

Summary
Replication initiation is a key event in the cell cycle of all organisms and oriC, the replication origin in Escherichia coli, serves as the prototypical model for this process. The minimal sequence required for oriC function was originally determined entirely from plasmid studies using cloned origin fragments, which have previously been shown to differ dramatically in sequence requirement from the chromosome. Using an in vivo recombineering strategy to exchange wt oriCs for mutated ones regardless of whether they are functional origins or not, we have determined the minimal origin sequence that will support chromosome replication. Nearly the entire right half of oriC could be deleted without loss of origin function, demanding a reassessment of existing models for initiation. Cells carrying the new DnaA box-depleted 163 bp minimal oriC exhibited little or no loss of fitness under slow-growth conditions, but were sensitive to rich medium, suggesting that the dense packing of initiator binding sites that is a hallmark of prokaryotic origins, has likely evolved to support the increased demands of multi-forked replication.
Introduction
Initiation of DNA replication in Escherichia coli occurs at a unique site, oriC. It was initially mapped using lambda phage genetics and isolated as an autonomously replicating sequence (ARS) driving minichromosome replication (von Meyenburg et al., 1977). By subcloning this ARS sequence into a ColE1-type plasmid, it became possible to analyse deletions of the oriC region, including lethal ones, leading to the establishment of the 245 bp minimal oriC (Oka et al., 1980). Cloning of oriC also made it possible for Kornberg and colleagues to carry out staged initiation experiments using purified proteins (Fuller et al., 1981; Bramhill and Kornberg, 1988). These in vitro studies led to a general model for replication initiation in which cooperative binding of an activated initiator protein, DnaA-ATP, to several cognate binding sites results in a higher-order nucleoprotein complex that induces helical distortion and duplex melting in an adjacent AT-rich DNA unwinding element (DUE). The DUE is composed of three highly conserved 13-mers and an additional non-specific AT-rich segment (AT-cluster; Fig. 1, top). Subsequent loading of helicase, primase and DNA polymerase into the DNA bubble leads to the formation of bidirectional replication forks. This basic model is still valid, and is applicable in concept to initiation systems in all organisms.
Fig. 1.

oriC deletion mutations. The oriC region with regulatory elements is shown drawn to scale. The position of the 10 9-mer DnaA binding sites R1–R5, I1–I3 and τ1–τ2 (blue rectangles); DNA unwinding elements AT-cluster and 13-mer repeats L, M and R (red rectangles); binding sites for IHF and FIS proteins (grey rectangles); putative 6-mer DnaA binding sites (small blue arrowheads); and GATC sequences (small white arrowheads) are indicated. The 12 oriC deletion alleles used in this study are shown (bottom). Right-end deletions (oriC227–oriC238) and left-end deletions (oriC233–oriC235) were generated by PCR mutagenesis (dashed lines indicate deleted regions). oriC239 and oriC243 were created by combining right- and left-end deletions. Growth rate of strains containing the mutant oriCs is indicated as viability (+++, fastest; –, non-viable).
Currently, there are 10 known 9-mer recognition sites for DnaA protein in the oriC sequence (Fig. 1, top). These consist of three high-affinity sites, R1, R2 and R4, and seven low-affinity sites, R3, R5, I1, I2, I3, τ1 and τ2 (Margulies and Kaguni, 1996; Grimwade et al., 2000; McGarry et al., 2004; Kawakami et al., 2005). R-sites bind active DnaA-ATP and inactive DnaA-ADP with equal affinity (Fuller et al., 1984), while I-sites and τ-sites preferentially bind DnaA-ATP (McGarry et al., 2004; Kawakami et al., 2005). Altering more than one of the DnaA-ATP discriminatory sites (I-boxes) on a minichromosome was not tolerated, suggesting that these sites are important for oriC function and might be responsible for coupling initiation rate and timing to DnaA-ATP levels (Grimwade et al., 2007). Supporting this idea, oriC plasmids that carried base changes in I2 and I3 that increase affinity for all forms of DnaA, perturbed growth of the host strain, presumably by imparting a competitive advantage over the wt chromosomal oriC (Grimwade et al., 2007; see Discussion). The three high-affinity sites are bound with DnaA for the majority of the cell cycle (Cassler et al., 1995), leading to the proposal that DnaA bound to these sites was equivalent to an origin recognition complex (ORC) in budding yeast (Grimwade et al., 2007). The lower-affinity DnaA boxes are filled at about the time of replication initiation (Ryan et al., 2004; Nievera et al., 2006), converting the complex to a pre-replicative state. Weak binding of DnaA is also reported at several 6-mer sequences within the DUE region (Speck and Messer, 2001). Although the physiological role of these interactions is unclear, binding was strongest when the DUE was single-stranded, suggesting that DnaA at 6-mer sites may stabilize the open complex.
In addition to DnaA, at least four other DNA bending proteins bind to oriC and facilitate the ordered filling of the seven low-affinity DnaA boxes. IHF (integration host factor) and Fis (factor for inversion stimulation) bind to recognition sites within oriC (Fig. 1), while HU (histone-like protein) binds non-specifically (Ryan et al., 2004 and references therein). In vivo studies showed that immediately prior to initiation, IHF binds to oriC, concomitant with a redistribution of bound DnaA to the weaker I-sites (Ryan et al., 2002). Fis binding to oriC, which is maximum at lower DnaA concentrations, represses filling of I-sites, and therefore likely acts as a negative regulator early in the cell-division cycle, assuring that initiation occurs quickly, and at all available origins, later in the cell cycle (Ryan et al., 2004). HU protein suppresses binding of DnaA specifically at I3, giving special credence to this site as a trigger for initiation (Ryan et al., 2002). The fourth DNA bending protein that facilitates proper DnaA binding is SeqA, which binds specifically and strongly to a cluster of GATC sites near the DUE element in oriC (Slater et al., 1995; reviewed in Waldminghaus and Skarstad, 2009). One major function of SeqA is to sequester newly replicated (hemi-methylated) origins from DnaA, preventing immediate reinitiations (Lu et al., 1994; Torheim and Skarstad, 1999). In vitro and in vivo data indicate that SeqA binding to the new (protein-free) origins has the effect of blocking DnaA binding to the lower-affinity DnaA boxes while allowing DnaA to reload onto the high affinity sites (Wold et al., 1998; Nievera et al., 2006). In this way, SeqA may reset origin structure to prepare them for the next round of initiation (Nievera et al., 2006).
In all cells, the genome is duplicated once and only once per mitotic cell cycle. Superimposed on this requirement, DNA replication must be completed and daughter chromosomes segregated prior to each cell division. Generally, replication is regulated at the level of initiation (Bell and Dutta, 2002; Mott and Berger, 2007; Nielsen and Lobner-Olesen, 2008). In many bacteria, including E. coli, there is an extra burden placed on initiation control during fast growth. Under these conditions, the cell division cycle is much shorter than the time it takes to complete replication of a chromosome (~45 min). To allow such fast growth, initiation occurs before the previous round of replication is finished – resulting in a multi-forked chromosomal structure. As each pair of forks converge on the terminus, a subsequent and coupled cell division event occurs, sometimes as frequent as every 15 min. All copies of oriC in a cell are initiated simultaneously and only once per cell division cycle, a phenomenon known as initiation synchrony (Skarstad et al., 1986). Regulation of initiation is thought to result from a balance of positive and negative factors acting at oriC. The level of the primary positive factor, DnaA-ATP, fluctuates during the cell cycle, reaching a maximum at the time of initiation (see Kaguni, 2006 for review, Fujimitsu et al., 2009). The major negative factor is SeqA protein, which, as described above, binds most strongly to newly replicated oriCs, presumably sequestering origins from DnaA. Despite these individual models, the control mechanism for the whole of initiation remains unclear, especially the complex role of individual protein binding elements within oriC.
