

Abstract
CD1d is expressed on APCs and presents glycolipids to CD1d-restricted NKT cells. For the first time, we demonstrate the ability of anti-CD1d mAbs to inhibit the growth of different CD1d-negative experimental carcinomas in mice. Anti-CD1d mAbs systemically activated CD1d+ APC, as measured by production of IFN-γ and IL-12. Tumor growth inhibition was found to be completely dependent on IFN-γ and IL-12 and variably dependent on CD8+ T cells and NK cells, depending upon the tumor model examined. Anti-CD1d mAb induced greater CD8+ T cell-dependent tumor suppression where regulatory CD1d-restricted type II NKT cells have been implicated, and were less effective in a NK cell-dependent manner against tumors where T regulatory cells were immunosuppressive. The ability of anti-CD1d mAbs to coincidently activate CD1d+ APCs to release IL-12 and inhibit CD1d-restricted type II NKT cells makes CD1d an exciting new target for immunotherapy of cancer based on tumor immunoregulation.
Dendritic cells (DCs)3 play an important role in the induction of an antitumor immune response (1). Depending on the activation signals received from either Toll-like receptors or interacting lymphocytes, DCs can differentiate and mature as characterized by up-regulation of costimulatory molecules and production of IL-12 and IFN-α(2, 3). Production of these immune activating cytokines often occurs in the context of cross-talk with lymphocytes such as conventional T cells, NKT cells (4), and NK cells (5), depending upon the initiating stimulus. However, in cancer, the interaction of DCs with tumors, tumor-derived factors, or other cells in the tumor microenvironment can result in DC differentiation into tolerogenic types (6). Given the importance of activated/matured DCs in the initiation and activation of tumor-specific T cells, many preclinical studies and clinical trials have examined the use of different agents to activate/mature DC (1).
To date, DCs have been activated/matured directly through the use of agonistic anti-CD40 mAbs (7) or Flt3L (8), or indirectly using CD1d-reactive glycolipid ligands such as α-galactosylceramide (α-GC), that engage cross-talk with CD1d-restricted type I NKT cells (9). However, toxicity issues have been associated with agonistic CD40 mAbs in recent clinical trials (10, 11), and a recent study has demonstrated that signaling of CD40 through endothelial cells can promote tumor growth (12). In addition, qualitative and quantitative defects of type I NKT cells in cancer patients can potentially hamper the use of glycolipid ligands to indirectly activate DCs via type I NKT cell TCR (13–15).
An overlooked alternative strategy to activate DCs involves the use of anti-CD1d mAbs. CD1d is expressed prominently and constitutively on professional APCs such as DCs, macrophages, and B cells (16). We recently demonstrated that anti-human CD1d mAbs matured DCs, resulting in the production of bio-active IL-12p70 (17). In addition to its agonistic activity, anti-CD1d mAbs may potentially block the activation of CD1d-restricted type II NKT cells (18). Two types of NKT cells are restricted by CD1d: type I and type II. Type I NKT cells have a host protective role in natural or α-GC-induced tumor immunity (9, 19, 20). In contrast, type II NKT cells have different tissue distribution in humans and mice and have been implicated in immunosuppression of effector T cell responses to cancer (18, 21, 22).
In this study, we assessed the antitumor efficacy of anti-CD1d mAbs in three different experimental tumor models: R331 renal carcinoma, 4T1 mammary carcinoma, and CT26L5 colon adenocarcinoma. These tumors do not express CD1d, and evidence suggests or shows type II NKT cells play a role in immune suppression in the 4T1 and CT26L5 tumor models (18), whereas regulatory T cells control immune effector response to the R331 tumor (M.J.S., unpublished data). We demonstrate, for the first time, the ability of anti-CD1d mAb monotherapy to inhibit the s.c. growth of three different aggressive experimental tumor models. Interestingly, anti-CD1d mAb therapy resulted in greater suppression of 4T1 and CT26 tumors than R331, and the mechanism of growth suppression was distinct. Given that our results implicate the ability of anti-CD1d mAb to coincidently activate APCs and block type II NKT cell activation, we believe there is a rationale to use similar agonistic mAb reactive with human CD1d in the treatment of human tumors immunoregulated by type II NKT cells.
