

Abstract
Oxidative DNA damage incidental to normal respiratory metabolism poses a particular threat to genomes of highly metabolic-long lived cells. We show that postmitotic brain has capacity to repair oxidatively damaged DNA ends, which are targets of the long patch (LP) base excision repair (BER) sub-pathway. LP-BER relies, in part, on proteins associated with DNA replication, including proliferating cell nuclear antigen (PCNA) and is inherent to proliferating cells. Nonetheless, repair products are generated with brain extracts, albeit at slow rates, in the case of 5′-DNA ends modeled with tetrahydrofuran (THF). THF at this position is refractory to DNA polymerase β 5′-deoxyribose 5-phosphate (dRP) lyase activity and drives repair into the long patch BER sub-pathway. Comparison of repair of 5′-THF-blocked termini in the postmitotic rat brain and proliferative intestinal mucosa, revealed that in mucosa, resolution of damaged 5′-termini is accompanied by formation of larger repair products. In contrast, adducts targeted by the single nucleotide (SN) BER are proficiently repaired with both extracts. Our findings reveal mechanistic differences in BER processes selective for the brain versus proliferative tissues. The differences highlight the physiological relevance of the recently proposed ‘Hit and Run’ mechanism of alternating cleavage/synthesis steps, in the PCNA-independent LP-BER process.
Keywords: brain, base excision repair (BER), oxidative DNA damage, DNA strand breaks, PCNA, postmitotic tissue
INTRODUCTION
Because the brain is protected from external insults by the cranium and blood brain barrier, arguably the most critical threat to genomic stability in neurons comes from endogenous insults. Endogenous DNA damage, which is incidental to normal cellular metabolism, includes DNA lesions that are readily generated by spontaneous decay, free radicals-mediated oxidation and strand breakage and other DNA transactions including base methylation and alkylation (De Bont & van Larebeke 2004, Lindahl 1993). Robust DNA repair processes are generally associated with DNA replication, which is central in renewal and maintenance of genetic information (Barnes & Lindahl 2004). Although terminally differentiated cells lack the replication machinery, to sustain their longevity, neurons must be equipped to effectively repair oxidative DNA damage (Liu et al. 1996, Lin et al. 2000, Moore et al. 2002, Wilson & McNeill 2007). Since in mammalian cells repair of oxidative DNA damage is largely carried out by the base excision repair (BER) pathway, we investigated how BER may differ in the functionally distinct postmitotic versus proliferative tissues.
BER is a stepwise process initiated by DNA glycosylases, which release aberrant bases. The abasic sites (AP) are subsequently cleaved and resultant DNA ends further processed to become substrates for the gap filling DNA synthesis and ligation to complete the repair process (Dianov et al. 1998, Wilson & Kunkel 2000). Depending on the type of DNA-end processing required, mammalian BER partitions into two major sub-pathways defined by the length of repair patch synthesis as short (single-nucleotide) or long (several-nucleotides) BER (Dogliotti et al. 2001, Frosina et al. 1996). Importantly, in addition to DNA ends produced as BER intermediates, direct single strand breaks (SSBs), which readily form in mammalian genomes, also may require processing of their 3′ and 5′-DNA ends to generate the 3′-hydroxyl and 5′-phosphate for DNA synthesis and ligation, respectively (Wilson 2007). Interestingly, the highest level of such lesions was detected in the normal brain DNA when compared to other tissues (Nakamura & Swenberg 1999). Nonetheless, active repair of oxidatively damaged DNA ends, which require further processing prior to the gap filling DNA repair synthesis, has been documented in a setting of ischemic brain injury in the mouse (Huang et al. 2000). However, processing of blocked DNA ends is often the rate limiting step in a repair flow and in the case of a 5′-blocked terminus, may channel repair to the long-patch (LP) BER sub-pathway (Narciso et al. 2007). LP-BER relies in part, on proteins associated with DNA replication including, the 5′-flap endonuclease (FEN1), ligase l and proliferating cell nuclear antigen (PCNA) (Sung & Demple 2006).
Since paucity of PCNA is characteristic of postmitotic cells and in the brain PCNA expression is limited to the lining of the lateral ventricle and progenitor cells (Ino & Chiba 2000, Lee et al. 2004), we compared handling of 5′-THF blocked DNA ends by the terminally differentiated brain tissue and by rapidly proliferating intestinal mucosa. We first demonstrated that simple single strand breaks, which are generally processed by SN-BER, were proficiently repaired by nuclear extracts from both, brain and mucosal tissues. Moreover, we showed that expected repair products were generated with brain extracts, albeit at significantly reduced rates also in the case of THF blocked 5′-termini, which are refractory to the DNA pol β dRPase activity and require alternate processing prior to ligation. Unexpectedly, with intestinal mucosa, these THF-blocked 5′-termini generated also larger products, not observed with the brain tissue or in in vitro reactions reconstituted with purified proteins. Together, these findings reveal characteristics of BER selective for the brain versus proliferative tissues and underscore the physiological relevance of the PCNA-independent LP-BER sub-pathway. Thus, PCNA-independent LP-BER may represent a specialized sub-pathway of BER that ensures resolution of complex adducts in post mitotic tissues.
