–5 | <10–5 | 1 |
| pGM774 | CI∼αcr620–777G732V | <10–5 | <10–5 | 1 |
| pGM775 | CI∼αcr620–777T675M | <10–5 | <10–5 | 1 |
| pGM726 | CI∼Cnr | <10–5 | <10–5 | 1 |
aThe plasmids are carried by strain CSH50.
bThe P4 α protein residues fused to the N-terminal region of the λ CI repressor are indicated. The amino acid substitutions caused by αcr mutations are indicated. Expression of the fusion proteins from plac was induced by addition of 1 mM IPTG in the top agar.
cEfficiency of plating was measured as indicated in Materials and Methods. λ, wild-type; λcY17, immunity sensitive; λ668, virulent mutant.
dThe CI∼Rop fusion, which is able to dimerize (26), was used as positive control.
Detection of Cnr–Cnr interactions in vivo
In order to test whether Cnr–Cnr interactions occurred, the Cnr protein was fused to the λ CI DNA-binding domain (pGM726). Expression of the CI∼Cnr hybrid repressor conferred immunity to λ infection (Table 4), indicating that Cnr proteins can interact with each other.
Cnr interferes with α–α interactions
We have shown that the interaction domain of the α protein is localized in its C-terminal part, in which most αcr mutations are mapped. This suggests that the same region of the α protein could be involved in both α–α and Cnr–α interactions and that Cnr could interfere with α–α interaction. To test this hypothesis, we used a competition assay, based on the λ immunity system (27). In this assay, the CI∼α fusion proteins were expressed from a low copy number plasmid and the competitor Cnr protein was fused to a mutant CI repressor (CI*), unable to bind DNA, and expressed in the same strain from a high copy number plasmid (pGM727). Interaction of CI*∼Cnr fusion protein with CI∼α would compete with CI∼α–CI∼α interactions, thus preventing expression of λ immunity.
CI∼α hybrid proteins carrying the α620–777, α664–777 and α741–777 regions were expressed from low copy number plasmids (pGM776, pGM738 and pGM740, respectively: the latter was used as a negative control). The hybrid CI∼Rop protein, expressed from pC168, was used as a positive control (27). The results are reported in Table 5A. As expected, both pGM776 and pGM738 conferred λ immunity to the CSH50 host when induced with IPTG, whereas pGM740 did not.
Table 5. Cnr interference with α–α interaction.
| Plasmida | Protein expressed from the plasmidb | Efficiency of platingc | |||||
| |
Low copy number |
High copy number |
Low concentration |
High concentration |
λ |
λcY17 |
λ668 |
| A | pGM776 | – | CI∼α620–777 | – | <10–5 | <10–5 | 1 |
| pGM738 | – | CI∼α664–777 | – | <10–5 | <10–5 | 1 | |
| pGM740 | – | CI∼α741–777 | – | 1 | 1 | 1 | |
| pC168 | – | CI∼Rop | – | <10–5 | <10–5 | 1 | |
| B | pGM776 | pGM727 | CI∼α620–777 | CI*∼Cnr | 1 | 1 | 1 |