The sequence requirement for oriC differs dramatically depending on whether the origin is chromosomal or on a plasmid (Asai et al., 1998), and mutations of individual regulatory elements within oriC that render cloned origins non-functional generally have minor effects on chromosomal oriC. Replacement of individual DnaA boxes R2, R3 or R5 with scrambled sequences (Weigel et al., 2001; Riber et al., 2009), single base changes in I2 or I3 (McGarry et al., 2004; Riber et al., 2009), scrambling the binding site for IHF or Fis (Weigel et al., 2001; Riber et al., 2009), or blocking transcription from the oriC proximal genes, gidA and mioC (Lobner-Olesen and Boye, 1992; Bates et al., 1997), did not significantly affect cell growth or initiation frequency from chromosomal oriC in minimal medium. Each of these mutations had severe if not lethal consequences for oriC plasmids. Deletion or scrambling of DnaA box R4 had the greatest effect of any mutation, with a reduction in cellular DNA content and loss of initiation synchrony (Bates et al., 1995; Weigel et al., 2001; Riber et al., 2009). Thus, preventing protein binding to any individual oriC regulatory element, except for DnaA box R4, has modest to little effect on chromosomal initiation. These experiments do not address, however, what effect these mutations have in combination, nor what the minimal origin sequence is for chromosomal initiation.
To determine the minimal oriC sequence that could function on the chromosome, we generated multiple deletions on either side of the minimal oriC sequence and replaced the endogenous origins via a specialized in vivo transfer system. By optionally transferring mutant origins into a constitutive stable DNA replication (cSDR) strain utilizing a bypass replication system that is independent of oriC and DnaA, we had the ability to introduce potentially non-functional oriC mutations and then assay those cells for oriC function in a controlled manner. This approach revealed that most of the right half of oriC could be deleted without loss of origin function. Other findings suggest that the role of nonessential oriC elements is to facilitate multi-forked replication. Our data emphasize the necessity to employ a relevant assay system when analysing oriC function and allow a reassessment of the significance and role of the different elements in oriC.
Results
A comprehensive set of oriC deletions
To determine the smallest oriC fragment that could support chromosome replication initiation and allow cell growth, we created a set of deletion constructs that were truncated from one or both ends of the minimal 245 bp oriC region. Deletion end-points were selected so that each successive construct deleted one or more protein binding sites as shown in Fig. 1. Mutations were generated by PCR then cloned into the oriC plasmid, pDB201. The ability of these resulting mutant oriCs to support plasmid replication was examined by the polA assay (Table 1), which takes advantage of the fact that the vector's replication origin (ColE1) requires the product of the gene polA (DNA Polymerase I) while oriC does not. Therefore, the origin activity of a mutant oriC plasmid can be determined by its ability to transform a strain deficient in DNA pol I activity. With the exception of pDB233, none of the plasmids carrying mutant oriCs were able to transform a polA1 host, suggesting that the entire 245 bp oriC region is required to initiate DNA replication on a plasmid (Table 1). Even pDB233, carrying a deletion of the AT-cluster just outside of the minimal oriC, exhibited a 75-fold reduction in transformation efficiency of a polA1 host compared with pDB201. One possible explanation for the inability of mutant oriC plasmids to grow in vivo is that there is competition for initiation proteins between cloned oriC and the host oriC (Skarstad and Lobner-Olesen, 2003). Theoretically, a mutant extrachromosomal oriC might rarely, or never, initiate replication in a wt host cell because the host origin binds DnaA more efficiently. To test this possibility, we attempted to transform each mutant oriC plasmid into a strain carrying the matching oriC allele created by our in vivo replacement system (see below). None of the plasmids gained a significant ability to transform a polA1 strain when the host cell replicated using the same mutant oriC as the plasmid (Table 1). We conclude that the oriC plasmid requirement for a complete 245 bp origin is not due to competition between cloned and chromosomal origins (see Discussion).
Table 1.
Viability of mutant oriC plasmids.
| Transformants (wild-type oriC host) |
Transformants (matching mutant oriC host) |
||||
|---|---|---|---|---|---|
| oriC plasmid | Deletion (bp) | polA+ | polA− | polA+ | polA− |
| pDB201 | 0 | 1.5 × 104 | 7.2 × 103 | 3.7 × 104 | 2.2 × 104 |
| pDB227 | R-39 | 9.2 × 103 | 0 | 2.2× 104 | 0 |
| pDB228 | R-55 | 1.6 × 104 | 0 | 2.1 × 104 | 17 |
| pDB229 | R-64 | 1.6 × 104 | 0 | 7.8 × 104 | 36 |
| pDB230 | R-90 | 1.2 × 104 | 0 | 5.0 × 104 | 0 |
| pDB231 | R-98 | 1.1 × 104 | 0 | 3.1 × 104 | 0 |
| pDB237 | R-16 | 1.4 × 104 | 0 | N/A | N/A |
| pDB238 | R-195 | 1.8 × 104 | 0 | N/A | N/A |
| pDB233 | L-15 | 1.3 × 104 | 84 | 7.5 × 103 | 303 |
| pDB234 | L-32 | 9.6 × 103 | 0 | 2.9 × 103 | 0 |
| pDB235 | L-47 | 1.2 × 104 | 0 | N/A | N/A |
| pDB239 | R-64,L-15 | 1.5 × 104 | 0 | 05.3 × 104 | 0 |
| pDB243 | R-64,L-32 | 9.9 × 103 | 0 | N/A | N/A |
Plasmid names correspond to oriC allele numbering shown in Fig. 1. Deletion size in base pairs is shown with end orientation (L = left, R = right).
Transformation frequencies of polA+ and polA1 host strains are indicated as total number of transformants per μg of plasmid DNA per 109 cells.
Each oriC plasmid was also transformed into host strains containing the same mutant oriC allele (matching oriC). (N/A; oriC237, 238, 235, 243 host strains are inviable, and cSDR derivatives require polA+). Values are averages of two independent experiments (standard deviation ≤ 2.2 × 103).
To test the effects of chromosomal oriC mutations, we transferred the oriC alleles onto the E. coli chromosome using an in vivo replacement system previously described (Bates et al., 1995; outlined in Fig. S1). Briefly, modified oriCs were transferred from a plasmid to a specialized lambda transducing phage by homologous recombination using a linked asnA∷cat marker (see map in Fig. 1). Recombinant phage was then purified and used to infect wild-type E. coli cells under conditions that did not permit viral replication or stable integration. The asnA∷cat marker and modified oriC is transferred to the chromosome by homologous recombination. Mutant oriCs were moved into strain MG1655 by P1 transduction, and the presence of the modified oriC (and absence of a wild-type origin) was confirmed by Southern blot hybridization (Fig. S2) and sequencing (data not shown). Using this technique, we were able to obtain viable recombinants for 8 of 12 mutant oriC constructs (Fig. 1). Surprisingly, deletions of up to 98 bp on the right side of oriC supported chromosome replication. The largest functional deletion (oriC231) removed four DnaA boxes (R2, R3, R4 and I3) and the binding site for FIS protein. This region was generally considered to be critical for oriC function, especially DnaA box R3, which is the last DnaA box to be filled before initiation in vitro and in vivo and has been hypothesized to mediate the trigger mechanism for initiation (Ryan et al., 2002 and references therein). We could not obtain oriC237 recombinants, missing the three tightly grouped DnaA boxes τ2, I1 and I2. The left side of oriC was less tolerant of deletion, with a 32 bp deletion including the AT-cluster and left 13-mer being the largest functional deletion. The additional deletion of the middle 13-mer was not permitted (Fig. 1). In combination, we were able to remove the AT-cluster and the right-most three DnaA boxes, R3, I3 and R4 (oriC239) without loss of origin function.