MATERIALS AND METHODS
BrdU incorporation and immunohistochemistry
Sprague-Dawley rats (male, 3 months) were handled according to the Institutional Animal Care and Utilization Committee. The thymidine analog BrdU (Sigma) was injected intra peritoneally at 50 mg/kg as we described (Varedi et al. 1999). Three injections were given at 12-hour intervals starting 48 hours prior to sacrifice. One group (n=3) was sacrificed at 2 hours after the first injection to capture pulse labeled proliferating intestinal crypt cells, and the second group at 48 hours after two additional injections at 12-hour intervals. Brain and duodenal tissues were fixed in freshly prepared PBS-buffered 4% paraformaldehyde, embedded in paraffin and sectioned. Sections (5 micron) were deparaffinized, rehydrated and stained following a 10-minute treatment with 2 N HCl and neutralization with 0.1 M borate buffer pH 8.5. Staining was with the anti BrdU (Roche) monoclonal antibody.
Tissue harvest and preparation of nuclear extracts
Intestinal mucosal scrapings: Approximately 4 cm long segments of the mid-duodenum were dissected. To enrich for mucosal crypt layer and remove loose villi, duodenal tubes were vigorously rinsed and agitated with ice cold PBS and then gently scraped in order to separate mucosa from the muscle layer. Mucosal scrapings were snap frozen in liquid nitrogen and stored at −80°C for preparation of nuclear extracts as described below. Brain: cerebral cortex was dissected, snap frozen and stored for protein extraction. Homogenization was in cold hypotonic buffer (10 mM Hepes, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 2 μg/ml Pepstatin, and Complete Protease Inhibitor Cocktail Tablet from Roche) followed by incubation on ice for 30 minutes. Homogenates were spun at 800 g for 3 minutes to obtain nuclear pellets. Pellets were gently washed 3 times, resuspended in a high-salt buffer (420 mM NaCl) and incubated at 4°C for 30 minutes with gentle shaking. Extracts were cleared by centrifugation (18,000 g, 10 min) made 15% with glycerol and stored at −80°C. Sprague-Dawley rats (male, 3 months) were used for both preparations.
Western blotting
Protein extracts were resolved by SDS-PAGE, electrotransferred to PVDF membranes and probed and reprobed once or twice; reprobing was carried out following gentle stripping with 5% H2O2 for 10 minutes at 25°C. The following antibodies were used: monoclonal anti-PCNA (sc-56) and polyclonal anti Fen-1 (sc-13051) were from Santa Cruz (Santa Cruz, CA), polymerase β from Alpha Diagnostic (San Antonio, TX), ligase lll monoclonal (#611876) from BD Transduction Laboratories and b-actin monoclonal was from Sigma (A5316).
Structure and sequence of oligonucleotide substrates
Substrate oligonucleotide sequences were designed previously (Karimi-Busheri et al. 1998) and used here as described, i.e., 1-nucleotide gapped DNA with 3′-hydroxyl (OH) or 3′-phosphate (P) upstream terminus, or modified to produce nicked or gapped substrates with a 5′-tetrahydrofuran (THF) downstream terminus. To generate the different modifications, substrates were assembled by annealing the 45-oligonucleotide lower strand with various complementary 5′-radiolabled upstream- and downstream-oligonucleotides. Oligonucleotides were synthesized by The Midland Certified Reagent Company (Midland, TX). Substrates sequences and design are provided in Table 1.
Table 1.
Structure and sequence of oligonucleotide substrates
| Strand break | Substrate design: sequences and modifications |
|---|---|
| nick [5′-phosphate(P)] |  |
| 1 nt-gap [5′-phosphate(P)] |  |
| 1 nt-gap [3′-P and 5′-P] | 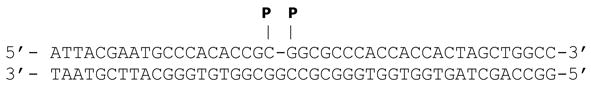 |
| nick [5′-THF] | 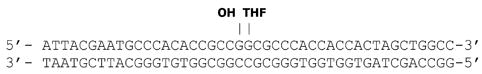 |
| 1-nt-gap [5′-THF] |  |
| 9-nt-gap |  |
nt-nucleotide, THF-tetrahydrofuran, P-phosphorylated terminus
Repair assays
Gap filling synthesis and ligation reactions were assembled with purified recombinant human proteins and/or with nuclear extracts from mucosal and brain tissues. Double-stranded oligonucleotide substrates are listed in Table 1. Substrates were prepared by annealing the 5′-end labeled upstream oligonucleotide, labeled with T4 polynucleotide kinase in the presence of [γ-32P] ATP, purified through a G25 spin column and annealed with the different combinations of complementary oligomers as indicated. Repair reactions were assembled in 20 μl with 10 μg nuclear extracts and/or purified recombinant proteins, 80 fmol end-labeled substrate, 10 mM ATP, 10 μM dNTPs and 20 ng carrier DNA (λ) with reaction buffer (50 mM Tris-HCl pH 8.0, 10 mM KCl, 10 mM MgCl2, 1 mM DTT, 1% glycerol). Assays assembled with purified recombinant proteins were adjusted with respect to reaction time and protein concentration to ascertain linear range: the human recombinant DNA pol β was used at 20 nM and FEN1 at 10 nM (Trevigen, Gaithesburg, MD), ligase l at 4 nM, ligase lll at 20 nM, and PCNA at 100 nM were from Enzymax (Lexington, KY). The relatively high concentration of PCNA, which was required to observe stimulatory effects, is consistent with the previously reported PCNA concentrations used with linear substrates (Maga et al. 2007). Incubations were at 37°C and 5 μl aliquots were removed at indicated times; reactions were terminated with loading buffer (96% formamide, 20 mM EDTA pH 8, 0.025% bromophenol blue, 0.025% xylene cyanol) and heated at 98°C for 10 minutes. Reaction aliquots were resolved in 18% polyacrylamide-7 M urea gels in Tris-Borate buffer pH 8.3 at 14 mA for 2.5 hours. Products were visualized by autoradiography and quantified on Phosphorimager (Molecular Dynamics) with ImageQuant software. Ligation products generated overtime are presented as mean±SEM of at least 3 independent reactions for each nuclear extract (n=3). Phosphorimager values representing the percent (product/[substrate+product]) of ligated substrate within linear range of each reaction were converted to product amounts and expressed as fmol/μg protein extract/time. For assays with pol β neutralizing antibody, extracts or purified pol β were preincubated 30 minutes on ice followed by 10 minutes at RT with pol β neutralizing antibody or with mixture of rabbit serum (Pierce) prior to assembly of repair assays. Pol β neutralizing polyclonal rabbit antibody was a gift from Dr. S.H. Wilson (Braithwaite et al. 2005).