| pGM776 | pC169 | CI∼α620–777 | CI*∼Rop | <10–5 | <10–5 | 1 | |
| pGM738 | pGM727 | CI∼α664–777 | CI*∼Cnr | 1 | 1 | 1 | |
| pGM738 | pC169 | CI∼α664–777 | CI*∼Rop | <10–5 | <10–5 | 1 | |
| pGM740 | pGM727 | CI∼α741–777 | CI*∼Cnr | 1 | 1 | 1 | |
| pGM740 | pC169 | CI∼α741–777 | CI*∼Rop | 1 | 1 | 1 | |
| pC168 | pGM727 | CI∼Rop | CI*∼Cnr | < 10–5 | < 10–5 | 1 | |
| pC168 | pC169 | CI∼Rop | CI*∼Rop | 1 | 1 | 1 | |
| C | pGM778 | pGM727 | CI∼αcr620–777G732W | CI*∼Cnr | 1 | 1 | 1 |
| pGM778 | pC169 | CI∼αcr620–777G732W | CI*∼Rop | <10–5 | <10–5 | 1 | |
| pGM779 | pGM727 | CI∼αcr620–777G732V | CI*∼Cnr | 1 | 1 | 1 | |
| pGM779 | pC169 | CI∼αcr620–777G732V | CI*∼Rop | <10–5 | <10–5 | 1 | |
| pGM780 | pGM727 | CI∼αcr620–777T675M | CI*∼Cnr | 1 | 1 | 1 | |
| pGM780 | pC169 | CI∼αcr620–777T675M | CI*∼Rop | <10–5 | <10–5 | 1 | |
| D | pGM776 | pGM283 | CI∼α620–777 | Cnr | 1 | 1 | 1 |
| pGM779 | pGM283 | CI∼αcr620–777G732V | Cnr | 1 | 1 | 1 | |
| pGM776 | pST106d | CI∼α620–777 | Cnrd | 0.25 | 0.25 | 1 | |
| pGM779 | pST106d | CI∼αcr620–777G732V | Cnrd | 0.25 | 0.25 | 1 | |
| pGM794 | pGM722 | CI*∼Cnr | CI∼α620–777 | <10–5 | <10–5 | 1 | |
| pGM794 | pGM774 | CI*∼Cnr | CI∼αcr620–777G732V | <10–5 | <10–5 | 1 | |
aThe plasmids are carried by strain CSH50. Low copy number plasmids are derivatives of pC168 (p15A origin); high copy number plasmids are derivatives of pC169, pC132 or pUC19 (ColE1 origin).
bThe P4 protein fused to the N-terminal region of either the wild-type λ repressor (CI) or the mutant repressor (CI*) are indicated. The α protein residues and the αcr amino acids substitutions are reported.
cEfficiency of plating was measured as indicated in Materials and Methods. λ, wild-type; λcY17, immunity sensitive; λ668, virulent mutant.
dLow copy number plasmid (derivative of pMS119EH; oriV) and low concentration of the protein expressed.
Coexpression of the CI*∼Cnr protein from a high copy number plasmid (pGM727) with either CI∼α620–777 or CI∼α664–777 restored λ sensitivity, whereas coexpression of the control protein CI*∼Rop from a high copy number plasmid (pC169) did not alter λ immunity (Fig. 2; Table 5B). On the other hand, CI*∼Cnr neither interfered with λ immunity expressed from pC168 (CI∼Rop) nor altered λ plating ability on CSH50/pGM740 (CI∼α741–777).
Figure 2.
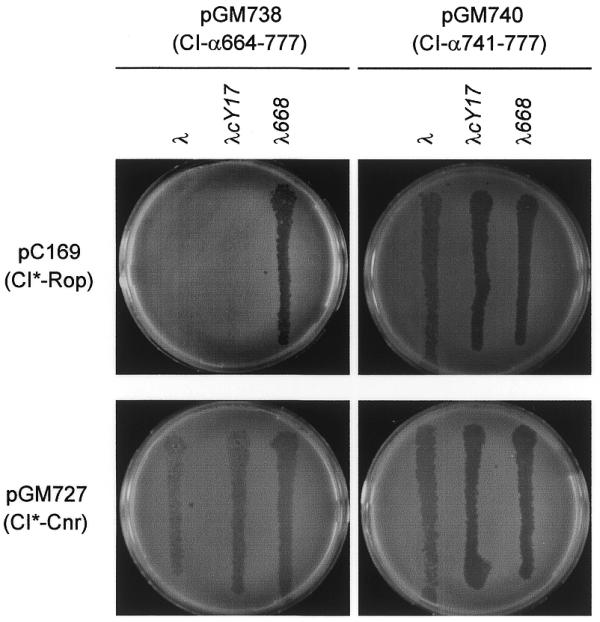
Competition assay for α–α interactions. Phage λ was plated on CSH50 carrying the indicated plasmids, as described in Materials and Methods. The fusion proteins expressed by the plasmids are indicated. The CI*∼Rop fusion protein, expressed by pC169, was used as a control (27). The λ phages were λ wild-type, the immunity sensitive λcY17 and the virulent mutant λ668, as indicated at the top of the Figure.
