

Abstract
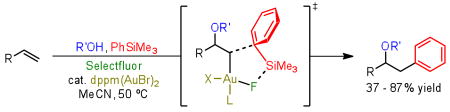
During continuing studies with a novel oxidative gold oxyarylation reaction, arylsilanes were found to be competent coupling partners, providing further evidence for an intramolecular electrophilic aromatic substitution mechanism. While providing complementary yields to the previously described boronic acid methods, the use of trimethylsilanes reduces the observation of homocoupling byproducts and allows for facile intramolecular coupling reactions.
With the development of transition metal-catalyzed cross-coupling reactions, significant effort has been expended in the development of inexpensive and reliable organometallic reagents as coupling partners.1 In this context, organosilanes present an attractive solution; however, there have been relatively few reports of transition metal-catalyzed reactions with the readily accessible aryltrimethylsilanes. Moreover, a stoichiometric additive such as fluoride or hydroxide is typically employed to activate the relatively unreactive silicon center for transmetallation.2 Alternatively, a variety of reactive silicon reagents have been designed and utilized in transition metal catalyzed cross-coupling reactions on milder conditions.3 During our recent examination of the Selectfluor mediated gold-catalyzed amino- and oxyarylation of terminal olefins with boronic acids,4 mechanistic experimentation and computational analysis provided evidence against transmetallation to the gold center. Instead, studies suggested a bimolecular reductive elimination reminiscent of an electrophilic aromatic substitution. While unactivated trimethylarylsilanes do not typically directly engage in cross-couplings,5 they can readily react as nucleophiles in aromatic substitution reactions.6 We therefore hypothesized that arylsilanes might be utilized as the terminating nucleophile in oxidative gold-catalyzed coupling with alkenes.
On the basis of this hypothesis, we were delighted to find that when olefin 1 was reacted with phenyltrimethylsilane under the previously described reaction conditions, the desired intermolecular oxyarylation product (2) was observed in 50% yield (Table 1, entry 1). No exogenous base or fluoride is required for silicon activation, and a wide variety of readily available silanes, silanols, and siloxanes are capable oxyarylation coupling partners. In attempting to identify the optimal silicon coupling regent, we found silanediols and –triols provided excellent yields (entries 5, 8), though rapid oligomerization lead to concerns regarding reproducibility. The more bench stable trisiloxane derivatives7 unfortunately resulted in significantly lower conversions (entry 7). Given their ease of synthesis, general stability, and orthogonality to a wide variety of conditions, trimethylsilanes provide the greatest potential benefits in terms of further reaction development. Moreover, the formation of biphenyl, a major byproduct formed from the reaction with boronic acids, is severely reduced by the use of aryltrimethylsilanes.8 Further optimization of the reaction conditions demonstrated that the yield can be increased by the use of 2 equivalents of Selectfluor (entry 2).9
Table 1.
Ethoxyarylation of 1 with Arylsilicon Reagentsa
 | |||
|---|---|---|---|
| entry | Ph-SiX3 | equiv | yield (%)b |
| 1 | PhSiMe3 | 1.5 | 50 |
| 2 | PhSiMe3 | 1.5 | 77c |
| 3 | PhMe2SiOH | 2.0 | 41 |
| 4 | Ph3SiOH | 1.0 | 33 |
| 5 | Ph2Si(OH)2 | 2.0 | 65 |
| 6 | PhMeSi(OH)2 | 2.0 | 22 |
| 7 | [(Ph2SiO)3] | 1.0 | 28 |
| 8 | PhSi(OH)3 | 2.0 | 83 |
| 9 | PhSi(OEt)3 | 2.0 | 74 |
Conditions: 1 (30 μmol), EtOH (300 μmol), Selectfluor (45 μmol), 5 mol % dppm(AuBr)2, 0.05 M in CD3CN, 50 °C for 14 h.
Yield determined by 1H NMR with nitrobenzene as internal standard.