Confirmation of non-functional origins
The four mutant oriCs that we could not successfully transfer to the chromosome to give viable cells were suspected to be non-functional. For these cases, we introduced the mutant oriCs into an rnhA224 strain that is capable of initiating replication independently of oriC. These cells initiate replication at stable R-loops, normally removed by the gene product of rnhA, RNase HI (Fig. 2B). This replication system, termed cSDR, is independent of oriC and DnaA protein and thus allows the introduction of non-functional oriC alleles onto the chromosome (Kogoma and von Meyenburg, 1983; Kogoma, 1997 for review). When transformed with a plasmid carrying an inducible rnhA+ gene, none of the four strains (oriC235, 237, 238, 243) could grow in the presence of the inducer ispropyl β-D-thiogalactopyranoside (IPTG), indicating that they were incapable of growth using the modified oriC (Fig. 2). A control strain containing the rnhA224 mutation and a viable oriC allele (oriC231) was unaffected by rnhA expression.
Fig. 2.
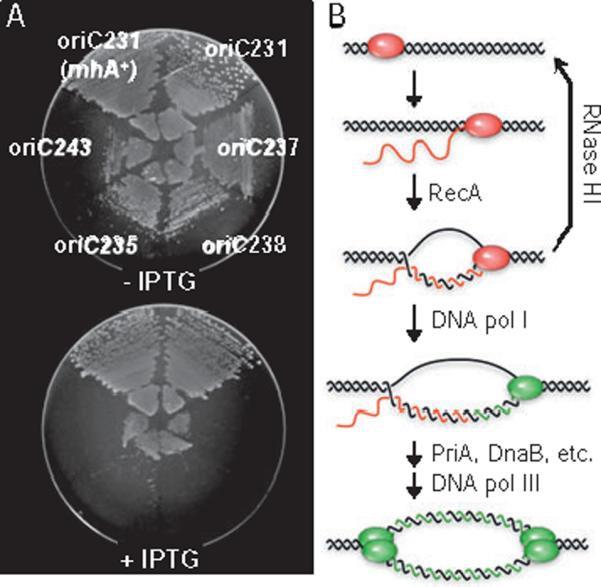
Strains carrying a non-functional oriC require cSDR for growth.
A. Four candidate oriC− mutations (oriC237, 238, 235, 243) were introduced into the chromosome of an rnhA224 strain and assayed for dependence on the bypass initiation system, cSDR, for viability. Each strain was transformed with an rnhA+ expression plasmid, pRNH-Km, plated onto M9 glucose medium with and without IPTG inducer, and incubated overnight at 37°C. An oriC+ mutation, oriC231, in either a wt or an rnhA224 background, was not affected by rnhA+ expression. Transformation with a vector control had no effect on growth of any strain (data not shown).
B. R-loop model for cSDR (adapted from Kogoma, 1997). Nascent mRNA (red strands) hybridizes to DNA template (black strands) when RNA polymerase (red oval) pauses at specific sites on the chromosome, oriK sites (hybridized RNA is nsormally removed by the product of the rnhA gene, RNase HI). DNA pol I (single green oval) then loads into the R-loop, synthesizing new DNA (green strands) from 3′ end of the mRNA. PriA mediates the formation of two replisomes (double green ovals) resulting in bidirectional replication.
Testimate to the advantage of using cSDR to confirm non-functional origins, one of the oriC alleles (oriC231) could not initially be transferred onto the chromosome of a wt cell, but turned out to be functional after we introduced it into an rnhA strain and tested it for dependency on cSDR. oriC231 supported chromosomal replication after repeated P1 transduction into wt strains, and cells were confirmed by Southern blot hybridization and sequencing to contain the mutant oriC allele and no suppressors. The reason that this allele did not initially transfer into wt cells may be due to replication difficulties caused during transition from a wt oriC to a much less efficient mutant one. In support of this idea, transfer efficiency decreased with increasing severity of the oriC mutation being transferred, and was significantly higher when rnhA cells were used as a recipient strain (Table S1). Subsequent P1 transduction of oriC231 into an rnhA+ strain utilized non-growing stationary phase cells, and thus this transition was likely less detrimental. This example illustrates the limitations of oriC transfer systems, and points out the importance of having an assay to confirm candidate null alleles when conducting a systematic mutagenesis of a replication origin.
Viable oriC mutants exhibit replication initiation defects
The growth rates and DNA replication phenotypes of the eight viable oriC mutants growing in minimal glucose medium (~40 min doubling time for wt) were examined with several assays. Growth rates, as assayed by optical density increase, ranged from normal to about 50% slower than wt (Fig. 3B). Cell viability, as measured by colony formation units per unit of optical density (CFU/OD), varied by about fourfold (Fig. 3B). Nucleoid morphology and cell membranes (indicating division septa) were imaged by 4′,6-diamidino-2-phenylindole (DAPI) and FM 4–64 staining, respectively (Fig. 3A), with large variations in observed phenotypes. DNA replication was analysed by two flow cytometry assays: Analysis of untreated exponentially growing cells provides a DNA/mass value (Fig. 3B), which reflects the coupling between DNA replication and general cell growth. Analysis of cells treated with rifampicin (blocking replication initiation) and cephalexin (blocking cell division) then allowed to complete ongoing rounds of replication, provides the number of replication origins present in the cells at the time of drug addition, an accurate indicator of replication initiation frequency (Skarstad et al., 1986) (Fig. 3B). The rifampicin run-off DNA histograms (Fig. 3A) also indicate the degree of replication synchrony. All origins normally initiate at the same time, so that wt cells contain either one, two, four or eight chromosomes depending on growth rate.
Fig. 3.

Growth characteristics of viable oriC mutants.
A. Nucleoid morphology (DAPI), cell membrane staining (FM4-64), and DNA histograms after rifampicin/cephalexin run-off (see text) are shown. DNA histograms indicate replication synchrony, with asynchrony index (A.I.) given as the fraction of cells with 3, 5, 6 or 7 chromosomes to those with 2, 4 or 8 chromosomes.
B. Growth rate, CFU/OD, average DNA/mass and average numbers of origins/cell are shown. Values are averages of two independent experiments (± SD). DNA/mass was measured by flow cytometry of exponential cells without rifampicin treatment. All cells were grown in M9 glucose with casamino acids.
Surprisingly, a strain carrying only a deletion of DnaA box R4 exhibited poorer growth than two strains carrying larger deletions. oriC227 cells, carrying a 39 bp deletion including DnaA box R4, showed a near 50% reduction in growth rate and CFU/OD compared with wt cells. DNA/mass and origins/cell values were also about half of wt, indicating inefficient replication initiation. The cells were elongated, with large segregated nucleoids, possibly as a consequence of late initiations interfering with subsequent chromosome segregation/cell division events (as observed in the dnaA46 strain at the restrictive temperature; D. Bates, unpublished). In contrast, cells carrying a deletion of DnaA box I3 (oriC228) or both I3 and R3 (oriC229) in addition to an R4 deletion performed much better than the strain carrying only the R4 deletion, both regarding growth rate, viability, DNA content and nucleoid morphology. Therefore, in the absence of DnaA box R4, the I3 box is inhibitory to initiation. All strains carrying a deletion of R4 (or more) exhibited a severe asynchrony phenotype, suggesting that DnaA binding to R4 is particularly important for the temporal regulation of initiation.
Additional deletion (Fig. 1) of the Fis binding site (oriC230) and DnaA box R2 (oriC231) were tolerated, but resulted in a further decrease in fitness as measured by growth rate, DNA/mass and origins/cell (Fig. 3B). These strains had particularly low CFU/OD values, perhaps indicating that during growth, some non-viable cells are present. Both strains also exhibited a classic par phenotype (Fig. 3A), with chromosome segregation failures including large un-segregated nucleoids and frequent apparently guillotined nucleoids (see also additional micrographs, Figs S4–S12). Several other oriC mutants exhibited filamentous growth, and the degree of cell elongation reflected the severity of the replication defect of the oriC mutants. Cells with smaller deletions, although elongated, generally had well-segregated nucleoids. None of the oriC mutants had elevated expression of the cell division inhibitor, sulA, indicating that elongation did not result from SOS induction (Fig. S3). We conclude that cell elongation most likely resulted from inappropriately timed initiations that lead to delays/blockages in subsequent cell divisions. It is noteworthy that cell fitness was not improved upon deletion of the Fis binding site (oriC230 compared with oriC229), in some measure unexpected given that Fis is generally considered a negative regulator of DnaA binding (Ryan et al., 2004; see Discussion). oriC231 represents the smallest functional oriC fragment known. The 163 bp sequence contains the DUE region, six DnaA binding sites (two strong and four weak) and the IHF binding site.