RESULTS
Cell proliferation status in brain and intestinal mucosa
To identify differences between the base excision repair process in post mitotic and proliferative tissues, we chose to work with the brain and intestinal mucosa. The striking difference in cell proliferation status between brain and mucosa was visualized by in situ detection of cells incorporating the thymidine analog, 5-bromo-2′-deoxyuridine (BrdU). Rats were injected BrdU and incorporation was visualized immunohistochemically, either 2 hours after a single injection or following three consecutive injections at 12-hour intervals over a 48-hour period. As expected, immunostaining for BrdU after the 48-hour period showed only a few BrdU positive nuclei in the dentate gyrus hippocampal region, which is a major site of neurogenesis in the adult brain (Fig 1C). In contrast, high density of BrdU positive cells was seen in the crypt zone of rapidly proliferating mucosal cells. These cells subsequently migrate upwards to make up and replenish intestinal villi (Potten 1995, Varedi et al. 1999). Accordingly, while after a 2-hour pulse BrdU label, incorporation was seen in the proliferative zone of the crypt (Fig 1A), labeling over a 48-hour period resulted in widespread BrdU staining, reflective of the rapid cellular turnover and replacement in intestinal mucosa (Fig 1B).
Figure 1. Cell proliferation status in the brain and intestinal mucosa – in vivo BrdU incorporation.

Photomicrographs of immunohistochemical detection of BrdU incorporation into proliferating cell nuclei in the dentate gyrus hippocampal region and duodenal cross sections. A) Two hours after BrdU injection, positive nuclei are limited to the proliferative zone of duodenal crypts (brown). B) After a 48-hour incorporation period, BrdU positive cells accumulate and push upwards to replenish mucosal villi, reflective of the rapid turnover of mucosal cells. C) In contrast in the brain, BrdU administration over the 48-period resulted in limited incorporation; BrdU positive cells are indicated by arrows (dentate gyrus is a major site of neurogenesis). Insets show enlarged areas with BrdU positive cells (brown).
Efficient repair of strand breaks targeted by the single nucleotide (SN) BER sub-pathway in brain and mucosal tissues
Repair of nicked, gapped as well as 3′-phosphate gapped DNA substrates was efficiently carried out by nuclear extracts from either the brain or intestinal mucosa. Generation of repair products was reproduced in reactions assembled with purified BER proteins, DNA pol β, and ligase l or lll, which catalyze the gap filling DNA synthesis and ligation, respectively (Fig 2A). By substrate design (Table 1), in all cases the expected synthesis/ligation product was 45-nucleotides long. The yield of the 45-mer increased in a time-dependent manner, albeit at different rates, with both brain and mucosal extracts (Fig 2B, C). With mucosal extracts, a significant product amount was seen already by 8 minutes, with all substrate consumed by 32 minutes (lane 24). With brain extracts, the 45-mer was generated at a slower rate (Fig 2B, C, note that different time points are shown for brain and mucosa). Similar repair rates were measured for the different substrates, i.e., nicked and gapped DNA as well as gapped DNA with a 3′-phosphorylated terminus that requires processing to generate 3′-OH prior to the gap filling synthesis (Fig 2B, C). Processing of a 3′-phosphate can be catalyzed by polynucleotide kinase (PNK), abasic endonuclease (APE) or aprataxin, which are ubiquitous in mammalian tissues, including the brain (Ahel et al. 2006, Englander & Ma 2006, Wilson et al. 1996). Consistently, processing of the 3′-phosphate terminus did not significantly slow generation of the 45-mer (Fig 2B, C). Radioactivity of substrates and products was quantified by phosphorimager and used to calculate product yields; averaged yields were graphed as function of time (Fig 2C). The individual activities shown in Table 2 were calculated from the means of product yields in linear range of each reaction and expressed as mean± SEM fmol/microgram extract protein/min. Overall, processing of strand breaks was ~2.5-fold slower with the brain compared to mucosal extracts (Table 2). Taken together, the data show that strand breaks, which are most likely handled by the SN BER sub-pathway, are efficiently resolved not only in proliferative tissues but also in the postmitotic brain.
Figure 2. Single strand breaks-processing in brain and mucosal nuclear extracts.