2.0 equiv (60 μmol) of Selectfluor used.
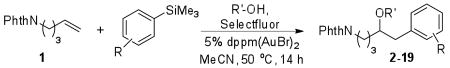
One limitation of the oxyarylation reaction with boronic acids is the incompatibility of nitrogen and oxygen containing coupling partners, due in part to competing reactions with Selectfluor.10,11 In contrast, we found that the scope of the gold-catalyzed methoxyarylation using arylsilanes was significantly expanded using aryltrimethylsilanes to afford the acetoxy-, trifloxy-, and sulfonamide substituted adducts in good to excellent yields. (Table 2, entries 1–3).12 In addition, the reaction remains functional for a wide variety of electron neutral and electron poor coupling partners, readily tolerating substitution in the para (entries 1–7), meta (entry 8), and ortho positions (entries 9,18).
Table 2.
 | ||||
|---|---|---|---|---|
| entry | R | R′ | product | yield (%)c |
| 1 | 4-OAc | Me | 3 | 83 |
| 2 | 4-OTf | Me | 4 | 53 |
| 3 | 4-N(Me)Ts | Me | 5 | 66 |
| 4 | 4-Me | Me | 6 | 73 |
| 5 | 4-Br | Me | 7 | 82 |
| 6 | 4-CHO | Me | 8 | 77 |
| 7 | 4-CO2Me | Me | 9 | 68 |
| 8 | 3-CO2Me | Me | 10 | 83 |
| 9 | 2-CH2CH2OH | Me | 11 | 69 |
| 10 | H | Me | 12 | 87 |
| 11 | H | Et | 2 | 83 |
| 12 | H | i-Pr | 13 | 81 |
| 13 | H | t-Bu | 14 | 37 |
| 14 | H | neopentyl | 15 | 64 |
| 15 | H | cyclopentyl | 16 | 68 |
| 16 | H | 2-methoxyethyl | 17 | 86 |
| 17 | H | H | 18 | 77 |
| 18 | 2-CH2CH2OH | H | 19 | 55 |
Conditions: 1 (100 μmol), R′OH (1.0 mmol), Selectfluor (200 μmol), ArSiMe3 (150 μmol), 5 mol % dppm(AuBr)2, 0.1 M in CH3CN, 50 °C for 14 h.
Catalyst addition in two portions over two hours.
Isolated yields.
As with the coupling of boronic acids, the reaction was general to a variety of alcohol nucleophiles, providing homobenzylic ethers in functional yields (Table 2). The reaction readily proceeded with secondary and bulky primary alcohols (entries 12, 14, and 15), although the reaction with a tertiary alcohol did not reach completion (entry 13). Additionally, the reluctance of gold(III) to undergo β-hydride elimination allowed for hydroxyarylation with water as the nucleophile (entries 17, 18) in contrast to the Wacker oxidation that is typically observed under palladium catalysis.13
As demonstrated in Figure 1, the olefin substrate scope remained broad, and the reaction tolerated a variety of functional groups, including esters, sulfonamides, carbamates, and halides. The addition was sensitive to the electronic environment, allowing for selective reaction at the less electron-deficient olefin (29). When pendent esters were employed in the reaction, either on the olefin (32 or 33) or the incoming silane (38), use of an alcohol nucleophile provided ether products, while water addition provided cyclized products following in situ lactonization (Scheme 1).
Figure 1.

Products of the oxyarylation of terminal olefins.
Scheme 1.

In situ lactonizations with pendant esters.