Cells carrying the two left-end oriC deletions, oriC233 and oriC234, also exhibited decreased fitness and DNA replication efficiency, but unlike right-end deletions, initiations remained relatively synchronous. oriC233 and oriC234 mutant cells had asynchrony indices of 0.17 and 0.24 respectively, compared with a value of ~0.8 for mutant cells carrying right-end deletions (Fig. 3A). This suggests that the co-ordination of initiation at individual origins is primarily controlled by an ordered binding of DnaA to oriC, while the AT-rich DUE region functions at the moment of strand opening after most of the regulation has already occurred. Deletion of the AT-cluster (oriC233) had only a minimal effect on origin function, while deletion of the AT-cluster and L 13-mer (oriC234) showed about 40% reduction in growth rate and DNA content. Thus oriC234 represents a very rare class of mutation that reduces the initiation rate while exhibiting reasonably good replication synchrony. This finding demonstrates that replication efficiency and synchrony are separable effects (see Discussion). Deletion of the M 13-mer resulted in a non-functional origin. To determine whether origins carrying DnaA box deletions functioned differently in the absence of part of the DUE, we combined the AT-cluster deletion with the right-end deletion of DnaA boxes R3, I3 and R4 (oriC239). This strain exhibited poorer growth than the DnaA box mutant alone (oriC229), suggesting that DnaA boxes and the DUE region function synergistically. Deletion of the L 13-mer was not tolerated in the absence of DnaA boxes R3, I3 and R4 (oriC243).
Mutant origin phenotypes are growth rate-dependent
Undoubtedly, one evolutionary advantage of having a strong initiation control system is the ability to regulate multiple origins simultaneously under fast growth conditions. To examine whether the phenotype of oriC mutants varied with growth rate, we measured growth rate of the viable oriC mutants growing in three different media of varying richness, M9 succinate, M9 glucose and Luria–Bertani (LB). Strikingly, all strains, including wt, had roughly equal growth rates in the poorest medium, M9 succinate, but there were up to 10-fold differences in growth rates between strains when grown in LB (Fig. 4, top, compare oriC231 with oriC201). M9 glucose-grown cells exhibited greater variation in growth rates than those in M9 succinate. Similar trends were seen in DNA/mass ratios (Fig. 4, bottom). It should also be noted that there was a good correlation between the growth rate measurements and the DNA/mass measurements, suggesting that it is DNA initiation capacity that determines the growth characteristics of these cells. The sensitivity to rich media was especially true for the largest oriC deletion strain, oriC231, which, in LB, grew ~1/3 as fast as the DnaA box R4 mutant oriC227. In fact, the ability of the oriC231-containing strain to proliferate in rich medium was so diminished that it actually grew more slowly and had a lower DNA/mass value in LB than in either minimal medium. These cells also acquired suppressors over long periods of growth in LB. Logically, fast growth places additional constraints on the cell cycle to produce an initiation event at all copies of oriC within a small time window (see Introduction). Therefore, the most straightforward interpretation of why cells carrying oriC231 are so sensitive to fast growth is that the mutant origin, which is missing four DnaA binding sites, cannot assemble initiation complexes at a rate sufficient to fire within an exceedingly short division cycle.
Fig. 4.
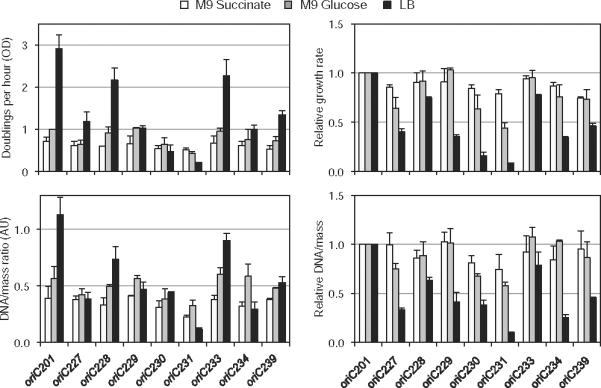
oriC mutants are sensitive to rich media. Growth rates (OD) and DNA/mass of viable oriC mutants were measured in three media as indicated. Values are averages of two independent experiments (± SD). Right panels indicate values relative to wt oriC201.
Deletion of DnaA box R4 suppresses replication elongation defects
Altered rates of DNA polymerization can lead to cell death via DNA double-strand breaks formed at stalled or collapsed replication forks (Simmons et al., 2004). It was previously shown that the temperature sensitivity of a mutant replisome subunit, dnaX(Ts), could be suppressed by dnaA(Cs,Sx) mutations (cold sensitivity, suppressors of DnaX) (Gines-Candelaria et al., 1995). It was later discovered that the suppression was due to decreased rates of replication initiation caused by the dnaA(Cs,Sx) mutation, presumably placing less stress on the poor DNA-polymerizing dnaX(Ts) mutant (Blinkova et al., 2003). To further confirm that the oriC deletion strains have decreased rates of replication initiation, as opposed to merely being asynchronous, we examined whether an R4− mutation (oriC227) could suppress a gyrB41 strain. gyrB41 encodes a temperature-sensitive gyrase protein, and exhibits defective DNA replication at high temperature due to a decreased ability to relieve positive supercoiling in front of the replisomes (Filutowicz and Jonczyk, 1983). Double mutant oriC227 gyrB41 cells indeed formed colonies at 40°C, whereas the single gyrB41 mutant did not (Fig. 5A), suggesting that decreased initiation frequency from oriC227 suppressed the replication elongation defect of gyrB41. Unexpectedly, the larger oriC231 mutation failed to suppress gyrB41 (Fig. 5A). The reason for this difference is not clear, although oriC231 cells exhibit atypical nucleoids (Fig. 3A) suggesting that they may have a chromosome structure problem that exacerbates the lack of gyrase.
Fig. 5.

oriC227 suppresses the replication elongation defect of gyrB41.
A. Colony forming ability of strains at 37°C and 40°C (non-permissive for gyrB41). Deletion of DnaA box R4 (oriC227) allowed growth of gyrB41 at high temperature, while a larger deletion (oriC231) did not.
B. DNA synthesis was measured in gyrB+ or gyrB41 cells carrying either a wt or an oriC227 origin. Exponential cells were treated with 3H-thymine for 30 min and incorporation into the acid-insoluble fraction was measured by liquid scintillation. Values are averages of two independent experiments (± SD). All cells grown in M9 glucose plus casamino acids.
To determine whether the suppression was due to enhanced DNA replication in the double mutant at high temperature, we measured DNA synthesis by thymine incorporation over a range of temperatures (Fig. 5B). Clearly, the rate of DNA synthesis of gyrB41 at higher temperatures compared with 29°C was greater when cells carried the oriC227 (R4-deleted) mutation (filled symbols). This data suggests that fewer replication forks initiated by the R4-less origin allow completion of replication elongation of an over-supercoiled chromosome, perhaps by freeing up existing gyrase or the similar but much less abundant Topo IV. Interestingly, DNA synthesis at high temperature compared with 29°C was greater in cells carrying the R4 deletion, even in a gyrB+ background (open symbols). We speculate that this is due to a compromised ability of the mutant origin to function at lower temperatures, thus giving it a higher relative rate of synthesis at high temperature than wt.