Materials and Methods
Mice
Inbred BALB/c mice were either bred at the Peter MacCallum Cancer Centre (Peter Mac) or purchased from The Jackson Laboratory or the Walter and Eliza Hall Institute of Medical Research. BALB/c SCID, BALB/c CD1d gene-targeted (CD1d−/−), and BALB/c Jα18 gene-targeted mice (Jα18−/−) (18) were bred and maintained at the Peter Mac or purchased from The Jackson Laboratory. Female mice were used for all experiments with the 4T1 tumor and all experiments were performed in accordance with guidelines set out by the Peter Mac animal ethics experimentation committee.
Tumor cell lines
BALB/c-derived CD1d-negative TRAIL-sensitive 4T1 mammary carcinoma (23, 24), the TRAIL-sensitive Renca variant R331 (23), and the carcinogen-induced colon carcinoma line Colon-26 (CT26L5) (18) were maintained as described previously.
Antibodies
We prepared and purified agonistic mAbs reactive with mouse CD1d (1B1) or purchased from BD Pharmingen, along with isotype control rat IgG2b. Depleting anti-mouse CD4 (GK1.5), anti-CD8 (53-6.7) mAb, neutralizing mAb to mouse IL-12 (C17.8) and IFN-γ (H22) (provided by Dr. Robert Schreiber, Washington University School of Medicine, St. Louis, MO) were prepared and used as previously described (25). Anti-asialo GM1 (anti-ASGM1) for depletion of NK cells was obtained from Wako Pure Chemical and used as previously described (26). Recombinant mouse IL-12 was provided by the Genetics Institute.
Measurement of cytokines derived from splenocytes or serum
Splenocytes (1 × 105/well) from BALB/c wild type (WT) or BALB/c CD1d−/− mice were stimulated with soluble or plate-bound anti-CD1d mAbs (10 μg/ml), isotype control (10 μg/ml), α-GC (100 ng/ml) (Kirin Pharma), or O111:B4 Escherichia coli LPS (10 ng/ml) (Sigma-Aldrich) in 96-well plates as previously described (17). Supernatants were collected at day 1 and 3 after stimulation and assayed for IFN-γ and IL-12. In some experiments, PE-labeled beads (Miltenyi Biotec) (1 μl per 200 μl cells) were use to prepurify F4/80+, class II+, CD3+, or CD19+ populations from mouse spleen as per the manufacturer’s instructions. These populations (>90% pure) were then cultured (105 per well) in a similar manner with plate bound anti-CD1d or LPS (and 10 ng/ml IFN-γ) for 24 h to measure active IL-12p70. For serum cytokines, groups of BALB/c WT (n = 4) or BALB/c CD1d−/− mice (n = 2) were i.p. injected with anti-CD1d mAb (50 μg) or isotype control (50 μg). Serum was harvested 24 h later and assayed for IFN-γ and IL-12. All cytokines were determined by at least triplicate samples by ELISA (PBL Biomedical Laboratory; R&D Systems). Limits of detection were ~1 pg/ml.
Therapy of transplanted tumors
Groups of five BALB/c WT, BALB/c CD1d−/−, BALB/c Jα18−/−, or BALB/c SCID mice were inoculated s.c. with either 1 × 105 CT26L5, 2 × 105 4T1, or 5 × 105 R331 tumor cells on day 0. On the days indicated after tumor inoculation, groups of mice were treated i.p. with anti-CD1d mAb (200 μg) or cIg (200 μg). In some experiments, mice were depleted of CD4+ (GK1.5) (100 μg), CD8+ (53.6.72) (100 μg), or NK cells (anti-ASGM1) (100 μg) on day 0 and then given twice weekly until day 20. In some experiments, mice were neutralized with IFN-γ (250 μg) or IL-12 mAbs (250 μg) starting on day 0 and then given twice weekly until day 20. Tumor size was measured with a caliper and recorded as the product of two perpendicular diameters (cm2).
Statistics
Statistical differences in between control Ig (cIg)-treated mice or mAb-treated mice injected with anti-CD1d mAbs were determined by Mann-Whitney Rank Sum test (*, p < 0.05).
