

Abstract
Histone deacetylases (HDACs) are enzymes that balance the acetylation activities of histone acetyltransferases on chromatin remodeling and play essential roles in regulating gene transcription. In the past several years, the role of HDACs in cancer initiation and progression, as well as the therapeutic effects of HDAC inhibitors in various types of cancer, has been well studied. Recent studies indicated that HDAC activity is also associated with the development and progression of some chronic diseases characterized by fibrosis, including chronic kidney disease, cardiac hypertrophy, and idiopathic pulmonary fibrosis. Here, we review what is known about HDACs in the progression of tissue fibrosis and the potential applications of HDAC inhibitors in the treatment of disorders associated with fibroblast activation and proliferation.
Introduction
Histone acetylation/deacetylation of the N-terminal tail is crucial in modulating gene expression. The balance between the acetylated/deacetylated states of histones is mediated by two different sets of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs preferentially acetylate specific lysine substrates on histones and some nonhistone proteins. Histone acetylation can lead to changes in chromatin structure and may decrease the histone-DNA interaction, promoting accessibility of the DNA for transcription activation. Acetylation of some nonhistone proteins, for example, transcriptional factors, can also affect their DNA binding properties and subsequently may regulate gene transcription (Boyes et al., 1998; Glozak et al., 2005). In addition, acetylation/deacetylation can occur in numerous cytoplasmic proteins, including tubulin and heat shock protein 90 and alter their functions (Kovacs et al., 2005; Catalano et al., 2007; Choudhary et al., 2009).
HDACs are a family of enzymes that remove acetyl groups from a ε-N-acetyl lysine amino acid on a histone and restore the positive charge to lysine residues (Thiagalingam et al., 2003; Acharya et al., 2005). HDAC proteins are also referred to as lysine deacetylases to more precisely describe their function rather than their targets, since they can catalyze deacetylation of many nonhistone proteins in addition to histones (Glozak et al., 2005). HDACs are classified into four groups based on their homology to yeast histone deacetylases: Class I (HDAC1, 2, 3 and 8) are related to yeast RPD3 gene and are mostly located in the nuclei; Class II (HDAC4, 5, 6, 7, 9 and 10) are related to yeast Hda1 gene and are primarily located in the cytoplasm but can shuttle to nucleus; Class III (SIRT1–7), also known as the sirtuins, are related to the Sir2 gene and are virtually unaffected by the HDAC inhibitors; and Class IV (HDAC11) has a conserved domain in the catalytic regions of both Class I and Class II enzymes. Class I HDACs, such as HDAC1 and HDAC2, seem to be important in the regulation of proliferation and survival of cancer cells (Fischle et al., 2002; Dokmanovic and Marks, 2005). Increased expression of some of the Class II HDAC enzymes (i.e., HDAC6) is linked to better survival in breast cancer and reduced expression of Class II HDAC enzymes HDAC 5 and HDAC10 is associated with poor prognosis in lung cancer patients (Osada et al., 2004). The first two groups of HDACs are considered “classical” HDACs and are the major targets of the present applications of HDAC inhibitors in therapy of cancer or other diseases.
Recent studies have shown that HDACs are critically involved in tissue fibrosis in multiple organs including kidney, heart, and lung. In this article, we review the role of HDACs in the development and progression of tissue fibrosis and discuss the potential applications of HDACs inhibitors in treatment of these disorders (Table 1).
TABLE 1.
Summery of HDAC inhibitor applications in experimental fibrotic disorders
| Disease Model | HDAC Inhibitors | Selectivity | Beneficial Effects after Treatment | Mechanism | References |
|---|---|---|---|---|---|
| Renal fibrosis | TSA | HDACI/II | Attenuates renal fibroblast proliferation, α-SMA expression and fibronectin deposition | Inhibits STAT3 activation induced by UUO. | Pang et al, 2009 |
| Sodium Valproate | HDACI | Attenuates macrophage infiltration and fibrotic changes. | Reduces CSF-1 expression induced by TNF-α in renal tubular cells. | Marumo et al, 2009 | |
| Diabetic nephropathy | TSA | HDACI/II | Decreases expressions of ECM components and prevents EMT in STZ-induced diabetic kidneys | Suppresses the ROS mediate TGF-β1-induced activation of HDAC-2. | Noh et al, 2009 |
| Valproic acid | HDACI | ||||
| SK-7041 | HDACI | ||||
| Cardiac hypotrophy and fibrosis | TSA | HDACI/II | Blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding; reverses atrial fibrosis, connexin40 remodeling and atrial arrhythmia vulnerability | Reduces expression of early growth response gene 1 (EGR-1), but detailed mechanism remains unclear. | Kee et al, 2006 |
| SK-7041 | HDACI | ||||
| Idiopathic pulmonary fibrosis | TSA | HDACI/II | Inhibits TGF-β1-mediated α-SMA type I collagen mRNA induction and contractile response in NHLFs. | Reduces AKT phosphorylation during the differentiation of NHLFs to myofibroblasts | Guo et al, 2009 |
| SAHA | pan-HDAC | Increases PGE2 synthesis in fibroblasts from IPF patients | Restores cyclooxygenase-2 (COX-2) mRNA and protein expression that has been repressed in IPF. | Coward et al, 2009 | |
| LBH589 | pan-HDAC | ||||
| System sclerosis | TSA | HDACI/II | Prevents SSc related tissue fibrosis by reducing the collagen I and fibronectin in SSc skin fibroblasts. | Inhibits the nuclear translocation and Smad binding to DNA. | Huber et al, 2007 |
| Cystic fibrosis | 4PBA | Pan-HDAC | Increases amounts of delta F508-CFTR and corrects cellular trafficking of CFTR. | Enables a greater fraction of delta F508-CFTR to escape degradation and appear at the cell surface. | Rubenstein et al, 1997, 2000 |
The Role of HDACs in Fibrosis-Related Kidney Diseases
HDACs in Renal Interstitial Fibrosis
Renal interstitial fibrosis is characterized by aberrant activation and growth of renal fibroblasts. The activated fibroblast, termed myofibroblast, demonstrate specific phenotypic changes, including the expression of α-smooth muscle actin (α-SMA) and increased production of extracellular matrix (ECM) components. The number of interstitial myofibroblasts directly correlates with the extent of tubulointerstitial scarring and functional outcome in clinical glomerulonephritis and IgA nephropathy (Arakawa et al., 2008).