To test if Cnr could interfere with αcr–αcr interactions, hybrid CI∼αcr proteins, carrying the 620–777 α region with either G732W, G732V or T675M mutations were cloned in the low copy number vector (pGM778, pGM779 and pGM780, respectively). Surprisingly, CI*∼Cnr efficiently competed with dimerization of all three CI∼αcr fusion proteins (Table 5C). Thus, it appears that the αcr mutations do not prevent interference of Cnr with αcr–αcr interactions.
It is possible that the above result depends on overexpression of CI*∼Cnr from the high copy number plasmid pGM727. Thus, we varied the concentration of Cnr relative to α by making use of compatible plasmids either in low or high copy number, and we compared the effects on λ immunity expressed from wild-type CI∼α620–777 and the mutant CI∼αcr620–777G732V. We found that: (i) the wild-type Cnr protein expressed from a high copy number plasmid (pGM283) efficiently competed both CI∼α and CI∼αcr dimerization; (ii) expression of Cnr from the low copy number plasmid pST106 caused a comparably weak interference with λ immunity with either α+ and αcr constructs; (iii) in both α+ and αcr constructs, CI∼α dimerization could not be competed by Cnr when CI∼α was expressed from a high copy number plasmid (Table 5D). Thus, although Cnr interference with λ immunity can be modulated, a similar effect was observed with both wild-type α and mutant αcr proteins. Our data indicate that Cnr is able to interfere with both α–α and αcr–αcr interactions.
DISCUSSION
Interaction between α protein and Cnr
Autonomous P4 DNA replication, which occurs both in the lytic cycle and in the plasmid state, depends on the α protein. In vitro, P4 DNA replication does not require other P4-encoded proteins (9). In vivo, the Cnr protein is essential for P4 maintenance in the plasmid state. In the absence of Cnr, as in P4 cnr deletion mutants, P4 over-replicates and plasmid propagation is impaired (17). Moreover, overexpression of Cnr inhibits P4 DNA replication. Thus, the cnr gene appears to encode a negative regulator that is required for plasmid copy number control.
P4 αcr mutants, isolated by their ability to grow on E.coli that overexpressed the Cnr protein (18), map in the C-terminal part of the α protein. This suggests that the negative control of Cnr is exerted through a direct interaction with α.
In this work, using the two-hybrid system in S.cerevisiae, we have shown that the α protein can interact with the Cnr protein. However, Cnr does not interact with αcr mutant proteins, with the exception of αcrT675M, in which a low level of activation of the LEU2 reporter gene was observed. This suggests that the P4 αcrT675M mutation, which maps ∼60 bp apart from the others, might be less relevant for the Cnr–α interaction.
The above results indicate that α is the target of the Cnr protein and suggest that Cnr–α interaction is required for negative regulation of DNA replication. Phage-plasmid P4 is the first example in which copy number control is carried out by a protein that interacts with the replication protein and inhibits its activity.
It has been shown that, in vitro, the Cnr protein increases α binding affinity to ori1 and crr (18). It may be hypothesized that Cnr interaction with α modifies its structure and increases its ability to bind DNA. In fact, Ziegelin et al. (19) observed that the truncated α C-terminal region has higher affinity for DNA than the complete α protein, suggesting that the N-terminus quenches the DNA binding potential of the α C-terminus and the interaction with other proteins may increase α DNA-binding activity.
We suggest that α–Cnr efficiently competes with α for ori1 and crr binding sites on P4 DNA and that the α–Cnr complex is not proficient for replication. It is not known which step of P4 replication is inhibited by Cnr. Preliminary in vitro results suggested that neither α primase nor α helicase activities are inhibited by Cnr (18); DNA unwinding, primer synthesis or replication fork progression remain potential candidates.