Results
Anti-CD1d mAb induce APC production of IFN-γ and IL-12
We first tested the ability of anti-CD1d mAb to activate CD1d+ APCs by testing for the production of IFN-γ and IL-12 (Fig. 1). Mouse splenocytes from BALB/c WT or BALB/c CD1d−/− mice were stimulated with plate-bound anti-CD1d mAb, isotype control, α-GC, or LPS for 1 (Fig. 1, A and C) or 3 days (Fig. 1, B and D). Supernatants derived from BALB/c WT splenocytes cultured on anti-CD1d mAbs resulted in more dramatic increases in both IL-12 and IFN-γ above those cultured with isotype control (Fig. 1, A–D). The increases in cytokine production were CD1d-dependent as only baseline levels of IL-12 and IFN-γ were detected from BALB/c CD1d−/− splenocytes stimulated with anti-CD1d mAbs (Fig. 1, A–D). The levels of cytokines produced by WT splenocytes activated by anti-CD1d mAb were similar to that induced by CD1d-dependent α-GC, but lower than those stimulated with the CD1d-independent TLR4 agonist LPS (Fig. 1, A–D). Soluble anti-CD1d also produced active IL-12p70, although cross-linking was optimal (Fig. 1E). Consistent with previous reports (27–30), when isolated into purified populations it was clear that F4/80+ macrophages and class II+ cells were stimulated to produce active IL-12p70 by plate bound anti-CD1d, whereas neither T nor B cells released IL-12p70 (Fig. 1F). We next tested the agonistic function of anti-CD1d mAbs in vivo. BALB/c WT or BALB/c CD1d−/− mice were treated with one dose of anti-CD1d mAb (50 μg) or isotype control and serum cytokines were assayed 24 h later. Similarly to in vitro results, increased levels of IL-12 and IFN-γ were detected in the serum of mice treated with anti-CD1d mAb (p = 0.0286, 0.0294) (Fig. 1, G and H). This induction was CD1d-specific, because background levels of these cytokines were observed in the serum derived from anti-CD1d mAb-treated BALB/c CD1d−/− mice. Similarly, baseline levels of IL-12 and IFN-γ were detected in isotype treated BALB/c WT mice (Fig. 1, G and H). These in vitro and in vivo data demonstrated that anti-CD1d mAb was agonistic and activated CD1d+ APC cytokine secretion, similarly to human CD1d mAb and APC in vitro.
FIGURE 1.

Anti-CD1d mAb induces IL-12 and IFN-γ production from CD1d+ splenocytes. A and B, Splenocytes from BALB/c WT or BALB/c CD1d−/− mice were stimulated with either soluble or plate-bound anti-CD1d mAbs (10 μg/ml), isotype control, α-GC (100 ng/ml), or LPS (10 ng/ml). Supernatants were collected one (A, C, E) and 3 days (B and D) after stimulation and cytokines assayed for IL-12p70 or IFN-γ by ELISA. F, Purified splenic F4/80+, class II+, CD3+, or CD19+ populations were cultured as shown with plate bound anti-CD1d or LPS for 24 h and active IL-12p70 measured by ELISA. G and H, Groups of BALB/c WT or BALB/c CD1d−/− mice (n = 2–4) were injected i.p with anti-CD1d mAb (50 μg) or cIg and serum harvested 24 h later to assay for IL-12 (G) and IFN-γ (H). All cytokines were determined by at least triplicate by ELISA. Results represents mean ± SDs. Representative of two experiments and time points. Statistical differences in cytokine production between isotype- and anti-CD1d mAb-treated WT mice were determined by Mann-Whitney Rank Sum test (*, p < 0.05).