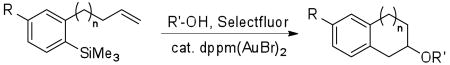
We were interested in performing the coupling reaction in an intramolecular fashion to provide bicyclic systems. Initial attempts utilizing boronic acids yielded only moderate success, likely due to dimerization during the reaction and problems with isolation and purification of the requisite boronic acids, which are prone to oligomerization. We hypothesized that the trimethylsilanes, which are readily synthesized and purified, would circumvent these issues. We were pleased to find that the use of silanes as intramolecular coupling partners in the gold-catalyzed oxidative process provided tetrahydronaphthalenes in excellent yields (Table 3). The intramolecular coupling has a similar scope to the three component reaction, tolerating a variety of ring substitutions. The formation of indanes (entry 4), however, provides significantly lower yields, possibly due to the geometric constraints imposed by the mechanism.
Table 3.
 | |||||
|---|---|---|---|---|---|
| entry | R | R′ | n | product | yield (%) |
| 1 | H | H | 1 | 41 | 66 |
| 2 | Me | 1 | 42 | 73 | |
| 3 | Et | 1 | 43 | 70 | |
| 4 | H | 0 | 44 | 15c | |
| 5 | 4-F | H | 1 | 45 | 47 |
| 6 | Et | 1 | 46 | 68 | |
| 7 | 4-Cl | H | 1 | 47 | 62 |
| 8 | Me | 1 | 48 | 65 | |
| 9 | 4-CF3 | H | 1 | 49 | 51 |
| 10 | Me | 1 | 50 | 59 | |
| 11 | 4-Ph | Me | 1 | 51 | 74 |
Conditions: Aryltrimethylsilane (100 μmol), Selectfluor (200 μmol), 5 mol % dppm(AuBr)2, 0.1 M in CH3CN/R′OH (10/1), 50 °C for 14 h.
Catalyst addition in two portions over two hours.
Yield based on 1H NMR versus nitrobenzene as internal standard.
Given the scarcity of cross-couplings with aryltrimethylsilanes, we performed several experiments to investigate the reaction mechanism. In our hands, trimethylsilanes proved inert to all attempts at transmetallation under relevant reaction conditions.14,15 In addition, the use of an external fluoride source generally had a negative impact on the reaction,16 suggesting that the gold fluoride itself is essential for the requisite activation of the silicon.17 We therefore propose a mechanism involving nucleophilic attack by ROH on the alkene promoted by a cationic gold(III) fluoride,18 followed by reductive aryl transfer from a pentavalent silicon species (Scheme 2). This mechanism is consistent with the observations from the gold-catalyzed oxidative alkene functionalization reactions with boronic acids.
Scheme 2.

Proposed mechanism for oxyarylation with arylsilanes
As the gold catalyzed alkoxyarylation does not proceed through a direct transmetallation, the reaction can be used to differentiate between two reactive metal species (Scheme 3). When the arylsilane 52 containing a MIDA boronate19 was reacted with 1 under the standard gold conditions, homobenzylic ether 53 was the sole product. However, when treated under palladium standard Suzuki-Miyaura cross-coupling conditions, the 4-trimethylsilylbiphenyl 54 was formed exclusively. More notable is the orthogonal reactivity observed in group 11 metal-catalyzed reaction with Selectfluor; 52 underwent silver-catalyzed fluorination of the boronate to provide 55,10 while the aryltrimethylsilyl selectively participated in the gold-catalyzed coupling.
Scheme 3.

Selective coupling of bifunctional arylsilane MIDA boronates.
In conclusion, we report the first examples of gold-catalyzed oxidative coupling reactions using organosilicon reagents. The reaction employs generally unreactive aryltrimethylsilanes as coupling partners, allows for the extension to phenol and aniline derived aryl groups, produces significantly less biphenyl byproducts, and provides a means to better access intramolecular couplings. Additionally, the reluctance of trimethylsilanes to undergo transmetallation with gold provides further evidence against a standard reductive elimination pathway and support for our previously proposed bimolecular reductive couplings from gold(III) fluorides.
Supplementary Material
Acknowledgments
We gratefully acknowledge NIHGMS (RO1 GM073932), Amgen, and Novartis for financial support and thank Johnson Matthey for a gift of AuCl3. J.-F. B. thanks the FQRNT for a postdoctoral research fellowship.