Activation and proliferation of renal fibroblasts are stimulated by a variety of growth factors and cytokines, such as transforming growth factor, platelet-derived growth factor, fibroblast growth factor, and interleukin-6. Several intracellular signaling pathways, including the signal transducer and activator of transcription3 (STAT3) pathways, are activated in response to those growth factors/cytokines. STAT3 belongs to a family of latent cytoplasmic transcription factors. Activated STAT3 forms homodimers or heterodimers and then translocates to the nucleus, where it binds with DNA and regulates gene transcription. Numerous targeted genes of STAT3 have been identified, including cyclin D1. An abundance of active STAT3 has been observed in renal interstitial fibroblasts in the unilateral ureteral obstruction (UUO) model of renal fibrosis (Kuratsune et al., 2007; Pang et al., 2009). Regulation of STAT3 activity is critical for the modulation of its biological functions. In addition to tyrosine phosphorylation, deacetylation is involved in regulation of STAT3 activity as shown in one of our recent publications (Pang et al., 2009).
In that article, Pang et al. (2009) examined the effect of trichostatin A (TSA), a HDAC I/II inhibitor, on the activation and proliferation of renal interstitial fibroblasts in vitro and in vivo. In the in vitro studies employing cultured rat renal interstitial fibroblasts (NRK-49F), TSA treatment inhibited fibroblast proliferation as indicated by decreasing cell numbers and suppressing the expression of cyclin D1. TSA also blocked fibroblast activation as shown by diminishing expression of α-SMA and fibronectin. STAT3 is phosphorylated when NRK-49F cells are activated and start to proliferate; this response was inhibited in the presence of TSA as well (Fig. 1): hydroxamates, cyclic peptides, aliphatic acids, and benzamides (Miller et al., 2003; Marks et al., 2004; Dokmanovic and Marks, 2005; Marks and Xu, 2009). The major mechanism of these HDAC inhibitors is to bind a critical Zn2+ ion required for catalytic function of the HDAC enzymes (Finnin et al., 1999).
Fig. 1.

HDAC inhibitors attenuate STAT3-mediated fibrosis. HDAC activity is required for STAT3 phosphorylation at tyrosine 705 and dimerization (activation). The dimerized STAT3 is translocated into the nucleus, where it regulates transcription of the target genes associated with development of tissue fibrosis, such as α-smooth muscle actin, fibronectin, and collagen I. HDAC inhibitors such as TSA can inhibit these actions of STAT3 and subsequently attenuate its effects in association of fibrosis.
Moreover, treatment with AG490 (tyrphostinAG 490 [(E)-2-cyano-3-(3,4-dihydrophenyl)-N-(phenylmethyl)-2-propenamide]), a STAT pathway inhibitor that is a synthetic tyrphostin derived from benzylidine malononitrile, suppressed all of the above events associated with renal fibroblast activation and proliferation, whereas a knockout of STAT3 reduced the inhibitory effect of TSA on proliferation of mouse embryonic fibroblasts. In an in vitro mouse model of the fibrotic kidney induced by UUO, administration of TSA also attenuated the proliferation of renal fibroblasts, expression of α-SMA, and deposition of fibronectin. In addition, STAT3 phosphorylation induced in this UUO model was completely blocked by TSA. The underlying mechanism by which inhibition of HDACs alters the tyrosine phosphorylation status is not clear, but these data indicate that HDAC activity is required for STAT3 tyrosine phosphorylation and activation. The data are also consistent with the idea that HDACs are involved in proliferation and activation of renal fibroblasts as well as development of tubulointerstitial fibrosis (Fig. 1).
In a separate study, Marumo et al. (2009) also observed an increase in the expression of HDAC1 and HDAC2 and a decrease in histone acetylation in kidneys injured by ureteral obstruction. Immunohistochemical analysis revealed that HDAC1 and HDAC2 were induced in renal tubular cells. Treatment with a TSA attenuated macrophage infiltration and fibrotic changes. The induction of colony stimulating factor-1 (CSF-1), a chemokine known to be involved in macrophage infiltration in tubulointerstitial injury, was reduced in injured kidneys of mice treated with TSA. TSA, valproate, and the knockdown of either HDAC1 or HDAC2 significantly reduced CSF-1 expression induced by tumor necrosis factor-α in renal tubular cells. These results suggest that increased expression of HDAC1 and HDAC2 contribute to the production of CSF-1, macrophage infiltration, and other profibrotic responses in response to injury and implicate a potential of HDAC inhibition in reducing inflammation and fibrosis in tubulointerstitial injury.