Reactions were assembled with radioactively labeled substrates (Table 1) and purified proteins or nuclear extracts as indicated. Products were resolved in denaturing polyacrylamide gels and visualized by autoradiography; representative autoradiograms are shown. Time points are indicated, and in each case, a substrate/BSA lane was included; mixtures of labeled oligonucleotides were resolved in parallel to provide length markers (M). A) Reactions assembled with purified proteins yielded a 45-mer product. B) To remain in linear range, different time points were used with mucosal (4, 8, 16 min) and brain (8, 16, 32 min) extracts. With mucosal extracts, a significant amount of product was seen already by 8 minutes, with all substrate consumed by 32 minutes (lane 24). C) Time courses: product yields were calculated from phosphorimager values using the ImageQuant software. Values for 3–4 independent assays for each substrate in the linear range of reaction were used to obtain means and SEM and plotted as a function of time. Yields for brain extracts are coded in red.
Table 2.
Repair activities
| Substrate | fmol product/μg protein/min | ratio | |
|---|---|---|---|
| Brain | Mucosa | Mucosa/Brain | |
| nick | 0.1354± 0.0041 | 0.3521 ± 0.0141 | 2.6 |
| gap | 0.1484 ± 0.0089 | 0.3406 ± 0.0256 | 2.3 |
| gap [3′-P] | 0.1287 ± 0.0188 | 0.3113 ± 0.0180 | 2.4 |
| nick [5′-THF] | 0.0017 ± 0.0001 | 0.0067 ± 0.0008 | 3.9 |
| gap [5′-THF] | 0.0019 ± 0.0001 | 0.0108 ± 0.0005 | 5.7 |
| Activity ratios | Brain | Mucosa | |
| nick/nick [5-THF] | 80 | 63 | |
| gap/gap [5′-THF] | 78 | 32 | |
Differential capacity for processing strand breaks with 5′-THF termini in brain and proliferative mucosa
Next, we examined brain and mucosal capacity to repair strand breaks, which require processing of blocked 5′-termini prior to ligation. We used THF to model damaged 5′-termini, which are refractory to the DNA polymerase β dRP lyase activity and, therefore, considered targets for the long-patch BER, i.e., gap filling displacement synthesis facilitated by FEN1 and other proteins typically associated with DNA replication. Nicked and gapped substrates with a 5′-THF residue (Table 1), were used to compare the capacity for repair by brain and mucosal nuclear extracts. As expected, THF-abutted nicked and gapped substrates were repaired at significantly slower rates than simple strand breaks with both, brain and mucosal extracts (Fig 3, Table 2). With brain extracts, formation of the 45-mer with nicked and gapped 5′-THF substrates increased linearly with time for at least 4 hours (lanes 1–3 and 8–10, bullet). In contrast, with mucosa, the 45-mer was detectable already after 8 minutes (bullet) and then gradually replaced (Fig 3A, lanes 4–6 and 11–13) by a larger product observed at 15 and 30 minutes (lanes 6 and 13, asterisk). Product yields for brain and mucosal extracts were calculated from radioactivity captured in products and plotted as a function of time (Fig 3B). Repair yields in linear range of each reaction with either the brain or mucosal extracts were expressed per microgram protein/min and the means ± SEM are shown in Table 2. Ratios between yields generated by brain and mucosal extracts reveal that differences in repair rates between mucosa and brain were significantly greater for LP (5.7-fold) than for the SN (2.5-fold) base excision repair process.
Figure 3. Processing of nicked and gapped DNA with 5′-THF blocked termini in brain and mucosal nuclear extracts.

Reactions were assembled with indicated substrates and extracts. A) Reaction products were resolved in denaturing polyacrylamide gels and visualized by autoradiography. Representative autoradiograms are shown and major products are indicated. To visualize products, different sets of time points were used with mucosa (8, 15 and 30 min) and brain (60, 120 and 240 min), and short and long exposures are shown for substrates and products, respectively. With brain extracts a single 45-mer product (bullet) was generated at a slow rate, linear up to at least 240 minutes. With mucosal extracts, trace amount of the 45-mer was seen already by 8 minutes, with an additional, larger product (asterisk) observed at 15 minutes with an increase by 30 minutes (lanes 5–6 and 12–13) for both nicked and gapped 5′-THF substrates. B) Time courses: for each substrate yields were calculated by plotting phosphorimager values. Radioactivity values for 4–5 independent assays for each time point were used to obtain means and SEM and plotted as a function of time.
Processing of gapped DNA with 5′-THF terminus in assays reconstituted with purified proteins
To confirm that the minimal set of repair proteins required to repair gapped DNA with a blocked/damaged 5′-terminus is compatible with our experimental setting, assays were assembled with the 1-nt-gap-[5′-THF] duplex oligonucleotide substrate (Table 1) and different combinations of purified repair proteins. In the case of DNA pol β alone catalyzed reaction, the major product corresponded to incorporation of one nucleotide [+1] with a very minor [+2] product (Fig 4). This outcome is consistent with the recently proposed ‘Hit and Run’ mechanism (Liu et al. 2005) where at a 5′-THF margin, DNA pol β alone does not catalyze a significant displacement synthesis to generate an authentic flap. Addition of PCNA had no effect on outcome; however, addition of FEN1 facilitated robust pol β-mediated synthesis with a major [+3] product, corresponding to removal of two nucleotides, with additional weak products extending up to approximately [+10] nucleotides. The combination of pol β and either ligase l or ligase lll yielded very low levels of the 45-mer. Notably, none of the combinations of purified proteins, led to generation of the larger ligation product seen with mucosal extracts. FEN1 markedly augmented generation of the 45-mer when added to either the pol β/ligase l or pol β/ligase lll catalyzed reaction. In the absence of FEN1, addition of PCNA to the pol β/ligase l reaction had no significant effect. This cannot be simply attributed to the previously noted weak retention of PCNA on linear substrates (Biade et al. 1998), because we observe a stimulatory effect of PCNA on DNA pol β catalyzed 9-nt-gap filing synthesis (Fig 4B). This indicates that PCNA can exert an effect also with linear substrates. Notably, stimulation by PCNA did not increase any further on biotinylated 9-nt-gap substrates, blocked at both ends with streptavidin beads (not shown). PCNA augmented pol β synthesis across an apparent pause site in the center of a 9-nt-gap and facilitated some displacement synthesis beyond the gap. Processive short gap filling synthesis by pol β documented previously (Singhal & Wilson 1993) might be consistent with the ~[+5] pause site within the 9-nt gap observed here (Fig 4B). The stimulation observed here, might be also consistent with the reported direct interaction between PCNA and pol β (Kedar et al. 2002) and with effects of PCNA on adduct bypass synthesis on linear substrates (Maga et al. 2007).