Footnotes
Supporting Information Available: Experimental procedures, product characterization, and copies of the 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd. 1 and 2 Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]; (b) Negishi Ei., editor. Handbook of Organopalladium Chemistry for Organic Synthesis. Wiley-Interscience; New York: 2002. [Google Scholar]
- 2.For recent reviews on cross-coupling with silicon reagents, see reference 1 and: Denmark SE, Butler CR. Chem Commun. 2009:20. doi: 10.1039/b809676g.Denmark SE, Regens CS. Acc Chem Res. 2008;41:1486. doi: 10.1021/ar800037p.Denmark SE, Baird JD. Chem Eur J. 2006;12:4954. doi: 10.1002/chem.200600034.Nakoa Y, Sahoo AK, Imanaka H, Yada A, Hiyama T. Pure Appl Chem. 2006;78:435.Handy CJ, Manoso AS, McElroy WT, Seganish WM, DeShong P. Tetrahedron. 2005;61:12201.Spivey AC, Gripton CJG, Hannah JP. Curr Org Synth. 2004;1:211.
- 3.Selected silicon-based cross-coupling reactions: Hatanaka Y, Hiyama T. J Org Chem. 1988;53:920.Hatanaka Y, Fukushima S, Hiyama T. Chem Lett. 1989:1711.Mowery ME, DeShong P. J Am Chem Soc. 1999;64:1684. doi: 10.1021/jo982463h.Mowery ME, DeShong P. Org Lett. 1999:2137. doi: 10.1021/ol991186d.Hirabayashi K, Mori A, Kawashima J, Suguro M, Nishihara Y, Hiyama T. J Org Chem. 2000;65:5342. doi: 10.1021/jo000679p.Murata M, Shimazaki R, Watanabe S, Masuda Y. Synthesis. 2001:2231.Denmark SE, Sweis RF. J Am Chem Soc. 2001;123:6439. doi: 10.1021/ja016021q.Lee JY, Fu GC. J Am Chem Soc. 2003;125:5616. doi: 10.1021/ja0349352.Nakao Y, Takeda M, Matsumoto T, Hiyama T. Angew Chem Int Ed. 2010;49:4447. doi: 10.1002/anie.201000816.
- 4.For Au-catalyzed amino- and oxyarylation of alkenes: Zhang G, Cui L, Wang Y, Zhang L. J Am Chem Soc. 2010;132:1474. doi: 10.1021/ja909555d.Brenzovich WE, Jr, Benitez D, Lackner AD, Shunatona SP, Tkatchouk E, Goddard WA, III, Toste FD. Angew Chem, Int Ed. 2010;49:5519. doi: 10.1002/anie.201002739.Melhado AD, Brenzovich WE, Jr, Lackner AD, Toste FD. J Am Chem Soc. 2010;132:8885. doi: 10.1021/ja1034123.For other oxidative gold-catalyzed reactions see: Kar A, Mangu N, Kaiser HM, Beller M, Tse MK. Chem Commun. 2008;3:386. doi: 10.1039/b714928j.Hashmi ASK, Ramamurthi TD, Rominger F. J Organomet Chem. 2009;694:592.Zhang G, Peng Y, Cui L, Zhang L. Angew Chem, Int Ed. 2009;48:3112. doi: 10.1002/anie.200900585.Iglesias A, Muñiz K. Chem Eur J. 2009;15:10563. doi: 10.1002/chem.200901199.Ye L, Cui L, Zhang G, Zhang L. J Am Chem Soc. 2010;132:3258. doi: 10.1021/ja100041e.Hopkinson MN, Tessier A, Salisbury A, Giuffredi GT, Combettes LE, Gee AD, Gouverneur V. Chem Eur J. 2010;16:4739. doi: 10.1002/chem.201000322.