Dimerization of α protein
Using the two-hybrid system we found that α proteins can interact with each other and the presence of the αcrG732W mutation does not prevent formation of both homo- and heterodimers. Using fusions with the λ CI repressor DNA-binding domain, we could locate the α dimerization domain to the 4729–4851 P4 DNA region, corresponding to residues 707–747 of α. This region overlaps the cluster of αcr mutations. Nevertheless, none of the αcr mutations tested affected α–α interaction, as can be deduced by the ability to confer immunity to λ infection also when expressed at low concentration. Thus, it is possible that the residues changed by the αcr mutations are not directly involved in α–α interaction. However, it should be emphasized that the P4 αcr mutants were selected for their ability to replicate in the presence of high levels of Cnr. If α–α interactions are essential for P4 replication the selection constraints might have screened a specific subset of mutants affected in Cnr–α interaction that still conserve dimerization ability.
The system used in this work to identify protein–protein interactions is based on the expression of immunity to λ infection. It is known that the CI repressor not only binds DNA as a dimer, but also forms tetramers and higher order oligomers by cooperative binding via its C-terminal domain (29). Thus, λ immunity observed with CI∼α fusion proteins might indicate that α proteins are able to oligomerize.
In vivo, α–α interaction might occur between both free α proteins and DNA-bound α proteins. In this latter case, the α subunits may be bound to the same site (either ori1 or crr) or to different sites (both ori1 and crr). Looping of P4 DNA molecules between ori1 and crr sites bound to α has been observed by electron microscopy (8,19,30). This suggests that α proteins, bound to ori1 and crr, might interact with each other to form an ordered structure competent for replication initiation. If different α molecules are required to carry out the primase and helicase activities, interaction might be required to bring the α molecules to the origin of replication. Thus, α–α interaction might be an essential event in the process of P4 DNA replication. Making use of the dimerization assay it will be possible to isolate α mutants affected in dimerization and test their replication ability.
Cnr interferes with α–α interaction
We observed that λ immunity conferred by CI∼α–CI∼α interactions was efficiently inhibited by the Cnr protein. Competition occurs also with CI∼αcr mutant proteins. These data suggest that two different types of interaction are possible between α and Cnr. The first, revealed by the two-hybrid test, is impaired by αcr mutations. The second is highlighted by Cnr competition with α–α interactions in the λ system. This second type of interaction implies different contacts between the two proteins since, unlike the first one, it is not affected by αcr mutations.
It may be hypothesized that Cnr contacts α–α complexes and causes a structural change of the multiprotein complex, thus modifying its functional role. The two types of interaction could be sequential: Cnr first interacts and modifies α complexes, thus increasing α affinity for DNA, then binds to α and interferes with its replication ability. The latter event would not occur with αcr mutant proteins. This observation also implies that Cnr interaction with α complexes does not directly inhibit P4 DNA replication.
Alternatively, α–α interactions might be essential for the formation of an active replication complex, and the αcr mutations are a subset of mutants that still retain this ability. Thus, a simple model where Cnr interferes with α–α interaction and this inhibits replication is still tenable. This model could be tested, for example, by isolating mutants in either α–α or α–Cnr interactions by a two-hybrid system and/or λ dimerization assay. Such mutants could then be analyzed for their replication proficiency and the reciprocal α–Cnr and α–α interactions.
Acknowledgments
ACKNOWLEDGEMENTS
We thank R. Brent, R. Calendar, J. Eriksson and F. Gigliani for kindly providing the strains and plasmids used in this work. We are grateful to E. Boye, E. Haggård-Ljungquist and P. Plevani for helpful discussions and suggestions. This work was supported by grant no. 98.00458.CT04 of the Consiglio Nazionale delle Ricerche, Rome, Italy, and by grants from the Ministero dell’Università e della Ricerca Scientifica e Tecnologica, Rome, Italy.