Anti-CD1d induces optimal tumor suppression when CD1d-restricted type II NKT cells regulate growth
We next assessed the antitumor efficacy of anti-CD1d mAbs in three different s.c. tumor models, the renal carcinoma cell line R331 (Fig. 2A), the mammary carcinoma cell line 4T1 (Fig. 2B), and the colon adenocarcinoma cell line CT26L5 (Fig. 2C). Consistent with a previous report using CD1d-deficient mice that showed the growth of some tumors was regulated by CD1d-restricted type II NKT cells (18), we found that both 4T1 (Fig. 2B) and CT26L5 (Fig. 2C) tumors grew and regressed in CD1d−/− mice, whereas R331 tumor growth was unaffected (Fig. 2A). The contrasting growth of R331, 4T1, and CT26L5 in CD1d−/− mice was also consistent with the ability of anti-CD1d mAbs to suppress tumor growth, although clearly anti-CD1d treatment commencing on day 11 was not equivalent in absolute tumor suppression with host deficiency of CD1d from day 0. Given that CD1d is expressed widely, the dose of anti-CD1d mAb administered in our experiments (200 μg/injection) is unlikely to saturate all CD1d+ cells for extended periods, but our efforts to further increase the dose of anti-CD1d mAb (to 400 μg) did not improve antitumor efficacy (data not shown). Additional experiments have shown that depletion of regulatory CD25+ T cells markedly suppresses R331 tumor growth (data not shown), but has no effect on the growth of 4T1 or CT26L5 (18). Thus, the greater therapeutic activity of anti-CD1d mAb in the 4T1 and CT26L5 tumor models correlates with the proposed suppressor activity of CD1d-restricted type II NKT cells in these models.
FIGURE 2.

Tumor-dependent anti-CD1d mAb therapy. Groups of BALB/c WT or BALB/c CD1d−/− mice (n = 5) were inoculated subcutaneously with the renal carcinoma cell line, R331 (5 × 105) (A), the mammary carcinoma cell line 4T1 (2 × 105) (B), and the colon carcinoma cell line CT26L5 (1 × 105) (C). On days 11, 15, and 19 after tumor inoculation, WT mice were treated i.p. with anti-CD1d mAbs (200 μg) (■) or cIg (●) as indicated. Tumor sizes are represented as the mean ± SEM and tumor rejection rates are indicated in parentheses. Statistical differences in tumor sizes between cIg- and anti-CD1d mAb-treated WT mice or CD1d−/− mice were determined by Mann-Whitney Rank Sum test (*, p < 0.05).
Anti-CD1d mAb suppress established s.c. tumor growth
To examine the therapeutic efficacy of anti-CD1d mAb against tumors of various sizes, we varied the commencement of treatment of mice with anti-CD1d mAb until days 3, 7, or 11 after tumor inoculation, with each group of mice receiving three treatments every 4 days. Anti-CD1d mAb therapy commencing at day 3 in fact induced modest growth inhibition of R331 tumor compared with similar groups of tumor-bearing mice treated at day 3 with cIg (Fig. 3A). This therapeutic effect was lost as treatment was progressively delayed. In contrast, anti-CD1d mAb therapy of day 3 established 4T1 and CT26L5 tumors resulted in strong suppression of tumor growth compared with groups of tumor-bearing mice treated with cIg (Fig. 3, B and C). Impressively, tumor growth suppression was observed even when therapy was delayed until day 11 after tumor inoculation (~0.25 cm2) (p = 0.0079).
FIGURE 3.

Anti-CD1d mAb induced suppression of established tumors. Groups of BALB/c mice (n = 5) were inoculated s.c. with the renal carcinoma cell line R331 (5 × 105) (A), the mammary carcinoma cell line 4T1 (2 × 105) (B), and the colon carcinoma cell line CT26L5 (1 × 105) (C). After tumor inoculation, mice were treated i.p with anti-CD1d mAbs (200 μg) (squares) at the time points indicated in the parentheses or cIg (●). Tumor sizes are represented as the mean ± SEM. Statistical differences in tumor sizes between cIg- and anti-CD1d-mAb-treated mice were determined by Mann-Whitney Rank Sum test (*, p < 0.05).