Figure 4. Processing of gapped DNA with a 5′-THF blocked terminus in assays reconstituted with purified proteins.

A) The 5′-labeled gap-[5′-THF] duplex oligonucleotide substrate was assembled with indicated combinations of purified proteins. The different synthesis and ligation products are demarcated. FEN1 was required for efficient pol β-catalyzed longer than [+1] synthesis and efficient generation of the 45-mer in the presence of ligase l or lll. Labeled oligonucleotides provide size references (M). B) Pol β catalyzed gap filling synthesis on a 9-nt gap substrate (Table 1) was used to demonstrate an effect of PCNA when used on linear duplex oligonucleotide substrates.
Distinct features of repair of gapped DNA with 5′-THF terminus in brain versus mucosal extracts revealed by supplementation of purified repair proteins
Next, we examined effects of supplementation of the different BER proteins on repair of the 5′-THF gapped DNA with brain and mucosal extracts. THF-residue serves to drive repair into the long-patch pathway. As shown earlier (Fig 3, Table 2), gapped 5′-THF substrates were repaired at significantly slower rates with both, brain and mucosal extracts. With brain extracts, the expected 45-mer was detected after 45 but not 15-minute incubation (Fig 5, bottom, lane 2, bullet). In contrast, with mucosa, the 45-mer was detectable already at 8 minutes (not shown), persisted at 15 (top, lane 8, bullet) and was replaced by a larger product, which then persisted as a sole product at the 45-minute incubation time (Fig 5, lane 8, asterisk). It is of note that assays assembled with mucosal extracts, which have relatively high nucleolytic activities typical of proliferating cells, show some degradation of the 45-mer at the longer incubation times and therefore, unlike with brain extracts, cannot be carried out over extended periods of time (lanes 11–13).
Figure 5. Processing of gapped DNA with a 5′-THF terminus in brain and mucosal extracts supplemented with purified repair proteins.

Gapped 5′-THF substrate was incubated with extracts alone or with extracts supplemented with indicated purified repair proteins. Reaction aliquots were taken at different time points and resolved in denaturing polyacrylamide gels. Products generated after 15- and 45-minute incubations are shown. With brain extracts, the 45-mer was observed after 45 but not 15 minutes (lane 2, bullet). In contrast, with mucosa, the 45-mer was transiently seen at 15 but not 45 minutes (lane 8, bullet). With mucosa, a larger product appeared at 15 minutes and then persisted as a sole product at 45 minutes (lane 8, asterisk). With mucosa, nucleolytic activities cause some degradation of the 45-mer at longer incubation times (lane 11). Purified proteins were added as indicated; in the brain addition of pol β, ligases, PCNA or FEN1 stimulated generation of the 45-mer in time dependent manner (lanes 3–7). In the case of FEN1, generation of a [+2] and longer extension products was strongly stimulated (lane 7). In contrast, supplementation of pol-β or FEN1 (lanes 8–9) had no significant effect on levels of products generated by mucosal extracts.
To discern whether the different repair proteins affect rates of repair of the THF abutted gap in brain and mucosal extracts, assays were supplemented with proteins involved in the different repair processes as indicated (Fig 5, lanes 3–7 and 9–13).
We found that in the brain addition of pol β, ligases, PCNA or FEN1 stimulated generation of the 45-mer in a time-dependent manner (top to bottom, lanes 3–7). With the addition of FEN1, generation of a [+2] and larger extension products was strongly enhanced, reflective of a stepwise nucleotide removal and gap filling DNA synthesis (lane 7). In contrast with mucosa, supplementation of pol β or FEN1 (lanes 9–10, 12–13) had no significant effect, suggesting that an adequate complement of BER proteins may already exist in proliferative tissues. Ligase l had a stimulatory effect with both, brain (lane 5) and mucosa (lane 11).