References
- 1.Lindqvist B.H., Dehò,G. and Calendar,R. (1993) Mechanisms of genome propagation and helper exploitation by satellite phage P4. Microbiol. Rev., 57, 683–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghisotti D., Briani,F., Forti,F., Piazza,F., Polo,S., Sabbattini,P., Sturniolo,T., Terzano,S., Zangrossi,S., Zappone,M., Sironi,G. and Dehò,G. (1995) Multiple regulatory mechanisms controlling phage-plasmid P4 propagation. FEMS Microbiol. Rev., 17, 127–134. [DOI] [PubMed] [Google Scholar]
- 3.Briani F., Ghisotti,D. and Dehò,G. (2000) Antisense RNA-dependent transcription termination sites that modulate lysogenic development of satellite phage P4. Mol. Microbiol., 36, 1124–1134. [DOI] [PubMed] [Google Scholar]
- 4.Krevolin M.D., Inman,R.B., Roof,D., Kahn,M. and Calendar,R. (1985) Bacteriophage P4 DNA replication. Location of the P4 origin. J. Mol. Biol., 182, 519–527. [DOI] [PubMed] [Google Scholar]
- 5.Ziegelin G., Linderoth,N.A., Calendar,R. and Lanka,E. (1995) Domain structure of phage P4 α protein deduced by mutational analysis. J. Bacteriol., 177, 4333–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flensburg J. and Calendar,R. (1987) Bacteriophage P4 DNA replication. Nucleotide sequence of the P4 replication gene and the cis replication region. J. Mol. Biol., 195, 439–445. [DOI] [PubMed] [Google Scholar]
- 7.Tocchetti A., Galimberti,G., Dehò,G. and Ghisotti,D. (1999) Characterization of the oriI and oriII origins of replication in phage-plasmid P4. J. Virol., 73, 7308–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziegelin G., Scherzinger,E., Lurz,R. and Lanka,E. (1993) Phage P4 α protein is multifunctional with origin recognition, helicase and primase activities. EMBO J., 12, 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Díaz-Orejas R., Ziegelin,G., Lurz,R. and Lanka,E. (1994) Phage P4 DNA replication in vitro. Nucleic Acids Res., 22, 2065–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helinski D.R., Toukdarian,A.E. and Novick,R.P. (1996) Replication control and other stable maintenance mechanisms of plasmids. In Neidhardt,F.C., Curtiss,R., Ingraham,J.L., Lin,E.C.C., Low,K.B., Magasanik,B., Reaznikoff,W.S., Riley,M., Schaechter,M. and Umbarger,H.E. (eds), Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. American Society for Microbiology Press, Washington, DC, pp. 2295–2324.
- 11.del Solar G., Giraldo,R., Ruiz-Echevarria,M.J., Espinosa,M. and Díaz-Orejas,R. (1998) Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev., 62, 434–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McEachern M.J., Bott,M.A., Tooker,P.A. and Helinski,D.R. (1989) Negative control of plasmid R6K replication: possible role of intermolecular coupling of replication origins. Proc. Natl Acad. Sci. USA, 86, 7942–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tocchetti A., Serina,S., Terzano,T., Dehò,G. and Ghisotti,D. (1998) Identification of two replicons in phage-plasmid P4. Virology, 245, 344–352. [DOI] [PubMed] [Google Scholar]
- 14.Dehò G., Zangrossi,S., Ghisotti,D. and Sironi,G. (1988) Alternative promoters in the development of bacteriophage plasmid P4. J. Virol., 6, 1697–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dehò G., Zangrossi,S., Sabbattini,P., Sironi,G. and Ghisotti,D. (1992) Bacteriophage P4 immunity controlled by small RNAs via transcription termination. Mol. Microbiol., 6, 3415–3425. [DOI] [PubMed] [Google Scholar]
- 16.Polo S., Sturniolo,T., Dehò,G. and Ghisotti,D. (1996) Identification of a phage-coded DNA-binding protein that regulates transcription from late promoters in bacteriophage P4. J. Mol. Biol., 257, 745–755. [DOI] [PubMed] [Google Scholar]
- 17.Terzano S., Christian,R., Espinoza,F.H., Calendar,R., Dehò,G. and Ghisotti,D. (1994) A new gene of bacteriophage P4 that controls DNA replication. J. Bacteriol., 176, 6059–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziegelin G., Calendar,R., Ghisotti,D., Terzano,S. and Lanka,E. (1997) Cnr protein, the negative regulator of bacteriophage P4 replication, stimulates specific DNA binding of its initiator protein α. J. Bacteriol., 179, 2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziegelin G., Calendar,R., Lurz,R. and Lanka,E. (1997) The helicase domain of phage P4 α protein overlaps the specific DNA binding domain. J. Bacteriol., 179, 4087–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 21.Golemis E.A., Gyuris,J. and Brent,R. (1994) Two hybrid system/interaction traps. In Ausubel,F.M. and Struhl,K. (eds), Current Protocols in Molecular Biology. John Wiley and Sons, New York, NY, pp. 13.14.1–13.14.17.