Anti-CD1d mAb-induced tumor suppression is dependent on IL-12 and IFN-γ
To determine the cells and cytokines involved in anti-CD1d mAb-mediated tumor suppression, we inoculated groups of BALB/c WT mice, BALB/c SCID mice, or BALB/c Jα18 mice, with R331 or 4T1 or CT26L5 tumors and treated with anti-CD1d mAb on days 0, 3, 7, 11, and 15 (for R331) or days 3, 7, and 11 (for 4T1 and CT26L5) (Figs. 4, 5, and 6). Additionally, some groups of mice were depleted of CD8+ T cells and/or NK cells or neutralized with anti-IFN-γ and/or IL-12. Interestingly, depletion of NK cells, but not CD8+ T cells, almost completely abrogated the antitumor effect of anti-CD1d mAb against R331 tumor growth (p = 0.6752 vs p = 0.0119) (Fig. 4A) and depletion of both NK cells and CD8+ T cells was completely inhibitory (p = 0.0119) (Fig. 4A). Similar R331 tumor growth suppression by anti-CD1d mAb was observed in Jα18−/− and SCID mice, indicating that T cells, including type I NKT cells, were not required (Fig. 5A). Neutralization of either IL-12 and/or IFN-γ completely inhibited the ability of anti-CD1d mAb to suppress R331 tumor growth compared with similar groups of mice treated with cIg and anti-CD1d (p < 0.05) (Fig. 6A). Overall, these data suggested that anti-CD1d mAb stimulated IL-12 and subsequent NK cell activation and IFN-γ secretion was largely responsible for activity against s.c. R331. By contrast, while either IL-12 or IFN-γ neutralization also completely inhibited the antitumor activity of anti-CD1d mAb against 4T1 and CT26L5 tumors (p < 0.05) (Fig. 6, B and C), NK cells, type I NKT cells, and CD4+ T cells were dispensable (p > 0.4) (Fig. 4, B and C, and Fig. 5B). Interestingly, depletion of CD8+ T cells resulted in a markedly reduced ability of anti-CD1d mAb to suppress 4T1 and CT26L5 tumor growth (p = 0.0119, 0.0079) (Fig. 4, B and C). Thus, in models where type II NKT cells are postulated to suppress effector tumor-specific CD4+ and CD8+ T cells, anti-CD1d mAb predominantly mediates its antitumor effect via CD8+ T cells in an IFN-γ- and IL-12-dependent manner. Despite the importance of host IL-12, recombinant mIL-12 given at day 0 or 8 after tumor inoculation did not provide tumor protection equivalent to anti-CD1d mAb, indicating that the role of IL-12 was context dependent, rather than systemic (Fig. 6D).
FIGURE 4.

Role of CD8+ T cells and NK cells in tumor suppression by anti-CD1d mAb. Groups of BALB/c WT mice (n = 5) were inoculated s.c. with the renal carcinoma cell line R331 (5 × 105) (A), the mammary carcinoma cell line 4T1 (2 × 105) (B), and the colon carcinoma cell line CT26L5 (1 × 105) (C). On days 0, 3, 7, 11, and 15 (A) or 3, 7, and 11 (B and C) after tumor inoculation, mice were treated i.p. with anti-CD1d mAbs (200 μg) or cIg. Additionally, some groups of mice were also injected i.p. on day 0 and then twice weekly with depleting anti-CD4, anti-CD8, and/or anti-ASGM1 Abs. Tumor sizes are represented as the mean ± SEM, representative of two independent experiments. Statistical differences in tumor sizes between mice injected with cIg and anti-CD1d mAbs and mAb-treated mice injected with anti-CD1d mAbs were determined by Mann-Whitney Rank Sum test (*, p < 0.05).
FIGURE 5.