Involvement of DNA pol β in the SN and LP-BER in brain and mucosal tissues
The central role of pol β in BER in postmitotic as well as proliferative tissues was further demonstrated using the pol β neutralizing antibody (Braithwaite et al. 2005). Notably, while similar levels of pol β were detected by Western blotting in brain and mucosal nuclear extracts, PCNA was detected only in mucosa. In contrast, ligase lll and FEN1 proteins were detected in both tissues, with significantly higher levels of FEN1 in mucosa compared to the brain (n=4) (Fig 6C). In the case of gapped DNA incubated with purified DNA pol β (Fig 6A), the antibody blocked formation of the [+1] synthesis product (lane 3 versus 2) and subsequently the formation of the 45-mer. Likewise, synthesis of the [+1] product was blocked in both, the brain and mucosal extracts consistent with pol β-catalyzed gap filling synthesis in both tissues (Fig 6A, lanes 4–7). Also in the case of gapped DNA with a 5′-THF terminus (Fig 6B), the pol β neutralizing antibody blocked gap filling synthesis and formation of the 45-mer in both, brain and mucosal extracts, confirming the central role of DNA pol β in both BER sub-pathways. In addition, neutralizing antibody blocked also formation of the larger repair product (asterisk), which was generated only with mucosal extracts (Fig 6, lane 17 vs 18). This indicates that with mucosa, formation of larger products also occurs downstream of pol β-catalyzed DNA synthesis. Interestingly, however, the larger product was not generated when only the incoming nucleotide (as opposed to all four dNTPs) was supplied in the reaction (not shown).
Figure 6. Repair of gapped and gapped 5′-THF substrates in brain and mucosa is abolished by pol β neutralizing antibody.

Reactions with gapped (A, lanes 1–7) and gapped-5′-THF (B, lanes 8–18) substrates were assembled with brain and mucosal extracts or with purified pol β. Duplicate reactions were assembled w/o preincubation with the pol β neutralizing antibody. In all cases the antibody blocked gap filling synthesis. Inhibition was proportional to antibody amount: lane 10 shows a significant reduction of the [+1] product with 1 μl, while lane 12 shows a lesser reduction using 0.5 μl antiserum. With nuclear extracts a similar amount of antibody was sufficient to block gap filling synthesis and subsequent ligation (lanes 14, 16, 18). In the case of mucosa, both products (lane 17) were seen following 8- and 15-minute reactions. B) For the gap-5′-THF assays (lanes 8–18) substrates (bottom) and products (top) are shown at different exposures to clearly visualize elimination of the [+1] synthesis product by pol β neutralizing antibody. C) Western blotting analysis shows similar levels of pol β protein in nuclear extracts from the brain and mucosa, while PCNA is detected only in mucosal extracts. Ligase lll and FEN1 are detected in both extracts; higher levels of FEN1 were observed in mucosa compared to the brain. Reprobing for actin served as a loading control.
Immunoreactivity levels are depicted in bar graphs representing densitometric measurements of signals for brain relative to mucosal extracts and expressed as means ± SEM (n=4). ND-not detected.
DISCUSSION
Since generally robust DNA repair activities are associated with DNA replication in proliferating cells, it was of much interest to compare the capacity for repair of different types of DNA damage in the largely postmitotic brain versus rapidly proliferating intestinal mucosa. Under normal physiological conditions, intestinal mucosa is among the fastest, if not, the fastest proliferating tissue in mammalian body, with a 13-hour cell cycle and a 7-hour S phase in the proliferative crypt cell zone (Potten 1995, Radtke & Clevers 2005), while cell proliferation in the adult brain is quite limited (Zhao et al. 2008). This difference was readily demonstrated in our setting via abundant BrdU incorporation into mucosal crypt layer versus detection of only a few BrdU incorporating cells in the brain. To enrich for the proliferative mucosal layer, we followed a harvesting protocol designed to enrich for mucosal crypts. Enrichment was confirmed by Western detection of PCNA in mucosal extracts. We then demonstrated that simple SSBs, including nicked and gapped DNA that are typically resolved by the SN-BER sub-pathway are rapidly repaired, with repair rates only ~2.5-fold faster in mucosa compared to the brain. With both tissues, repair of gapped DNA with 3′-phosphate was slightly although not significantly slower than that of nicked or gapped DNA substrates, which do not require 3′-terminus processing prior to gap filling synthesis or nick ligation. This is consistent with the robust expression in mammalian tissues, including the brain (Ahel et al. 2006, Englander & Ma 2006, Wilson et al. 1996) of PNK, APE and aprataxin, which catalyze the 3′-phosphate removal. Notably, with gapped DNA substrates, gap filling synthesis and subsequent ligation were completely abolished with the pol β neutralizing antibody (Braithwaite et al. 2005) indicating that not only in the brain but also in proliferative mucosa, the process of single nucleotide gap filling synthesis is catalyzed primarily by DNA pol β. While the utility of the pol β-blocking antibody has been previously demonstrated in reconstituted assays and in vitro in cultured cells, here it is used in a physiological context of different types of tissue.