HDACs in Diabetic Nephropathy
Progressive accumulation of ECM in glomerular mesangium and tubulointerstitium is the hallmark of diabetic nephropathy (Mauer et al., 1984). It has been reported that myofibroblasts can be derived from tubular epithelial cells by epithelial to mesenchymal transition (EMT), a process that is involved in the loss of epithelial cell adhesion, expression of α-SMA and reorganization of actin, disruption of tubular basement membrane, and enhancement of cell migration and invasion. Recently, Noh et al. (2009) demonstrated that TSA decreased the expressions of ECM components and prevented EMT in streptozotocin-induced diabetic kidneys as well as rat kidney tubular epithelial cells (NRK-52E) exposed to transforming growth factor -β1 (TGF-β1). Similar effects were also observed in NRK-52E cells treated with another HDAC I/II inhibitor, valproic acid, or HDAC I-selective HDAC inhibitor 3-(4-substituted phenyl)-N-hydroxy-2-propenamide (SK-7041). This suggests that Class I HDACs play an essential role in initiation of EMT. Among the six HDACs tested (HDAC-1–5 and HDAC-8), HDAC-2 activity was significantly increased in the kidneys of streptozotocin-induced diabetic rats, db/db mice, and TGF-β1-treated NRK-52E cells. Knockdown of HDAC-2 with its specific siRNA decreased expression of fibronectin and α-SMA in NRK-52E cells. These findings suggest the importance of HDAC-2 in mediating accumulation of ECM and development of EMT (Noh et al., 2009). Further studies are needed to validate the role of HDACs in vivo models of diabetic kidney.
HDACs in Polycystic Kidney Disease
Polycystic kidney disease (PKD) is a common human genetic disease with mutation of the cilia-localized polycystin proteins 1 and 2 (PKD1 and PKD2) responsible for the significant majority of PKD patients. Pugacheva et al. (2007) demonstrated that activation of HDAC6, a tubulin deacetylase, promotes ciliary disassembly, whereas treatment with either TSA, a HDAC pan inhibitor, or tubacin, an inhibitor specifically targeting HDAC6, completely blocked serum-induced ciliary disassembly. Cao et al. (2009) recently demonstrated that inhibiting Class I HDACs, either by valproic acid or by knocking down HDAC1, suppressed kidney cyst formation and body curvature caused by PKD2 deficiency. In addition, they showed that valproic acid was effective in attenuating the progression of cyst formation and slowing the decline of kidney function in a mouse autosomal dominant PKD model. Taken together, these studies suggest that HDACs also play a critical role in the pathogenesis of PKD.
The Role of HDACs in Cardiac Hypotrophy and Fibrosis
Cardiac fibrosis is a classical feature of hypertrophy and is characterized by the expansion of the extracellular matrix due to the accumulation of collagen, particularly collagen types I and III (Manabe et al., 2002; Zhang et al., 2002). Treatment of cultured cardiac myocytes with HDAC inhibitors prevented pressure overload-induced hypertrophy after constriction of the thoracic aorta (sarcomere organization and activation of the fetal gene program normally evoked by hypertrophic agonists), suggesting that HDACs play dual roles as repressors and activators of cardiac hypertrophy (Antos et al., 2003). Because both TSA and SK-7041, two Class I HDAC-selective inhibitors, can block the development of cardiac hypertrophy, this suggests that Class I HDACs are prohypertrophic factors in cardiomyocyte (Kee et al., 2006). Besides, Class II HDACs may also be involved in the pathogenesis of cardiac hypertrophy. In a study investigating the action of natriuretic peptide receptor-A-induced cardiac hypertrophy and fibrosis, Ellmers et al. (2007) demonstrated that HDAC 7a mRNA expression was increased in natriuretic peptide receptor-A knockout mice at a more advanced stage, followed by increased TGF β1 and other structural molecules associated with cardiac fibrosis such as collagen I.
Atrial interstitial fibrosis is a significant factor driving arrhythmia genesis and is highly prevalent in heart failure and cardiac hypertrophy (Verheule et al., 2004). Atrial fibrosis influences the development of atrial fibrillation, particularly in the setting of structural heart disease where angiotensin-inhibition is partially effective in reducing atrial fibrosis and atrial fibrillation. Previous studies showed the involvement of the renin-angiotensin-aldosterone system in the development of atrial fibrosis (Sun et al., 1997; McEwan et al., 1998). Recent studies demonstrated that HDAC overactivation also causes atrial fibrosis, connexin 40 down-regulation, and atrial arrhythmia susceptibility in transgenic mice and that pharmacologic inhibition of HDAC suppresses deleterious atrial remodeling. In mice overexpressing homeodomain-only protein, TSA treatment reduced atrial arrhythmia duration and atrial fibrosis and normalized the expression and size distribution of connexin 40 gap junctions. These results clearly indicate that HDAC inhibition reverses atrial fibrosis, connexin 40 remodeling, and atrial arrhythmia vulnerability (Liu et al., 2008).