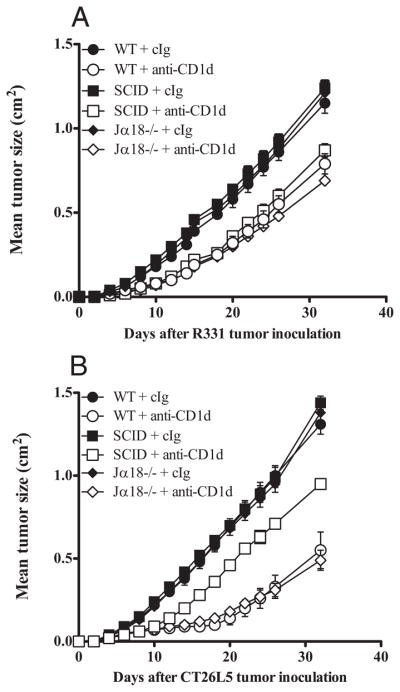
No role of type I NKT cells in tumor suppression by anti-CD1d mAb. Groups of BALB/c WT, SCID, or Jα18−/− mice (n = 5) were inoculated s.c. with the renal carcinoma cell line R331 (5 × 105) (A) or the colon carcinoma cell line CT26L5 (1 × 105) (B). On days 0, 3, 7, 11, and 15 (A) or 3, 7, and 11 (B) after tumor inoculation, mice were treated i.p. with anti-CD1d mAbs (200 μg) or cIg. Tumor sizes are represented as the mean ± SEM, representative of two independent experiments. Statistical differences in tumor sizes between mice injected with cIg and anti-CD1d mAbs and mAb-treated mice injected with anti-CD1d mAbs were determined by Mann-Whitney Rank Sum test (*, p < 0.05).
FIGURE 6.

Role of IFN-γ and IL-12 in tumor suppression by anti-CD1d mAb. Groups of BALB/c WT mice (n = 5) were inoculated s.c. with the renal carcinoma cell line R331 (5 × 105) (A), the mammary carcinoma cell line, 4T1 (2 × 105) (B), and the colon carcinoma cell line CT26L5 (1 × 105) (C). On days 0, 3, 7, 11, and 15 (A) or 3, 7, and 11 (B and C) after tumor inoculation, mice were treated i.p. with anti-CD1d mAbs (200 μg) or cIg. Additionally, some groups of mice were also injected i.p. on day 0 and then twice weekly with either neutralizing anti-IL-12 and/or anti-IFN-γ Abs or cIg. D, Groups of BALB/c WT mice (n = 5) were inoculated s.c. with the mammary carcinoma cell line 4T1 (2 × 105). Commencing on day 3 or 11 after tumor inoculation, mice received either PBS or 500 ng IL-12 for 5 or 4 successive days, respectively. Tumor sizes are represented as the mean ± SEM, representative of two independent experiments. Statistical differences in tumor sizes between mice injected with cIg and anti-CD1d mAbs and mAb-treated mice injected with anti-CD1d mAbs were determined by Mann-Whitney Rank Sum test (*, p < 0.05).
Discussion
This is the first report to document the potential antitumor activity and mechanism of action of anti-CD1d mAb. Anti-CD1d mAbs systemically activated CD1d+ APC, as measured by in vitro and in vivo production of IL-12 and IFN-γ. Tumor growth inhibition was found to be completely dependent on IFN-γ and IL-12 and variably dependent on CD8+T and NK cells, depending upon the tumor model examined. This ability confers obvious NK cell-mediated antitumor activity against some tumors, while the postulated ability of anti-CD1d mAb to block type II NKT cell activity, allows for additional potency mediated by downstream CD8+ T cells that are either released from dynamic/active type II NKT cell inhibition and/or stimulated by APC IL-12 release. The ability of anti-CD1d mAbs to coincidently activate CD1d+ APCs to release IL-12 and inhibit CD1d-restricted type II NKT cells makes CD1d an exciting new target for immunotherapy of cancer based on tumor immunoregulation.
Although the molecular and cellular characterization of mouse and human CD1d-restricted type II NKT cells remains elusive, they are described to have a variant TCR and activity in several disease models as suppressor populations (21, 22, 31–35). Further characterization of CD1d-restricted T cells should improve our understanding of how anti-CD1d mAb mediates its antitumor activity. The activity of anti-CD1d mAb may well allow for the rational targeting of cancers where type II NKT cells are thought to be involved. Because anti-CD1d mAb monotherapy has significant, but modest effects on established tumors, we will now seek to use these in combination with other immune-stimulating agents and mAb-based approaches to treat established cancer.
Acknowledgments
We thank Michelle Stirling and Nicole McLaughlin for technical assistance.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; α-GC, α-galactosylceramide; anti-ASGM1, anti-asialo GM1; WT, wild type; cIg, control Ig.