To examine how the R4 mutant origin interacted genetically with other initiation mutations, we introduced oriC227 into a seqAΔ mutant, exhibiting a hyper-initiation phenotype (Boye et al., 1996). SeqA protein negatively regulates replication initiation via direct binding to GATC sequences in the DUE region of oriC, which represses strand opening (Slater et al., 1995). seqA null mutants exhibit over-initiation, and during rapid cell growth, forks often fail to complete replication probably as a consequence of exhausted replication resources and/or collisions between adjacent forks (Lu et al., 1994; and Fig. 6B). As expected, the oriC227 seqAΔ double mutant grew faster (~30%) than seqAΔ alone (Fig. 6A, grey bars). Flow cytometry of rifampicin run-off samples indicated that the double mutant cells were able to complete all initiated replication forks, whereas the seqAΔ strain was not (Fig. 6B). Nucleoid morphology of seqAΔ was significantly improved by oriC227 with near-normal chromosome segregation.
Fig. 6.

oriC227 suppresses the over-initiation phenotype of seqAΔ.
A. Growth rates of wt or seqAΔ cells carrying the indicated oriC allele. Averages of two independent experiments (± SD) shown.
B. Nucleoid morphology and DNA content were assayed in seqAΔ cells carrying the indicated oriC alleles. DNA histograms were generated from rifampicin run-off cultures. Unresolved peaks in seqAΔ oriC+ cells indicate an inability to complete replication elongation. All cells were grown in M9 glucose plus casamino acids.
Interestingly, an oriC228 mutant, which was similarly asynchronous as an oriC227 mutant (Fig. 3A), but resulted in relatively normal DNA/mass and origins/cell values (Fig. 3B), did not suppress the growth defect (Fig. 6A) or replication run-out defect (Fig. 6B) of seqAΔ. Therefore, the inability of seqAΔ to complete replication elongation is only alleviated by a sufficiently reduced initiation rate, and not by improperly timed (asynchronous) initiations. Likewise, the much larger oriC231 mutation did not suppress either of these seqAΔ phenotypes (Fig. 6A and B), and the double mutant grew only slightly better than oriC231 alone. Given that oriC231 mutant cells showed a low initiation efficiency (Fig. 3), it is reasonable to conclude that the under-initiation defects of the mutant origin outweighed the over-replication defects resulting from a lack of SeqA. Unsurprisingly, each of the oriC mutations exacerbated the under-initiation phenotype of dnaA46 (data not shown).
Discussion
Differences between oriC plasmids and chromosomal oriC
None of the mutations that we introduced within the traditional 245 bp minimal oriC sequence could support replication of an oriC plasmid (Table 1), while 8 out of 11 mutations were fully functional origins on the chromosome (Fig. 1). This supports a growing body of evidence that oriCs cloned into a plasmid vector have far stricter sequence requirements than chromosomal oriC (Lobner-Olesen and Boye, 1992; Bates et al., 1995; 1997; Asai et al., 1998; Weigel et al., 2001; Riber et al., 2009), and is likely a universal feature of bacterial origins (Dziadek et al., 2002). The failure of mutant oriC plasmids to replicate in vivo is likely not due to competition for initiation proteins between cloned oriC and the host oriC; the plasmids did not gain a significant ability to grow when the host cell carried the same mutant origin (Table 1). This is in general agreement with earlier results with plasmids carrying scrambled DnaA boxes (Weigel et al., 2001). However, it is still possible that perturbed initiation in the mutant host strains presents a situation in which the cell cannot support multiple origin copies, thus preventing oriC plasmid transformation. An alternative explanation for the discrepancy between cloned and chromosomal oriC is differences in DNA topology between the two molecules. oriC plasmids are maintained poorly or not at all in cells that have altered DNA supercoiling including mutant topoisomerases and major nucleoid proteins (Leonard et al., 1985; Ogawa et al., 1989; Gille et al., 1991; Kano et al., 1991). Also, changing the local DNA supercoiling levels by blocking transcription from the two oriC-proximal genes mioC and gidA, has no detectable effect on chromosomal initiation, but reduces oriC plasmid efficiency by > 90% (Bates et al., 1997).
The minimal oriC
We found that the smallest oriC fragment capable of supporting chromosomal replication was a 163 bp sequence containing the DUE element, the left-most six binding sites for DnaA, and the IHF binding site (Fig. 1; oriC231). This origin is missing a total of four known DnaA binding sites; R2 and R4, which are two of the highest affinity DnaA boxes (Margulies and Kaguni, 1996; Grimwade et al., 2000) and remain bound to DnaA throughout the cell cycle (Cassler et al., 1995); and R3 and I3, which are both low-affinity sites and only become occupied at the time of initiation (Grimwade et al., 2007). Also missing in this minimal oriC is the binding site for Fis, which in wild-type oriC regulates the binding of IHF, which redistributes bound DnaA to fill the weaker `trigger' boxes at the time of initiation (Ryan et al., 2004), analogous to the eukaryotic origin transition from an ORC-bound complex to a pre-replicative state. Previous findings have shown that mutations within any one of these elements did not prevent chromosomal oriC from functioning (Weigel et al., 2001; Riber et al., 2009), leading to the idea that redundant elements could compensate for single omissions to form the canonical initiation complex. However, given that oriC231 can function in the absence of nearly half the full allotment of binding sites, it is clear that initiation is highly flexible with no one prerequisite DnaA complex as has been traditionally assumed. Instead, we propose that initiation complexes are inherently stochastic, and likely vary greatly between different growth conditions and from cell to cell. In this light, initiation can be viewed as a very simple process, consisting of some sufficient amount of initiator protein binding to a highly AT-rich stretch of DNA. This is analogous to eukaryotic initiation systems (below).
DnaA binding sites on the right end of oriC facilitate multi-forked replication
Right-end oriC deletions involving DnaA box R4 or more exhibited decreased growth rate, lower DNA/mass, fewer origins/cells and replication asynchrony in M9 glucose medium (Fig. 3). Growth defects were greatly exacerbated when cells were grown in rich medium (LB), and were suppressed when grown in poor medium (M9 succinate) (Fig. 4). This was especially true for the largest deletion strain, oriC231, which grew 13-fold more slowly than wt in LB, but only 1.4-fold more slowly than wt in M9 succinate. In fact, some oriC mutants exhibited a longer doubling time in LB than they did in either M9 succinate or M9 glucose (Fig. 4, oriC230 and oriC231). Interestingly, a mutant retaining near wt DNA/mass levels, but extremely asynchronous initiations (oriC228), exhibited only a slight sensitivity to rich medium. This strongly suggests that LB sensitivity resulted from inefficient initiations, not improperly timed ones. Given that under fast growth there are 8 or 16 copies of oriC in every cell that must fire within a short window of time, it should not be surprising that an inefficient initiating origin would have difficulties. Supporting the notion that mutant oriCs could not keep up with division, LB-sensitive strains exhibited cell elongation, and frequent chromosome segregation, failures (Fig. 3A), hallmarks of poor co-ordination between replication and the cell cycle (Boye and Nordstrom, 2003). In LB, these strains exhibited a more severe phenotype with abundant chromosome guillotining and anucleated cells (data not shown).
One prediction of this hypothesis is a DnaA boxdeficient origin should suppress mutations that lead to over-initiation, or mutations that result in slowed replication elongation. These defects lead to incompletion of replication elongation and the build-up of potentially lethal DSBs at stalled replication forks (Simmons et al., 2004). We indeed found that an R4 deletion (oriC227) suppressed the growth defects and over-initiation phenotype of a seqAΔ mutation (Fig. 6) and partially suppressed the temperature sensitivity and low DNA synthesis of a gyrB41 mutation (Fig. 5). Thus, conditions in which there are either too many replication forks (seqAΔ), or the replication forks that are present cannot be efficiently processed (gyrB41), are alleviated by an under-initiating DnaA box deletion mutation. Together, our results suggest that the clustered DnaA binding sites on the right half of oriC function primarily in rich medium to support the increased demands of multi-forked replication.