The Role of HDACs in Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive and lethal fibrotic lung disorder characterized by inflammatory injury and irreversible fibrosis of the lung parenchyma (Gross and Hunninghake, 2001). Among the identified fibrotic mediators, the cytokine TGF-β1 and the lipid mediator prostaglandin E2 (PGE2) have been identified as potent profibrotic and antifibrotic mediators, respectively, and therefore are critical in IPF pathogenesis (Atamas and White, 2003).
To investigate the molecular link between TGF-β1-mediated myofibroblast differentiation and HDAC activity, normal human lung fibroblasts (NHLFs) were treated with TSA. TSA inhibited TGF-β1-mediated expression of α-SMA and collagen I. Furthermore, inhibition of α-SMA by TSA decreased phosphorylation of AKT, a critical mediator in epithelial-mesenchymal transition (Lien et al., 2006; Guo et al., 2009). Additional studies identified HDAC4 as a HDAC that mediates those processes in TGF-β1-treated NHLFs. Taken together, these data suggest that differentiation of NHLFs to myofibroblasts is mediated by HDAC4-dependent activation of AKT signaling pathway (Guo et al., 2009).
PGE2 synthesis is associated with cyclooxygenase-2 activation, and decreased cyclooxygenase-2 activity contributes to a lower production of prostaglandin E2. A recent study showed that COX-2 expression declined in the lung of the patients with IPF, whereas treatment with HDAC inhibitors suberoylanilide hydroxamic acid (SAHA) and LBH589 (panobinostat) was effective in the restoration of cyclooxygenase-2 expression that had been repressed in IPF. The mechanism by which HDAC regulates expression of cyclooxygenase-2 remains unclear but may be associated with epigenetic abnormality caused by histone hypoacetylation in IPF (Wilborn et al., 1995; Coward et al., 2009). In another recent study, Wang et al. (2009) showed that SAHA abrogated TGF-β1-induced transdifferentiation of lung fibroblasts into myofibroblasts and inhibited serum-induced fibroblast proliferation, suggesting a therapeutic potential of HDAC inhibitors in pulmonary fibrosis.
The Role of HDACs in Other Diseases Involving Fibrotic Injuries
Systemic Sclerosis
Systemic sclerosis (SSc) is characterized by severe fibrosis in the skin and various other organs (Derk and Jimenez, 2003). Several profibrotic cytokines, including TGF-β1, interleukin-4, and platelet-derived growth factor (Gay et al., 1989; Higley et al., 1994; Blobe et al., 2000; Distler et al., 2006), as well as the downstream signaling cascades of TGF-β1, have been implicated in the pathogenesis of SSc (Mori et al., 2003; Ishida et al., 2006). It appears that HDAC activity is also required for initiation and development of SSc. This is evident by the fact that inhibition of HDAC activity by TSA attenuated expression of collagen I and fibronectin in both normal and SSc skin fibroblasts and reduced the accumulation of total collagen proteins in SSc skin fibroblasts in response to various cytokines (Huber et al., 2007). Moreover, in a mouse model of skin fibrosis, Hemmatazad et al. (2009) showed that TSA treatment prevented the dermal accumulation of extracellular matrix.
Primary Myelofibrosis
Primary myelofibrosis is a chronic malignant hematological disorder with fibrotic pathogenic changes (Tefferi, 2005). Wang et al. (2008) reported that some Class I/II isoforms of HDACs (HDAC1, 2, 4, 5, 6, 8, 10) and all Class III HDACs were significantly elevated in patients with primary myelofibrosis relative to those with other myeloproliferative neoplasms and normal volunteers, suggesting that HDACs may be involved in the pathogenesis of primary myelofibrosis. Further studies are needed to identify the role of individual HDACs involved in this process.
HDACs are able to increase the stability of hypoxia-inducible factor-1α through deacetylation (Jeong et al., 2002), suggesting the possibility that HDACs are indirectly involved in increased angiogenesis occurring in primary myelofibrosis (Arora et al., 2004). HDACs are also found to interact with the nuclear factor-κB (NF-κB) pathway by acetylating and deacetylating NF-κB, a transcriptional factor that is associated with the pathogenesis of primary myelofibrosis (Komura et al., 2005). HDAC1 and HDAC3 bind to the inhibitor of NF-κBα, IκBα, and as a result, NF-κB expression levels are increased (Kiernan et al., 2003; Viatour et al., 2003). This study has laid the groundwork for suggesting a role for HDAC inhibitors in primary myelofibrosis treatment (Wang et al., 2008). A clinical trial for treatment of primary myelofibrosis with HDAC inhibitors is underway (Hemavathy and Wang, 2009).
Cystic Fibrosis
Cystic fibrosis (CF) is a common hereditary disease most commonly characterized by infection-induced inflammation followed by pulmonary fibrosis. It stems from mutations in a chloride channel responsible for transepithelial salt and water transport, the cystic fibrosis transmembrane conductance regulator (CFTR) (Dörk et al., 1991; Jensen et al., 1995). The mutant CFTR is retained in the endoplasmic reticulum and degraded, resulting in the loss of CFTR in patients with CF (Dörk et al., 1991; Jensen et al., 1995).