Disclosures
The authors have no financial conflict of interest.
We thank the National Health and Medical Research Council (NHMRC) of Australia for the support of a Program Grant, a Research Fellowship (to M.J.S.), and a Doherty Fellowship (to M.W.L.T.). We also thank the Cancer Council of Victoria for Project Grant Support. M.A.E. was supported by NIH DK066917.
References
- 1.Palucka AK, Ueno H, Fay JW, Banchereau J. Taming cancer by inducing immunity via dendritic cells. Immunol Rev. 2007;220:129–150. doi: 10.1111/j.1600-065X.2007.00575.x. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 3.Steinman RM. Linking innate to adaptive immunity through dendritic cells. Novartis Found Symp. 2006;279:101–109. [PubMed] [Google Scholar]
- 4.Fujii S, Shimizu K, Hemmi H, Steinman RM. Innate Vα14+ natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev. 2007;220:183–198. doi: 10.1111/j.1600-065X.2007.00561.x. [DOI] [PubMed] [Google Scholar]
- 5.Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol. 2005;5:112–124. doi: 10.1038/nri1549. [DOI] [PubMed] [Google Scholar]
- 6.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 7.Tong AW, Stone MJ. Prospects for CD40-directed experimental therapy of human cancer. Cancer Gene Ther. 2003;10:1–13. doi: 10.1038/sj.cgt.7700527. [DOI] [PubMed] [Google Scholar]
- 8.Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat Med. 1997;3:625–631. doi: 10.1038/nm0697-625. [DOI] [PubMed] [Google Scholar]
- 9.Swann JB, Coquet JM, Smyth MJ, Godfrey DI. CD1-restricted T cells and tumor immunity. Curr Top Microbiol Immunol. 2007;314:293–323. doi: 10.1007/978-3-540-69511-0_12. [DOI] [PubMed] [Google Scholar]
- 10.Vonderheide RH, Dutcher JP, Anderson JE, Eckhardt SG, Stephans KF, Razvillas B, Garl S, Butine MD, Perry VP, Armitage RJ, Ghalie R, Caron DA, Gribben JG. Phase I study of recombinant human CD40 ligand in cancer patients. J Clin Oncol. 2001;19:3280–3287. doi: 10.1200/JCO.2001.19.13.3280. [DOI] [PubMed] [Google Scholar]
- 11.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25:876–883. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 12.Chiodoni C, Iezzi M, Guiducci C, Sangaletti S, Alessandrini I, Ratti C, Tiboni F, Musiani P, Granger DN, Colombo MP. Triggering CD40 on endothelial cells contributes to tumor growth. J Exp Med. 2006;203:2441–2450. doi: 10.1084/jem.20060844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, Krasovsky J. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. 2003;197:1667–1676. doi: 10.1084/jem.20021650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong C, Park SH. Application of natural killer T cells in antitumor immunotherapy. Crit Rev Immunol. 2007;27:511–525. doi: 10.1615/critrevimmunol.v27.i6.20. [DOI] [PubMed] [Google Scholar]
- 15.Tahir SM, Cheng O, Shaulov A, Koezuka Y, Bubley GJ, Wilson SB, Balk SP, Exley MA. Loss of IFN-γ production by invariant NK T cells in advanced cancer. J Immunol. 2001;167:4046–4050. doi: 10.4049/jimmunol.167.7.4046. [DOI] [PubMed] [Google Scholar]
- 16.Dougan SK, Kaser A, Blumberg RS. CD1 expression on antigen-presenting cells. Curr Top Microbiol Immunol. 2007;314:113–141. doi: 10.1007/978-3-540-69511-0_5. [DOI] [PubMed] [Google Scholar]