Repair of complex strand breaks, which require processing of damaged 5′-ends and typically are channeled for repair by the LP-BER sub-pathway, was markedly slower with either the brain or mucosal extracts. This is expected, as the DNA pol β dRP lyase-catalyzed removal of a BER intermediate, the 5′-deoxyribose phosphate (dRP) group, which may result from APE cleavage of frequent abasic sites generated by monofunctional glycosylases, is clearly rate limiting and significantly slows the BER process (Chagovetz et al. 1997, Prasad et al. 2005, Srivastava et al. 1998, Wong & Demple 2004). Here, repair of breaks with THF blocked 5′-termini was substantially slower in both extracts, with rates reduced by ~30–80-fold compared to the corresponding simple strand breaks, which do not require end-processing (Table 2). With the brain, repair rates were reduced about 80-fold for both 5′-THF nicked and gapped DNA compared to the corresponding non-THF substrates. Likewise, rates were reduced in mucosa, but while for the nick [5′-THF] the ratio was reduced ~60-fold, for the gap [5′-THF], it was reduced only about 30-fold. This might be consistent with a more efficient recruitment of pol β to the site of damage in the context of gapped DNA (Klungland & Lindahl 1997), a process which might be augmented in mucosa via physical interactions with replication proteins that are present in proliferating cells (Kedar et al. 2002). Here, effects of PCNA are readily detectable in assays assembled with purified proteins and in assays with extracts supplemented with PCNA. Since PCNA is abundant in intestinal mucosa (Smartt et al. 2007) it is likely to participate in the repair process. Surprisingly however, while only expected length of repair products was seen with brain extracts, in the case of mucosa, larger products were also observed. Importantly, also the larger products were abolished with the pol β neutralizing antibody, indicating that they were also dependent on pol β-catalyzed gap filling synthesis. Furthermore, when only the incoming nucleotide was included in repair assays, the larger repair product was eliminated (not shown). The apparent length of the extended product was 48 nucleotides, i.e., about three nucleotides longer than the 45-mer expected based on substrate design. Although speculative, since generation of both products appears to be catalyzed by pol β, it is plausible that in the milieu of abundant replication proteins in intestinal mucosa, extension products may form looped intermediates, which might be ligated yielding the larger products. A sequence-dependent expansion of a DNA template was recently documented in the context of oxidative DNA damage repair initiated by the removal of an oxidized guanine in mouse model of Huntington disease (Kovtun et al. 2007), whereas in an in vitro setting, frameshift mutations were induced during BER by overexpression of DNA pol β (Chan et al. 2007). The fact that our substrate picks up a difference between brain and mucosal activities, underscores inherent dissimilarities between proliferative and non-proliferative tissues.
Since an important feature of postmitotic tissues is paucity of PCNA, we view the emerging evidence for PCNA independent, pol β/FEN1 catalyzed LP-BER via the ‘Hit and Run’ mechanism (Liu et al. 2005) with much interest. Reports show that PCNA expression in the mature mammalian brain is marginal and confined to progenitor cells, lining of the lateral ventricle and olfactory bulb, where cell proliferation continues also in the mature brain (Ino & Chiba 2000, Lee et al. 2004, Zhao et al. 2008). FEN1 expression (Otto et al. 2001) as well as robust expression of pol β in the brain (Waser et al. 1979, Englander 2008) have been reported. It is therefore, plausible that postmitotic cells, including neurons, rely on PCNA-independent BER for repair of oxidative damage, such as SSBs with damaged termini, which require processing prior to DNA repair synthesis and ligation. Thus, the notion that these complex lesions might be repaired in a PCNA-independent manner is of major physiological relevance and central to our understanding of mechanisms for repair of oxidatively damaged DNA in postmitotic cells. Our data show that damaged DNA termini can be accurately, albeit slowly repaired in the brain.
In view of these new findings, it is plausible that under normal physiological conditions, slow DNA repair transactions in the brain are sufficient for handling endogenous oxidative damage and therefore, consistent with neuronal longevity. However, under compromising conditions, brain repair systems may lack adequate capacity to counteract damaging cascades and preserve genomic integrity. In this case, the inherently slower repair rates may lead to accumulation of un-repaired or partially repaired intermediates and ultimately contribute to neuronal dysfunction. Thus, our findings provide new insights into tissue specific limitations of systems for maintenance of genomic integrity, which may underlie, in part, brain vulnerability to insults associated with disrupted oxygenation and excessive oxidative stress.
Acknowledgments
We are grateful to Dr. William Beard for critical suggestions and comments on the manuscript. We thank Eileen Figueroa and Steve Schuenke for editorial assistance. This work was supported by National Institutes of Health Grants NS039449 and ES014613 and Shriners Research Grant SHG8670 to E.W.E.
References
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. DNA polymerase lambda mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts. J Biol Chem. 2005;280:18469–18475. doi: 10.1074/jbc.M411864200. [DOI] [PubMed] [Google Scholar]
- Chagovetz AM, Sweasy JB, Preston BD. Increased activity and fidelity of DNA polymerase beta on single-nucleotide gapped DNA. J Biol Chem. 1997;272:27501–27504. doi: 10.1074/jbc.272.44.27501. [DOI] [PubMed] [Google Scholar]