- 5.For examples of trimethylsilyl cross-coupling reactions involving an intermediate metal species, see: Farinola GM, Fiandanese V, Mazzone L, Naso F. J Chem Soc, Chem Commun. 1995:2523.Smith AB, III, Kim WS, Tong R. Org Lett. 2010;12:588. doi: 10.1021/ol902784q.
- 6.For silicon reagents in electrophilic aromatic substitution reactions, see Chan TH, Fleming I. Synthesis. 1979:761. and references therein.
- 7.Endo M, Sakurai T, Ojima S, Katayama T, Unno M, Matsumoto H, Kowase S, Sano H, Kosugi M, Fugami K. Synlett. 2006:749. [Google Scholar]
- 8.The reaction of PhB(OH)2 with dppm(AuBr)2 and Selectfluor provides biphenyl in about 80% yield, while PhSiMe3 yields < 10 %.
- 9.For full details on reaction optimization and catalyst screening, see Supporting Information.
- 10.For metal-mediated reactions of Selectfluor with arylboronic acids, see Furuya T, Kaiser HM, Ritter T. Angew Chem, Int Ed. 2008;47:5993. doi: 10.1002/anie.200802164.Furuya T, Ritter T. Org Lett. 2009;11:2860. doi: 10.1021/ol901113t.Tang P, Furuya T, Ritter T. J Am Chem Soc. 2010;132:12150. doi: 10.1021/ja105834t.
- 11.For Selectfluor induced oxidations of electron-rich arenes, see Zupan M, Iskra J, Stavber S. J Fluor Chem. 1995;70:7.Gaj Stavber G, Zupan M, Jereb M, Stavber S. Org Lett. 2004;6:4973. doi: 10.1021/ol047867c.Stavber G, Zupan M, Stavber S. Tetrahedron Lett. 2007;48:2671.
- 12.Use of the more electron-rich silanes, such as 4-(methoxyphenyl)-trimethylsilane, yielded no product.
- 13.For a recent review on the mechanism of the Wacker oxidation, see: Keith JA, Henry PA. Angew Chem, Int Ed. 2009;48:9038. doi: 10.1002/anie.200902194.
-
14.Under the action of CsF in MeCN, Ph3PAuBr readily transmetallates with boronic acids and silanols but not trimethylsilanes:
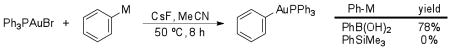 See: Partyka David V, Zeller M, Hunter AD, Gray TG. Angew Chem Int Ed. 2006;45:8188. doi: 10.1002/anie.200603350.Hashmi ASK, Ramamurthi TD, Rominger F. J Organomet Chem. 2009;694:592.
See: Partyka David V, Zeller M, Hunter AD, Gray TG. Angew Chem Int Ed. 2006;45:8188. doi: 10.1002/anie.200603350.Hashmi ASK, Ramamurthi TD, Rominger F. J Organomet Chem. 2009;694:592.
- 15.In investigations with the sila-Cope rearrangement, no transmetallation to gold(I) was observed, see Horino Y, Luzung MR, Toste FD. J Am Chem Soc. 2006;128:11364. doi: 10.1021/ja0636800.
- 16.Addition of fluoride sources, such as CsF and KF, significantly retarded the reaction. See Supporting Information for more details.
- 17.Mankad N, Toste FD. J Am Chem Soc. 2010;132:12859. doi: 10.1021/ja106257n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaLonde RL, Brenzovich WE, Jr, Benitez D, Tkatchouk E, Kelley K, Goddard WA, III, Toste FD. Chem Sci. 2010;1:226. doi: 10.1039/C0SC00255K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For synthetic applications of MIDA boronates, see Gillis EP, Burke MD. Aldrichimica Acta. 2009;42:17.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961. doi: 10.1021/ja901416p.Gillis EP, Burke MD. J Am Chem Soc. 2008;130:14084. doi: 10.1021/ja8063759.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.