The DUE promotes efficient initiations but not synchrony
In contrast to mutations on the right end of oriC, deletions in the DUE region exhibited reduced initiation efficiency, but remained more or less synchronous. oriC234 mutants, carrying a deletion of the L-13mer and adjacent AT-cluster (Fig. 1) showed reduced growth rate, DNA/mass, and origins/cell, but did not exhibit the severe replication asynchrony phenotype indicative of the right-end deletions (Fig. 3, note low frequency of three origin cells). We imagine that deletions within the DUE region, where strand unwinding occurs, likely assemble a timely initiation complex but fail to efficiently convert to the open complex due to loss of AT sequence. The role and significance of replication synchrony has remained unclear. It is possible that it is merely a by-product of a highly efficient initiation system or that it facilitates organization of multiple replication forks into replication hyperstructures (Morigen et al., 2009). It is noteworthy that while the asynchronous but efficient DnaA box mutant oriC228 showed only slight sensitivity to rich medium, the generally synchronous but inefficient DUE mutant oriC234 exhibited significant sensitivity to rich medium (Fig. 4), suggesting that replication synchrony is not the determining factor for multi-forked replication. The fact that initiation efficiency and initiation synchrony are separable phenotypes, may shed new light on the strong asynchrony phenotype of dnaA(Ts) mutants, which was assumed to stem merely from a low initiation ability (Boye et al., 1996).
Bacterial versus eukaryotic origins
Replication origins of eubacteria, and especially the enteric bacteria, are exceptionally well conserved. Most origins consist of three AT-rich 13-mer sequences adjacent to multiple binding sites for DnaA, often with similar spacing to that of oriC in E. coli (Zyskind et al., 1983; Kogoma, 1998). Eukaryotic origins, on the other hand, tend to be very loosely defined, with little or no consensus origin sequences. Saccharomyces cerevisiae has the best-characterized origins, each containing a single essential 17 bp ARS consensus sequence and a nearby adenine-rich region (Breier et al., 2004; Chen et al., 2008). In meta-zoans, origins do not share any known consensus sequence, and mapping typically cannot narrow down replication start sites to less than a few kb (Gilbert, 2001; DePamphilis, 2005). Some evidence suggests that essentially any AT-rich sequence of sufficient length can function as a replication origin (Dai et al., 2005). Why are bacterial and eukaryotic origins apparently so different? As shown in this study, removing binding sites for the DnaA initiator protein from oriC is extremely detrimental under fast growth conditions, but had almost no effect under slow growth conditions. Having multiple binding sites for DnaA may allow prokaryotes to couple initiation rate to changes in physiological conditions. Eukaryotes, on the other hand, utilize a sophisticated replication licensing system, designed primarily to throttle down replication initiation (Diffley, 2004; Robinson and Bell, 2005). In addition, a highly specific replicator like oriC may ensure replication synchrony, which is a feature specific to bacteria.
Experimental procedures
Media and bacterial strains
Cells were grown at 37°C, with aeration by shaking, in minimal M9 medium supplemented with 0.1% carbon source as indicated, or LB medium supplemented with 0.1% glucose. Antibiotics were added at the following concentrations: chloramphenicol (50 μg ml−1), tetracycline (20 mgl−1) and kanamycin (55 μg ml−1). Where indicated, casamino acids were added at 0.5%.
All strains shown are derivatives of MG1655, with the exception of competent cells for the polA assay (Table 1), in which one set of strains (wild-type oriC host) are derivatives of AQ634 (Ogawa et al., 1984) and the other set of strains (matching mutant oriC host) are polA1 (P1 transduced selecting for Tn10 TetR) derivatives of MG1655. gyrB41 (Filutowicz and Jonczyk, 1983) linked to zid-501::Tn10 (this work) was introduced into oriC mutants by P1 transduction. seqAΔ (Boye et al., 1996) is an in-frame deletion mutation that has wild-type expression of the downstream pgm gene, and was introduced by the recombineering method of Yu et al. (2000).
oriC mutagenesis
Left- and right-end oriC deletions were generated by selective PCR amplification of chromosomal DNA. oriC co-ordinates are according to Meijer and Beck (Meijer et al., 1979). To create right-end deletions, a single forward primer was used complimentary to the 5′ upstream end of oriC overlapping the SmaI (−47) site (TTGAAGCCCGGGCCGTGGATTCTACT) and various reverse primers were used, complimentary to locations within oriC and containing an AccI tail (ATACTAGTATAC) to facilitate cloning. The sequences of the reverse primers were as follows (minus the AccI tail); oriC227-TTGGATCAACCGGTAG, oriC228-TTATCCAAAG AACAACTG, oriC229-GAACAACTGTTGTTCAG, oriC230-TGTGTATAACCCCTCAT, oriC231-ACCCCTCATTCTGATC, oriC237-TTTCCAGGTTGTTGATC, oriC238-TTATCCACAG GGCAGTG. The resulting PCR products were then cloned into the oriC plasmid pDB201 (Bates et al., 1997) between the SmaI (−47) and AccI (+285) sites. The oriC regions of the resulting plasmids (Table 1) were then sequenced to confirm deletions and transferred onto the chromosome by the in vivo transfer system. The left-end deletions were constructed in a similar manner, except the reverse primer was anchored at the HindIII (+244) site (TCAGGAAGCTTGGATCAACC), and the forward primers, which contained a BclI (5′-TTAAT TGATCA-3′) tail, were varied depending on the desired deletion. The sequences of the 5′ primers (minus the BclI tail) are as follows; oriC133-AGATCTATTTATTTAGAGATCT, oriC135-GTGATCTCTTATTAGGATCGC. oriC134 was made by deleting the BglII (+22–+28) fragment from the oriC133 PCR product. These PCR fragments were then ligated to a SacII (−164) to BclI (+1 BamHI converted) fragment and then cloned into pDB201 for sequencing and transfer to the chromosome. To create oriC239 and oriC243, left and right-end oriC deletions (oriC233 + oriC229, oriC234 + oriC229 respectively) were combined by restriction endonuclease cloning.
In vivo transfer procedure
Modified oriC sequences constructed on plasmids were introduced onto the chromosome by a two-step procedure described elsewhere (Bates et al., 1995; see Fig. S1). Briefly, mutant oriCs were first transferred from plasmids to a specialized lambda phage vector containing an oriC fragment with a gidA∷kan marker (λ10.2) by homologous recombination. Recombinant phage was selected for the ability to transduce a strain to chloramphenicol but not kanamycin resistance. The modified oriC was then transferred from the purified phage to the chromosome of AQ7664 (Bates et al., 1995) by infection selecting for chloramphenicol resistance under conditions not allowing for phage lysogenization (42°C growth in presence of cI857 mutation). oriC deletions were subsequently moved into MG1655 by P1 transduction and verified by Southern blot hybridization (Fig. S2).
In cases where no chloramphenicol-resistant and kanamycin-sensitive AQ7664 recombinants were obtained, it was considered likely that the mutant oriC was non-functional. In such cases, the modified oriCs were transferred from the phage to the chromosome of AQ10033 (Bates et al., 1995), which carries the rnhA224 mutation. This mutation allows for a bypass replication system to occur, cSDR, which is independent of oriC and DnaA (Kogoma, 1997; Fig. 2). Mutant oriC regions were verified by Southern blot hybridization, and confirmed for non-functional oriC by sensitivity to expression of RNase HI from an inducible plasmid.
DNA replication analysis
To determine DNA content in the oriC mutant strains, cells were analysed by flow cytometry as follows. Cells were grown in M9 glucose medium maintaining exponential growth for a minimum of 10 generations by successive dilutions into fresh media. An aliquot of cells were centrifuged and resuspended in cold TE, then added to cold ethanol (70% final concentration) and stored overnight at 4°C. Cells were then washed twice in PBS and resuspended in PBS containing RnaseA (10 mg l−1), and incubated at room temperature for 15 min to digest cellular RNA. Cells were stained with SYTOX Green (Invitrogen) at 2.5 μM for a minimum of 15 min. Samples were analysed in a BD Biosciences LSR II flow cytometer equipped with a forward-scatter PMT. DNA/mass values were determined for each sample as the average SYTOX green fluorescence divided by average forward light scatter, roughly equivalent to cell mass. To determine origins per cell, cultures were treated with rifampicin (0.15 mg ml−1) and cephalexin (10 μg ml−1) and incubated with shaking for an additional 2.5 h before fixation as above. Flow cytometry data were analysed using Cyflogic software (Finland). DNA synthesis rates were determined by 3H-thymine incorporation as previously described (Kogoma and von Meyenburg, 1983).