- 22.Kunkel T.A., Roberts,J.D. and Zakour,R.A. (1987) Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol., 154, 367–382. [DOI] [PubMed] [Google Scholar]
- 23.Sherman F., Fink,G.R. and Hicks,J.H. (1986) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 24.Battaglia,P.A., Longo,F., Ciotta,C., Del Grosso,M.F., Ambrosini,E. and Gigliani,F. (1994) Genetic tests to reveal Tat homodimer formation and select Tat homodimer inhibitor. Biochem. Biophys. Res. Commun., 201, 701–708. [DOI] [PubMed] [Google Scholar]
- 25.Bailone A. and Galimbert,F. (1980) Nucleotide sequence of the operators of λ ultravirulent mutants. Nucleic Acids Res., 8, 2147–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castagnoli L., Vetriani,C. and Cesareni,G. (1994) Linking an easily detectable phenotype to the folding of a common structural motif. J. Mol. Biol., 237, 378–387. [DOI] [PubMed] [Google Scholar]
- 27.Longo,F., Marchetti,M.A., Castagnoli,L., Battaglia,P.A. and Gigliani,F. (1995) A novel approach to protein-protein interaction: complex formation between the p53 tumor suppressor and the HIV Tat proteins. Biochem. Biophys. Res. Commun., 201, 701–708. [DOI] [PubMed] [Google Scholar]
- 28.Hu J.C., O’Shea,E.K., Kim,P.S. and Sauer,R.T. (1990) Sequence requirements for coiled-coils: analysis repressor-GCN4 leucine zipper fusions. Science, 250, 1400–1403. [DOI] [PubMed] [Google Scholar]
- 29.Ptashne M. (1992) A Genetic Switch, 2nd edn. Cell Press and Blackwell Scientific Publications, Cambridge, MA.
- 30.Ziegelin G. and Lanka,E. (1995) Bacteriophage P4 replication. FEMS Microbiol. Rev., 17, 99–107. [DOI] [PubMed] [Google Scholar]
- 31.Chang A.C. and Cohen,S.N. (1978) Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol., 134, 1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolivar F., Rodriguez,R.L., Greene,P.J., Betlach,M.C., Heynecker,H.L., Boyer,H.W., Crosa,J.H. and Falkow,S. (1977) Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene, 2, 95–113. [PubMed] [Google Scholar]
- 33.Lessl M., Balzer,D., Lurz,R., Waters,V.L., Guyney,D.G. and Lanka,E. (1992) Dissection of IncP conjugative plasmid transfer: definiton of the transfer region Tra2 by mobilization of the Tra1 region in trans. J. Bacteriol., 174, 2493–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strack B., Lessl,M., Calendar R. and Lanka,E. (1992). A common sequence motif, -E-G-Y-A-T-A, identified within the primase domains of plasmid-encoded I- and P-type DNA primases and the α protein of the Escherichia coli satellite phage P4. J. Biol. Chem., 267, 13062–13072. [PubMed] [Google Scholar]
- 35.Vieira J. and Messing,J. (1982) The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene, 19, 259–268. [DOI] [PubMed] [Google Scholar]
- 36.Halling C., Calendar,R., Christie,G.E., Dale,E.C., Dehò,G., Finkel,S., Flensburg,J., Ghisotti,D., Kahn,M.L., Lane,K.B., Lin,C.-S., Lindqvist,B.H., Pierson,L.S.,III, Six,E.W., Sunshine,M.G. and Ziermann,R. (1990) DNA sequence of satellite bacteriophage P4. Nucleic Acids Res., 18, 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lucchini G., Hinnebusch,A.G., Chen,C. and Fink,G.R. (1984) Positive regulatory interactions of the HIS4 gene of Saccharomyces cerevisiae. Mol. Cell. Biol., 4, 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]