- 17.Yue SC, Shaulov A, Wang R, Balk SP, Exley MA. CD1d ligation on human monocytes directly signals rapid NF-κB activation and production of bioactive IL-12. Proc Natl Acad Sci USA. 2005;102:11811–11816. doi: 10.1073/pnas.0503366102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, Godfrey DI, Ostrand-Rosenberg S, Smyth MJ, Berzofsky JA. A nonclassical non-Vα14Jα18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med. 2005;202:1627–1633. doi: 10.1084/jem.20051381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, Kawano T, Pelikan SB, Crowe NY, Godfrey DI. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishikawa H, Kato T, Tawara I, Takemitsu T, Saito K, Wang L, Ikarashi Y, Wakasugi H, Nakayama T, Taniguchi M, et al. Accelerated chemically induced tumor development mediated by CD4+ CD25+ regulatory T cells in wild-type hosts. Proc Natl Acad Sci USA. 2005;102:9253–9257. doi: 10.1073/pnas.0503852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Exley MA, Koziel MJ. To be or not to be NKT: natural killer T cells in the liver. Hepatology. 2004;40:1033–1040. doi: 10.1002/hep.20433. [DOI] [PubMed] [Google Scholar]
- 22.Terabe M, Berzofsky JA. NKT cells in immunoregulation of tumor immunity: a new immunoregulatory axis. Trends Immunol. 2007;28:491–496. doi: 10.1016/j.it.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Takeda K, Yamaguchi N, Akiba H, Kojima Y, Hayakawa Y, Tanner JE, Sayers TJ, Seki N, Okumura K, Yagita H, Smyth MJ. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med. 2004;199:437–448. doi: 10.1084/jem.20031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol. 2002;168:1356–1361. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- 25.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 26.Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13:459–463. doi: 10.1093/intimm/13.4.459. [DOI] [PubMed] [Google Scholar]
- 27.Trinchieri G. Proinflammatory and immunoregulatory functions of interleukin-12. Int Rev Immunol. 1998;16:365–396. doi: 10.3109/08830189809043002. [DOI] [PubMed] [Google Scholar]
- 28.Reis e Sousa C, Diebold SD, Edwards AD, Rogers N, Schulz O, Sporri R. Regulation of dendritic cell function by microbial stimuli. Pathol Biol. 2003;51:67–68. doi: 10.1016/s0369-8114(03)00099-3. [DOI] [PubMed] [Google Scholar]
- 29.Dzionek A, Inagaki Y, Okawa K, Nagafune J, Rock J, Sohma Y, Winkels G, Zysk M, Yamaguchi Y, Schmitz J. Plasmacytoid dendritic cells: from specific surface markers to specific cellular functions. Hum Immunol. 2002;63:1133–1148. doi: 10.1016/s0198-8859(02)00752-8. [DOI] [PubMed] [Google Scholar]
- 30.Xing Z. Current understanding of macrophage type 1 cytokine responses during intracellular infections. Histol Histopathol. 2000;15:199–205. doi: 10.14670/HH-15.199. [DOI] [PubMed] [Google Scholar]
- 31.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, et al. Transforming growth factor-β production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durante-Mangoni E, Wang R, Shaulov A, He Q, Nasser I, Afdhal N, Koziel MJ, Exley MA. Hepatic CD1d expression in hepatitis C virus infection and recognition by resident proinflammatory CD1d-reactive T cells. J Immunol. 2004;173:2159–2166. doi: 10.4049/jimmunol.173.3.2159. [DOI] [PubMed] [Google Scholar]
- 33.Terabe M, Khanna C, Bose S, Melchionda F, Mendoza A, Mackall CL, Helman LJ, Berzofsky JA. CD1d-restricted natural killer T cells can down-regulate tumor immunosurveillance independent of interleukin-4 receptor-signal transducer and activator of transcription 6 or transforming growth factor-β. Cancer Res. 2006;66:3869–3875. doi: 10.1158/0008-5472.CAN-05-3421. [DOI] [PubMed] [Google Scholar]
- 34.Renukaradhya GJ, Khan MA, Vieira M, Du W, Gervay-Hague J, Brutkiewicz RR. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood. 2008;111:5637–5645. doi: 10.1182/blood-2007-05-092866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JH, Choi EY, Chung DH. Donor bone marrow type II (non-Vα14Jα18 CD1d-restricted) NKT cells suppress graft-versus-host disease by producing IFN-γ and IL-4. J Immunol. 2007;179:6579–6587. doi: 10.4049/jimmunol.179.10.6579. [DOI] [PubMed] [Google Scholar]