Microscopy
To image cell nucleoid and membrane morphology, cells were grown to mid-exponential phase (OD450 = 0.4) and fixed in 2% paraformaldehyde for 10 min at room temperature, then 10 min on ice. Cells were washed twice in PBS by low-speed centrifugation (5000 g for 1 min) and resuspended in PBS containing 0.2 μg ml−1 DAPI and 1 mM FM 4–64 (Invitrogen). Images were acquired using a Zeiss AxioImager Z1 fluorescence microscope equipped with a Hamamatsu Electron Multiplier CCD camera. DAPI and FM 4–64 dyes were imaged using 31013v2 and SP103v2 filter sets (Chroma).
Supplementary Material
Acknowledgements
We would like to acknowledge the assistance of the Cytometry and Cell Sorting Core facility at Baylor College of Medicine, and Christophe Herman and Jade Wang for critical reading of the manuscript. We dedicate this paper to the memory of Dr Tokio Kogoma.
Footnotes
Supporting information Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Asai T, Bates DB, Boye E, Kogoma T. Are minichromosomes valid model systems for DNA replication control? Lessons learned from Escherichia coli. Mol Microbiol. 1998;29:671–675. doi: 10.1046/j.1365-2958.1998.00901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DB, Asai T, Cao Y, Chambers MW, Cadwell GW, Boye E, Kogoma T. The DnaA box R4 in the minimal oriC is dispensable for initiation of Escherichia coli chromosome replication. Nucleic Acids Res. 1995;23:3119–3125. doi: 10.1093/nar/23.16.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DB, Boye E, Asai T, Kogoma T. The absence of effect of gid or mioC transcription on the initiation of chromosomal replication in Escherichia coli. Proc Natl Acad Sci USA. 1997;94:12497–12502. doi: 10.1073/pnas.94.23.12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Blinkova A, Hermandson MJ, Walker JR. Suppression of temperature-sensitive chromosome replication of an Escherichia coli dnaX (Ts) mutant by reduction of initiation efficiency. J Bacteriol. 2003;185:3583–3595. doi: 10.1128/JB.185.12.3583-3595.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye E, Nordstrom K. Coupling the cell cycle to cell growth. EMBO Rep. 2003;4:757–760. doi: 10.1038/sj.embor.embor895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye E, Stokke T, Kleckner N, Skarstad K. Coordinating DNA replication initiation with cell growth: differential roles for DnaA and SeqA proteins. Proc Natl Acad Sci USA. 1996;93:12206–12211. doi: 10.1073/pnas.93.22.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramhill D, Kornberg A. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell. 1988;52:743–755. doi: 10.1016/0092-8674(88)90412-6. [DOI] [PubMed] [Google Scholar]
- Breier AM, Chatterji S, Cozzarelli NR. Prediction of Saccharomyces cerevisiae replication origins. Genome Biol. 2004;5:R22. doi: 10.1186/gb-2004-5-4-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassler MR, Grimwade JE, Leonard AC. Cell cycle-specific changes in nucleoprotein complexes at a chromosomal replication origin. EMBO J. 1995;14:5833–5841. doi: 10.1002/j.1460-2075.1995.tb00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Speck C, Wendel P, Tang C, Stillman B, Li H. The architecture of the DNA replication origin recognition complex in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2008;105:10326–10331. doi: 10.1073/pnas.0803829105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Chuang RY, Kelly TJ. DNA replication origins in the Schizosaccharomyces pombe genome. Proc Natl Acad Sci USA. 2005;102:337–342. doi: 10.1073/pnas.0408811102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePamphilis ML. Cell cycle dependent regulation of the origin recognition complex. Cell Cycle. 2005;4:70–79. doi: 10.4161/cc.4.1.1333. [DOI] [PubMed] [Google Scholar]
- Diffley JF. Regulation of early events in chromosome replication. Curr Biol. 2004;14:R778–R786. doi: 10.1016/j.cub.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Dziadek J, Rajagopalan M, Parish T, Kurepina N, Greendyke R, Kreiswirth BN, Madiraju MV. Mutations in the CCGTTCACA DnaA box of Mycobacterium tuberculosis oriC that abolish replication of oriC plasmids are tolerated on the chromosome. J Bacteriol. 2002;184:3848–3855. doi: 10.1128/JB.184.14.3848-3855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filutowicz M, Jonczyk P. The gyrB gene product functions in both initiation and chain polymerization of Escherichia coli chromosome replication: suppression of the initiation deficiency in gyrB-ts mutants by a class of rpoB mutations. Mol Gen Genet. 1983;191:282–287. doi: 10.1007/BF00334827. [DOI] [PubMed] [Google Scholar]
- Fujimitsu K, Senriuchi T, Katayama T. Specific genomic sequences of E. coli promote replicational initiation by directly reactivating ADP-DnaA. Genes Dev. 2009;23:1221–1233. doi: 10.1101/gad.1775809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller RS, Kaguni JM, Kornberg A. Enzymatic replication of the origin of the Escherichia coli chromosome. Proc Natl Acad Sci USA. 1981;78:7370–7374. doi: 10.1073/pnas.78.12.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller RS, Funnell BE, Kornberg A. The dnaA protein complex with the E. coli chromosomal replication origin (oriC) and other DNA sites. Cell. 1984;38:889–900. doi: 10.1016/0092-8674(84)90284-8. [DOI] [PubMed] [Google Scholar]
- Gilbert DM. Making sense of eukaryotic DNA replication origins. Science. 2001;294:96–100. doi: 10.1126/science.1061724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille H, Egan JB, Roth A, Messer W. The FIS protein binds and bends the origin of chromosomal DNA replication, oriC, of Escherichia coli. Nucleic Acids Res. 1991;19:4167–4172. doi: 10.1093/nar/19.15.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gines-Candelaria E, Blinkova A, Walker JR. Mutations in Escherichia coli dnaA which suppress a dnaX (Ts) polymerization mutation and are dominant when located in the chromosomal allele and recessive on plasmids. J Bacteriol. 1995;177:705–715. doi: 10.1128/jb.177.3.705-715.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwade JE, Ryan VT, Leonard AC. IHF redistributes bound initiator protein, DnaA, on supercoiled oriC of Escherichia coli. Mol Microbiol. 2000;35:835–844. doi: 10.1046/j.1365-2958.2000.01755.x. [DOI] [PubMed] [Google Scholar]
- Grimwade JE, Torgue JJ, McGarry KC, Rozgaja T, Enloe ST, Leonard AC. Mutational analysis reveals Escherichia coli oriC interacts with both DnaA-ATP and DnaA-ADP during pre-RC assembly. Mol Microbiol. 2007;66:428–439. doi: 10.1111/j.1365-2958.2007.05930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaguni JM. DnaA: controlling the initiation of bacterial DNA replication and more. Annu Rev Microbiol. 2006;60:351–375. doi: 10.1146/annurev.micro.60.080805.142111. [DOI] [PubMed] [Google Scholar]
- Kano Y, Ogawa T, Ogura T, Hiraga S, Okazaki T, Imamoto F. Participation of the histone-like protein HU and of IHF in minichromosomal maintenance in Escherichia coli. Gene. 1991;103:25–30. doi: 10.1016/0378-1119(91)90386-p. [DOI] [PubMed] [Google Scholar]
- Kawakami H, Keyamura K, Katayama T. Formation of an ATP-DnaA-specific initiation complex requires DnaA Arginine 285, a conserved motif in the AAA+ protein family. J Biol Chem. 2005;280:27420–27430. doi: 10.1074/jbc.M502764200. [DOI] [PubMed] [Google Scholar]
- Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev. 1997;61:212–238. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T. Origins of chromosome replication. In: de Bruijn FJ, Lupski JR, Weinstock GM, editors. Bacterial Genomes: Physical Structure and Analysis. Chapman & Hall; New York: 1998. pp. 67–77. [Google Scholar]
- Kogoma T, von Meyenburg K. The origin of replication, oriC, and the dnaA protein are dispensable in stable DNA replication (sdrA) mutants of Escherichia coli K-12. EMBO J. 1983;2:463–468. doi: 10.1002/j.1460-2075.1983.tb01445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AC, Whitford WG, Helmstetter CE. Involvement of DNA superhelicity in minichromosome maintenance in Escherichia coli. J Bacteriol. 1985;161:687–695. doi: 10.1128/jb.161.2.687-695.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobner-Olesen A, Boye E. Different effects of mioC transcription on initiation of chromosomal and minichromosomal replication in Escherichia coli. Nucleic Acids Res. 1992;20:3029–3036. doi: 10.1093/nar/20.12.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Campbell JL, Boye E, Kleckner N. SeqA: a negative modulator of replication initiation in E. coli. Cell. 1994;77:413–426. doi: 10.1016/0092-8674(94)90156-2. [DOI] [PubMed] [Google Scholar]
- McGarry KC, Ryan VT, Grimwade JE, Leonard AC. Two discriminatory binding sites in the Escherichia coli replication origin are required for DNA strand opening by initiator DnaA-ATP. Proc Natl Acad Sci USA. 2004;101:2811–2816. doi: 10.1073/pnas.0400340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies C, Kaguni JM. Ordered and sequential binding of DnaA protein to oriC, the chromosomal origin of Escherichia coli. J Biol Chem. 1996;271:17035–17040. doi: 10.1074/jbc.271.29.17035. [DOI] [PubMed] [Google Scholar]
- Meijer M, Beck E, Hansen FG, Bergmans HE, Messer W, von Meyenburg K, Schaller H. Nucleotide sequence of the origin of replication of the Escherichia coli K-12 chromosome. Proc Natl Acad Sci USA. 1979;76:580–584. doi: 10.1073/pnas.76.2.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenburg K, Hansen FG, Nielsen LD, Jorgensen P. Origin of replication, oriC, of the Escherichia coli chromosome: mapping of genes relative to R.EcoRI cleavage sites in the oriC region. Mol Gen Genet. 1977;158:101–109. doi: 10.1007/BF00455124. [DOI] [PubMed] [Google Scholar]
- Morigen I, Odsbu and Skarstad K. Growth rate dependent numbers of SeqA structures organize the multiple replication forks in rapidly growing Escherichia coli. Genes Cells. 2009;14:643–657. doi: 10.1111/j.1365-2443.2009.01298.x. [DOI] [PubMed] [Google Scholar]
- Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol. 2007;5:343–354. doi: 10.1038/nrmicro1640. [DOI] [PubMed] [Google Scholar]
- Nielsen O, Lobner-Olesen A. Once in a lifetime: strategies for preventing re-replication in prokaryotic and eukaryotic cells. EMBO Rep. 2008;9:151–156. doi: 10.1038/sj.embor.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nievera C, Torgue JJ, Grimwade JE, Leonard AC. SeqA blocking of DnaA–oriC interactions ensures staged assembly of the E. coli pre-RC. Mol Cell. 2006;24:581–592. doi: 10.1016/j.molcel.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, Pickett GG, Kogoma T, Kornberg A. RNase H confers specificity in the dnaA-dependent initiation of replication at the unique origin of the Escherichia coli chromosome in vivo and in vitro. Proc Natl Acad Sci USA. 1984;81:1040–1044. doi: 10.1073/pnas.81.4.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, Wada M, Kano Y, Imamoto F, Okazaki T. DNA replication in Escherichia coli mutants that lack protein HU. J Bacteriol. 1989;171:5672–5679. doi: 10.1128/jb.171.10.5672-5679.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Sugimoto K, Takanami M, Hirota Y. Replication origin of the Escherichia coli K-12 chromosome: the size and structure of the minimum DNA segment carrying the information for autonomous replication. Mol Gen Genet. 1980;178:9–20. doi: 10.1007/BF00267207. [DOI] [PubMed] [Google Scholar]
- Riber L, Fujimitsu K, Katayama T, Lobner-Olesen A. Loss of Hda activity stimulates replication initiation from I-box, but not R4 mutant origins in Escherichia coli. Mol Microbiol. 2009;71:107–122. doi: 10.1111/j.1365-2958.2008.06516.x. [DOI] [PubMed] [Google Scholar]
- Robinson NP, Bell SD. Origins of DNA replication in the three domains of life. FEBS J. 2005;272:3757–3766. doi: 10.1111/j.1742-4658.2005.04768.x. [DOI] [PubMed] [Google Scholar]
- Ryan VT, Grimwade JE, Nievera CJ, Leonard AC. IHF and HU stimulate assembly of pre-replication complexes at Escherichia coli oriC by two different mechanisms. Mol Microbiol. 2002;46:113–124. doi: 10.1046/j.1365-2958.2002.03129.x. [DOI] [PubMed] [Google Scholar]
- Ryan VT, Grimwade JE, Camara JE, Crooke E, Leonard AC. Escherichia coli prereplication complex assembly is regulated by dynamic interplay among Fis, IHF and DnaA. Mol Microbiol. 2004;51:1347–1359. doi: 10.1046/j.1365-2958.2003.03906.x. [DOI] [PubMed] [Google Scholar]
- Simmons LA, Breier AM, Cozzarelli NR, Kaguni JM. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Mol Microbiol. 2004;51:349–358. doi: 10.1046/j.1365-2958.2003.03842.x. [DOI] [PubMed] [Google Scholar]
- Skarstad K, Lobner-Olesen A. Stable co-existence of separate replicons in Escherichia coli is dependent on once-per-cell-cycle initiation. EMBO J. 2003;22:140–150. doi: 10.1093/emboj/cdg003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K, Boye E, Steen HB. Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J. 1986;5:1711–1717. doi: 10.1002/j.1460-2075.1986.tb04415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater S, Wold S, Lu M, Boye E, Skarstad K, Kleckner N. E. coli SeqA protein binds oriC in two different methyl-modulated reactions appropriate to its roles in DNA replication initiation and origin sequestration. Cell. 1995;82:927–936. doi: 10.1016/0092-8674(95)90272-4. [DOI] [PubMed] [Google Scholar]
- Speck C, Messer W. Mechanism of origin unwinding: sequential binding of DnaA to double- and single-stranded DNA. EMBO J. 2001;20:1469–1476. doi: 10.1093/emboj/20.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torheim NK, Skarstad K. Escherichia coli SeqA protein affects DNA topology and inhibits open complex formation at oriC. EMBO J. 1999;18:4882–4888. doi: 10.1093/emboj/18.17.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldminghaus T, Skarstad K. The Escherichia coli SeqA protein. Plasmid. 2009;61:141–150. doi: 10.1016/j.plasmid.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Weigel C, Messer W, Preiss S, Welzeck, Morigen M, Boye E. The sequence requirements for a functional Escherichia coli replication origin are different for the chromosome and a minichromosome. Mol Microbiol. 2001;40:498–507. doi: 10.1046/j.1365-2958.2001.02409.x. [DOI] [PubMed] [Google Scholar]
- Wold S, Boye E, Slater S, Kleckner N, Skarstad K. Effects of purified SeqA protein on oriC-dependent DNA replication in vitro. EMBO J. 1998;17:4158–4165. doi: 10.1093/emboj/17.14.4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyskind JW, Cleary JM, Brusilow WS, Harding NE, Smith DW. Chromosomal replication origin from the marine bacterium Vibrio harveyi functions in Escherichia coli: oriC consensus sequence. Proc Natl Acad Sci USA. 1983;80:1164–1168. doi: 10.1073/pnas.80.5.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.