However, in the presence of sodium 4-phenylbutyrate (4-PBA), a HDAC inhibitor, a greater fraction of ΔF508-CFTR escapes from degradation and appears at the cell surface of the primary cultures of nasal polyp epithelia from CF patients and the CF bronchial epithelial cell line IB3-1. These data suggest that inhibition of HDAC may be able to correct the CF phenotype in patients carrying mutated CFTR (Rubenstein et al., 1997). In addition, 4-PBA treatment was reported to correct cellular trafficking of CFTR (Rubenstein and Zeitlin, 2000). Mechanistic studies have shown that the activation of intestinal CFTR by histone acetylation was mediated by transcription factors hepatic nuclear factor 1α, Cdx2, and Tcf4, which converge to modify chromatin architecture. These studies suggest a therapeutic potential for inhibition of HDAC activity in increasing CFTR expression (Paul et al., 2007).
HDAC Inhibitors and Their Applications
HDAC inhibitors are a new group of agents that can regulate gene expression, induce apoptosis, and arrest cell cycle of cancer cells by altering the acetylation status of chromatin and other nonhistone proteins. They are divided into four categories based on their structures (Table 2).
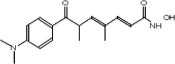
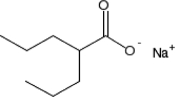
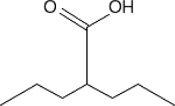
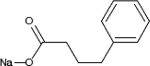
TABLE 2.
Chemical structure of HDAC inhibitors
| TSA | Sodium Valproate | Valproic Acid (Depakote) | 4-PBA |
|---|---|---|---|
 |
 |
 |
 |
| SAHA (Vorinostat) | LBH589 (Panobinostat) | SK-7041 |
|---|---|---|
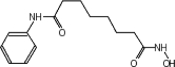 |
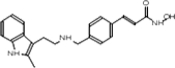 |
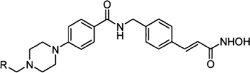 |
TSA (Taunton et al., 1996); sodium valproate (Gottlicher et al., 2004); valproic acid (Depakote) (Zelent et al., 2005); 4-PBA (Wiech et al., 2009); SAHA (vorinostat) (Marks and Breslow, 2007); LBH589 (panobinostat) (Atadja et al., 2009); SK-7041(Kim et al., 2003).
Genetic deletion of the Class I genes HDAC1 or HDAC2 results in embryonic or perinatal lethality, respectively (Lagger et al., 2002; Trivedi et al., 2007). In many tumor cell lines, inhibition or down-regulation of HDACs also leads to cell-cycle arrest by up-regulation of cell cycle gene p21, and blockade of the cyclin D/cyclin-dependent kinase 4 complex (Richon et al., 2000; Sandor et al., 2000). In addition, HDAC inhibitors can suppress the tumor cells survival by accelerating degradation of proangiogenic transcription factor, hypoxia-inducible factor 1α, decreasing the expression of vascular endothelial growth factor receptor, or/and increasing generation of intracellular reactive oxygen species (Deroanne et al., 2002; Jeong et al., 2002; Carew et al., 2008).
As described above, numerous investigations have been conducted to address the mechanisms of anti-tumor actions of HDAC inhibitors. Preclinical and early clinical trials of HDAC inhibitors have achieved variable efficacy (Lane and Chabner, 2009; Ma et al., 2009; Marks and Xu, 2009), and SAHA (vorinostat) and romidepson have recently been approved for treatment of refractory cutaneous T-cell lymphoma (Monneret, 2007; Grant et al., 2010). Although treatment of cancer has been the primary target for the clinical development of HDAC inhibitors, administration of HDAC inhibitors has also shown beneficial effects in some noncancer disorders, such as sickle cell anemia, muscular dystrophy, neurodegenerative diseases, and inflammatory disorders (Wiech et al., 2009). In addition, an inhibitory effect of TSA on hepatic fibrosis has been reported by Niki et al. (1999). Recently, we reported that inhibition of the HDAC activity with TSA also decreased activation of myofibroblast and excessive expression of ECM components such as fibronectin (Pang et al., 2009). In addition, increased HDAC activity was required for TGF-β1-induced myofibroblastic differentiation (Glenisson et al., 2007; Guo et al., 2009). Thus, understanding the molecular events responsible for activation and proliferation of renal fibroblasts may lead to new approaches for slowing the progression of chronic kidney disease and chronic diseases associated with fibrosis in other organs.
Conclusion and Future Directions
Recent studies have demonstrated that treatment with HDAC inhibitors inhibits activation and proliferation of cultured fibroblasts and attenuates fibrosis in multiple organs in vivo animal models. This suggests that HDAC activity is required for the development and progression of tissue fibrosis. The mechanisms of HDAC-mediated profibrotic actions remain largely unknown but may be associated with expression of some fibrosis-related genes and activation of some cellular molecules that mediate tissue fibrosis. For example, HDAC inhibition can decrease transcription of profibrotic genes such as CSF-1 (Marumo et al., 2009) and reduce activation of some transcriptional factors, such as STAT3 and NF-κB (Hu and Colburn, 2005; Pang et al., 2009). In addition, HDAC inhibitors have been reported to suppress TGF-β, a major profibrotic cytokine involved in induced activation and proliferation of fibroblasts and deposition of ECM, suggesting the involvement of HDACs in regulating TGF-triggered fibrotic signaling; however, the targeted proteins have not been identified. Currently, the profile of HDAC-modulated proteins in the setting of fibrosis is not clear. A global analysis of protein lysine acetylation in response to HDAC inhibition using proteomic approach would resolve this issue. This knowledge is not only important for further understanding of the mechanism by which HDACs induce tissue fibrosis but also helpful to develop HDAC inhibition as a novel therapeutic strategy for diseases with fibrosis as their pathogenesis.