- Chan K, Houlbrook S, Zhang QM, Harrison M, Hickson ID, Dianov GL. Overexpression of DNA polymerase beta results in an increased rate of frameshift mutations during base excision repair. Mutagenesis. 2007;22:183–188. doi: 10.1093/mutage/gel070. [DOI] [PubMed] [Google Scholar]
- De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Piotrowski J, Bohr VA. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J Biol Chem. 1998;273:33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- Dogliotti E, Fortini P, Pascucci B, Parlanti E. The mechanism of switching among multiple BER pathways. Prog Nucleic Acid Res Mol Biol. 2001;68:3–27. doi: 10.1016/s0079-6603(01)68086-3. [DOI] [PubMed] [Google Scholar]
- Englander EW. Brain capacity for repair of oxidatively damaged DNA and preservation of neuronal function. Mech Ageing Dev. 2008 Feb 14; doi: 10.1016/j.mad.2008.02.001. [epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander EW, Ma H. Differential modulation of base excision repair activities during brain ontogeny: Implications for repair of transcribed DNA. Mech Ageing Dev. 2006;127:64–69. doi: 10.1016/j.mad.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- Huang D, Shenoy A, Cui J, Huang W, Liu PK. In situ detection of AP sites and DNA strand breaks bearing 3′-phosphate termini in ischemic mouse brain. Faseb J. 2000;14:407–417. doi: 10.1096/fasebj.14.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino H, Chiba T. Expression of proliferating cell nuclear antigen (PCNA) in the adult and developing mouse nervous system. Brain Res Mol Brain Res. 2000;78:163–174. doi: 10.1016/s0169-328x(00)00092-9. [DOI] [PubMed] [Google Scholar]
- Karimi-Busheri F, Lee J, Tomkinson AE, Weinfeld M. Repair of DNA strand gaps and nicks containing 3′-phosphate and 5′-hydroxyl termini by purified mammalian enzymes. Nucleic Acids Res. 1998;26:4395–4400. doi: 10.1093/nar/26.19.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase beta and proliferating cell nuclear antigen. J Biol Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HM, Hu Z, Ma H, Greeley GH, Jr, Wang C, Englander EW. Developmental changes in expression and subcellular localization of the DNA repair glycosylase, MYH, in the rat brain. J Neurochem. 2004;88:394–400. doi: 10.1046/j.1471-4159.2003.02164.x. [DOI] [PubMed] [Google Scholar]
- Lin LH, Cao S, Yu L, Cui J, Hamilton WJ, Liu PK. Up-regulation of base excision repair activity for 8-hydroxy-2′-deoxyguanosine in the mouse brain after forebrain ischemia-reperfusion. J Neurochem. 2000;74:1098–1105. doi: 10.1046/j.1471-4159.2000.741098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair, and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- Moore N, Okocha F, Cui JK, Liu PK. Homogeneous repair of nuclear genes after experimental stroke. J Neurochem. 2002;80:111–118. doi: 10.1046/j.0022-3042.2001.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59:2522–2526. [PubMed] [Google Scholar]
- Narciso L, Fortini P, Pajalunga D, et al. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc Natl Acad Sci U S A. 2007;104:17010–17015. doi: 10.1073/pnas.0701743104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto CJ, Almqvist E, Hayden MR, Andrew SE. The “flap” endonuclease gene FEN1 is excluded as a candidate gene implicated in the CAG repeat expansion underlying Huntington disease. Clin Genet. 2001;59:122–127. doi: 10.1034/j.1399-0004.2001.590210.x. [DOI] [PubMed] [Google Scholar]
- Potten CS. Structure, function and proliferative organisation of mammalian gut. In: Potten CS, Hendry JH, editors. Radiation and Gut. Elsevier; New York: 1995. pp. 1–30. [Google Scholar]
- Prasad R, Batra VK, Yang XP, Krahn JM, Pedersen LC, Beard WA, Wilson SH. Structural insight into the DNA polymerase beta deoxyribose phosphate lyase mechanism. DNA Repair (Amst) 2005;4:1347–1357. doi: 10.1016/j.dnarep.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science. 2005;307:1904–1909. doi: 10.1126/science.1104815. [DOI] [PubMed] [Google Scholar]
- Singhal RK, Wilson SH. Short gap-filling synthesis by DNA polymerase beta is processive. J Biol Chem. 1993;268:15906–15911. [PubMed] [Google Scholar]
- Smartt HJ, Guilmeau S, Nasser SV, et al. p27kip1 Regulates cdk2 activity in the proliferating zone of the mouse intestinal epithelium: potential role in neoplasia. Gastroenterology. 2007;133:232–243. doi: 10.1053/j.gastro.2007.04.043. [DOI] [PubMed] [Google Scholar]
- Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- Sung JS, Demple B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. FEBS J. 2006;273:1620–1629. doi: 10.1111/j.1742-4658.2006.05192.x. [DOI] [PubMed] [Google Scholar]
- Varedi M, Greeley GH, Jr, Herndon DN, Englander EW. A thermal injury-induced circulating factor(s) compromises intestinal cell morphology, proliferation, and migration. Am J Physiol. 1999;277:G175–182. doi: 10.1152/ajpgi.1999.277.1.G175. [DOI] [PubMed] [Google Scholar]
- Waser J, Hubscher U, Kuenzle CC, Spadari S. DNA polymerase beta from brain neurons is a repair enzyme. Eur J Biochem. 1979;97:361–368. doi: 10.1111/j.1432-1033.1979.tb13122.x. [DOI] [PubMed] [Google Scholar]
- Wilson DM., 3rd Processing of nonconventional DNA strand break ends. Environ Mol Mutagen. 2007;48:772–782. doi: 10.1002/em.20346. [DOI] [PubMed] [Google Scholar]
- Wilson DM, 3rd, McNeill DR. Base excision repair and the central nervous system. Neuroscience. 2007;145:1187–1200. doi: 10.1016/j.neuroscience.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- Wilson TM, Rivkees SA, Deutsch WA, Kelley MR. Differential expression of the apurinic/apyrimidinic endonuclease (APE/ref-1) multifunctional DNA base excision repair gene during fetal development and in adult rat brain and testis. Mutat Res. 1996;362:237–248. doi: 10.1016/0921-8777(95)00053-4. [DOI] [PubMed] [Google Scholar]
- Wong D, Demple B. Modulation of the 5′-deoxyribose-5-phosphate lyase and DNA synthesis activities of mammalian DNA polymerase beta by apurinic/apyrimidinic endonuclease 1. J Biol Chem. 2004;279:25268–25275. doi: 10.1074/jbc.M400804200. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]