This work was supported by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK071997].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- HAT
- histone acetyltransferase
- HDAC
- histone deacetylase
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CSF-1
- colony stimulating factor-1
- ECM
- extracellular matrix
- EMT
- epithelial to mesenchymal transition
- IPF
- idiopathic pulmonary fibrosis
- NRK-49F
- rat renal interstitial fibroblasts
- NRK-52E
- rat kidney tubular epithelial cells
- NHLF
- normal human lung fibroblast
- PDGF
- platelet-derived growth factor
- PGE2
- prostaglandin E2
- PKD
- polycystic kidney disease
- SAHA
- suberoylanilide hydroxamic acid
- SK-7041
- 3-(4-substituted phenyl)-N-hydroxy-2-propenamide
- |ga-SMA
- |ga-smooth muscle actin
- STAT3
- signal transducer and activator of transcription3
- UUO
- unilateral ureteral obstruction
- SSc
- systemic sclerosis
- TGF-|gb1
- transforming growth factor-|gb1
- TSA
- trichostatin A
- 4-PBA
- sodium 4-phenylbutyrate
- LBH589
- panobinostat
- AG490
- tyrphostinAG 490 [(E)-2-cyano-3-(3,4-dihydrophenyl)-N-(phenylmethyl)-2-propenamide]
References
- Acharya MR, Sparreboom A, Venitz J, Figg WD. ( 2005) Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol 68: 917– 932 [DOI] [PubMed] [Google Scholar]
- Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. ( 2003) Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem 278: 28930– 28937 [DOI] [PubMed] [Google Scholar]
- Arakawa T, Masaki T, Hirai T, Doi S, Kuratsune M, Arihiro K, Kohno N, Yorioka N. ( 2008) Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol Dial Transplant 23: 3418– 3426 [DOI] [PubMed] [Google Scholar]
- Arora B, Mesa R, Tefferi A. ( 2004) Angiogenesis and anti-angiogenic therapy in myelofibrosis with myeloid metaplasia. Leuk Lymphoma 45: 2373– 2386 [DOI] [PubMed] [Google Scholar]
- Atadja P. ( 2009) Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett 280: 233– 241 [DOI] [PubMed] [Google Scholar]
- Atamas SP, White B. ( 2003) Cytokine regulation of pulmonary fibrosis in scleroderma. Cytokine Growth Factor Rev 14: 537– 550 [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. ( 2000) Role of transforming growth factor beta in human disease. N Engl J Med 342: 1350– 1358 [DOI] [PubMed] [Google Scholar]
- Boyes J, Byfield P, Nakatani Y, Ogryzko V. ( 1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 396: 594– 598 [DOI] [PubMed] [Google Scholar]
- Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R, Sun Z. ( 2009) Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci USA 106: 21819– 21824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew JS, Giles FJ, Nawrocki ST. ( 2008) Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett 269: 7– 17 [DOI] [PubMed] [Google Scholar]
- Catalano MG, Poli R, Pugliese M, Fortunati N, Boccuzzi G. ( 2007) Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocr Relat Cancer 14: 839– 845 [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. ( 2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325: 834– 840 [DOI] [PubMed] [Google Scholar]
- Coward WR, Watts K, Feghali-Bostwick CA, Knox A, Pang L. ( 2009) Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol 29: 4325– 4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derk CT, Jimenez SA. ( 2003) Systemic sclerosis: current views of its pathogenesis. Autoimmun Rev 2: 181– 191 [DOI] [PubMed] [Google Scholar]
- Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens BV, et al. ( 2002) Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 21: 427– 436 [DOI] [PubMed] [Google Scholar]
- Distler JH, Jüngel A, Caretto D, Schulze-Horsel U, Kowal-Bielecka O, Gay RE, Michel BA, Müller-Ladner U, Kalden JR, Gay S, et al. ( 2006) Monocyte chemoattractant protein 1 released from glycosaminoglycans mediates its profibrotic effects in systemic sclerosis via the release of interleukin-4 from T cells. Arthritis Rheum 54: 214– 225 [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Marks PA. ( 2005) Prospects: histone deacetylase inhibitors. J Cell Biochem 96: 293– 304 [DOI] [PubMed] [Google Scholar]
- Dörk T, Wulbrand U, Richter T, Neumann T, Wolfes H, Wulf B, Maass G, Tümmler B. ( 1991) Cystic fibrosis with three mutations in the cystic fibrosis transmembrane conductance regulator gene. Hum Genet 87: 441– 446 [DOI] [PubMed] [Google Scholar]
- Ellmers LJ, Scott NJ, Piuhola J, Maeda N, Smithies O, Frampton CM, Richards AM, Cameron VA. ( 2007) Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J Mol Endocrinol 38: 245– 257 [DOI] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. ( 1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401: 188– 193 [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. ( 2002) Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 9: 45– 57 [DOI] [PubMed] [Google Scholar]
- Gay S, Jones RE, Jr., Huang GQ, Gay RE. ( 1989) Immunohistologic demonstration of platelet-derived growth factor (PDGF) and sis-oncogene expression in scleroderma. J Invest Dermatol 92: 301– 303 [DOI] [PubMed] [Google Scholar]
- Glenisson W, Castronovo V, Waltregny D. ( 2007) Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim Biophys Acta 1773: 1572– 1582 [DOI] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, Seto E. ( 2005) Acetylation and deacetylation of non-histone proteins. Gene 363: 15– 23 [DOI] [PubMed] [Google Scholar]
- Gottlicher M. ( 2004) Valproic acid: an old drug newly discovered as inhibitor of histone deacetylases. Ann Hematol 83 ( Suppl 1): S91– S92 [DOI] [PubMed] [Google Scholar]
- Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, Gardner ER, Figg WD, Bates SE. ( 2010) Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther 10: 997– 1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross TJ, Hunninghake GW. ( 2001) Idiopathic pulmonary fibrosis. N Engl J Med 345: 517– 525 [DOI] [PubMed] [Google Scholar]
- Guo W, Shan B, Klingsberg RC, Qin X, Lasky JA. ( 2009) Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol Lung Cell Mol Physiol 297: L864– L870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemavathy K, Wang JC. ( 2009) Epigenetic modifications: new therapeutic targets in primary myelofibrosis. Curr Stem Cell Res Ther 4: 281– 286 [DOI] [PubMed] [Google Scholar]
- Hemmatazad H, Rodrigues HM, Maurer B, Brentano F, Pileckyte M, Distler JH, Gay RE, Michel BA, Gay S, Huber LC, et al. ( 2009) Histone deacetylase 7, a potential target for the antifibrotic treatment of systemic sclerosis. Arthritis Rheum 60: 1519– 1529 [DOI] [PubMed] [Google Scholar]
- Higley H, Persichitte K, Chu S, Waegell W, Vancheeswaran R, Black C. ( 1994) Immunocytochemical localization and serologic detection of transforming growth factor beta 1. Association with type I procollagen and inflammatory cell markers in diffuse and limited systemic sclerosis, morphea, and Raynaud's phenomenon. Arthritis Rheum 37: 278– 288 [DOI] [PubMed] [Google Scholar]
- Hu J, Colburn NH. ( 2005) Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res 3: 100– 109 [DOI] [PubMed] [Google Scholar]
- Huber LC, Distler JH, Moritz F, Hemmatazad H, Hauser T, Michel BA, Gay RE, Matucci-Cerinic M, Gay S, Distler O, et al. ( 2007) Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum 56: 2755– 2764 [DOI] [PubMed] [Google Scholar]
- Ishida W, Mori Y, Lakos G, Sun L, Shan F, Bowes S, Josiah S, Lee WC, Singh J, Ling LE, et al. ( 2006) Intracellular TGF-beta receptor blockade abrogates Smad-dependent fibroblast activation in vitro and in vivo. J Invest Dermatol 126: 1733– 1744 [DOI] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. ( 1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83: 129– 135 [DOI] [PubMed] [Google Scholar]
- Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. ( 2002) Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 111: 709– 720 [DOI] [PubMed] [Google Scholar]
- Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, et al. ( 2006) Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113: 51– 59 [DOI] [PubMed] [Google Scholar]
- Kiernan R, Brès V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. ( 2003) Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem 278: 2758– 2766 [DOI] [PubMed] [Google Scholar]
- Kim DK, Lee JY, Kim JS, Ryu JH, Choi JY, Lee JW, Im GJ, Kim TK, Seo JW, Park HJ, et al. ( 2003) Synthesis and biological evaluation of 3-(4-substituted-phenyl)-N-hydroxy-2-propenamides, a new class of histone deacetylase inhibitors. J Med Chem 46: 5745– 5751 [DOI] [PubMed] [Google Scholar]
- Komura E, Tonetti C, Penard-Lacronique V, Chagraoui H, Lacout C, Lecouédic JP, Rameau P, Debili N, Vainchenker W, Giraudier S. ( 2005) Role for the nuclear factor kappaB pathway in transforming growth factor-beta1 production in idiopathic myelofibrosis: possible relationship with FK506 binding protein 51 overexpression. Cancer Res 65: 3281– 3289 [DOI] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. ( 2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18: 601– 607 [DOI] [PubMed] [Google Scholar]
- Kuratsune M, Masaki T, Hirai T, Kiribayashi K, Yokoyama Y, Arakawa T, Yorioka N, Kohno N. ( 2007) Signal transducer and activator of transcription 3 involvement in the development of renal interstitial fibrosis after unilateral ureteral obstruction. Nephrology 12: 565– 571 [DOI] [PubMed] [Google Scholar]
- Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, et al. ( 2002) Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J 21: 2672– 2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chabner BA. ( 2009) Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 27: 5459– 5468 [DOI] [PubMed] [Google Scholar]
- Lien SC, Usami S, Chien S, Chiu JJ. ( 2006) Phosphatidylinositol 3-kinase/Akt pathway is involved in transforming growth factor-beta1-induced phenotypic modulation of 10T1/2 cells to smooth muscle cells. Cell Signal 18: 1270– 1278 [DOI] [PubMed] [Google Scholar]
- Liu F, Levin MD, Petrenko NB, Lu MM, Wang T, Yuan LJ, Stout AL, Epstein JA, Patel VV. ( 2008) Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J Mol Cell Cardiol 45: 715– 723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Ezzeldin HH, Diasio RB. ( 2009) Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs 69: 1911– 1934 [DOI] [PubMed] [Google Scholar]
- Manabe I, Shindo T, Nagai R. ( 2002) Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res 91: 1103– 1113 [DOI] [PubMed] [Google Scholar]
- Marks PA, Breslow R. ( 2007) Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 25: 84– 90 [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Miller T, Kelly WK. ( 2004) Histone deacetylase inhibitors. Adv Cancer Res 91: 137– 168 [DOI] [PubMed] [Google Scholar]
- Marks PA, Xu WS. ( 2009) Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem 107: 600– 608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T. ( 2009) Histone deacetylase modulates the pro-inflammatory and fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol 298: F133– F141 [DOI] [PubMed] [Google Scholar]
- Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC. ( 1984) Structural-functional relationships in diabetic nephropathy. J Clin Invest 74: 1143– 1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan PE, Gray GA, Sherry L, Webb DJ, Kenyon CJ. ( 1998) Differential effects of angiotensin II on cardiac cell proliferation and intramyocardial perivascular fibrosis in vivo. Circulation 98: 2765– 2773 [DOI] [PubMed] [Google Scholar]
- Miller TA, Witter DJ, Belvedere S. ( 2003) Histone deacetylase inhibitors. J Med Chem 46: 5097– 5116 [DOI] [PubMed] [Google Scholar]
- Monneret C. ( 2007) Histone deacetylase inhibitors for epigenetic therapy of cancer. Anticancer Drugs 18: 363– 370 [DOI] [PubMed] [Google Scholar]
- Mori Y, Chen SJ, Varga J. ( 2003) Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum 48: 1964– 1978 [DOI] [PubMed] [Google Scholar]
- Niki T, Rombouts K, De Bleser P, De Smet K, Rogiers V, Schuppan D, Yoshida M, Gabbiani G, Geerts A. ( 1999) A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology 29: 858– 867 [DOI] [PubMed] [Google Scholar]
- Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, Lee HB. ( 2009) Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am J Physiol Renal Physiol 297: F729– F739 [DOI] [PubMed] [Google Scholar]
- Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T, Takahashi T. ( 2004) Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer 112: 26– 32 [DOI] [PubMed] [Google Scholar]
- Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, Zhuang S. ( 2009) Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 297: F996– F1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul T, Li S, Khurana S, Leleiko NS, Walsh MJ. ( 2007) The epigenetic signature of CFTR expression is coordinated via chromatin acetylation through a complex intronic element. Biochem J 408: 317– 326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. ( 2007) HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129: 1351– 1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. ( 2000) Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA 97: 10014– 10019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein RC, Egan ME, Zeitlin PL. ( 1997) In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest 100: 2457– 2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. ( 2000) Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol 278: C259– C267 [DOI] [PubMed] [Google Scholar]
- Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE. ( 2000) P21-dependent G(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer 83: 817– 825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ramires FJ, Weber KT. ( 1997) Fibrosis of atria and great vessels in response to angiotensin II or aldosterone infusion. Cardiovasc Res 35: 138– 147 [DOI] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, Schreiber SL. ( 1996) A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272: 408– 411 [DOI] [PubMed] [Google Scholar]
- Tefferi A. ( 2005) Pathogenesis of myelofibrosis with myeloid metaplasia. J Clin Oncol 23: 8520– 8530 [DOI] [PubMed] [Google Scholar]
- Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. ( 2003) Histone deacetylases: unique players in shaping the epigenetic histone code. Ann NY Acad Sci 983: 84– 100 [DOI] [PubMed] [Google Scholar]
- Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, et al. ( 2007) Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med 13: 324– 331 [DOI] [PubMed] [Google Scholar]
- Verheule S, Sato T, Everett T, 4th, Engle SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. ( 2004) Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res 94: 1458– 1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Legrand-Poels S, van Lint C, Warnier M, Merville MP, Gielen J, Piette J, Bours V, Chariot A. ( 2003) Cytoplasmic IkappaBalpha increases NF-kappaB-independent transcription through binding to histone deacetylase (HDAC) 1 and HDAC3. J Biol Chem 278: 46541– 46548 [DOI] [PubMed] [Google Scholar]
- Wang JC, Chen C, Dumlao T, Naik S, Chang T, Xiao YY, Sominsky I, Burton J. ( 2008) Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk Lymphoma 49: 2321– 2327 [DOI] [PubMed] [Google Scholar]
- Wang Z, Chen C, Finger SN, Kwajah S, Jung M, Schwarz H, Swanson N, Lareu FF, Raghunath M. ( 2009) Suberoylanilide hydroxamic acid: a potential epigenetic therapeutic agent for lung fibrosis? Eur Respir J 34: 145– 155 [DOI] [PubMed] [Google Scholar]
- Wiech NL, Fisher JF, Helquist P, Wiest O. ( 2009) Inhibition of histone deacetylases: a pharmacological approach to the treatment of non-cancer disorders. Curr Top Med Chem 9: 257– 271 [DOI] [PubMed] [Google Scholar]
- Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. ( 1995) Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 95: 1861– 1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelent A, Petrie K, Lotan R, Waxman S, Gore SD. ( 2005) Clinical translation of epigenetics in cancer: eN-CORe–a report on the second workshop. Mol Cancer Ther 4: 1810– 1819 [DOI] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. ( 2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110: 479– 488 [DOI] [PMC free article] [PubMed] [Google Scholar]