

Abstract
Background & Aims
There is little data available from genome-wide association studies (GWAS) of liver histology in patients with non-alcoholic fatty liver disease (NAFLD). We conducted a pilot GWAS in patients with NAFLD, characterized by histology, who were enrolled in the NASH CRN Database Study.
Methods
We studied clinical, laboratory, and histological data from 236 non-Hispanic Caucasian women with NAFLD. We analyzed 324,623 single nucleotide polymorphisms (SNPs) from the 22 autosomal chromosomes. Multivariate-adjusted logistic regression analyses were conducted for binary outcomes and linear regression analysis was applied for quantitative traits. A P-value < 1×10−6 was considered to be significant.
Results
In multivariate models adjusted for age, body mass index, diabetes, waist:hip ratios, and levels of hemoglobin A1c, the NAFLD activity score was associated with the SNP rs2645424 on chromosome 8 in farnesyl diphosphate farnesyl transferase 1 (FDFT1) (P=6.8×10−7). The degree of fibrosis was associated with the SNP rs343062 on chromosome 7 (P=2.7×10−8). SNPs associated with lobular inflammation included SNP rs1227756 on chromosome 10 in COL13A1 (P=2.0×10−7), rs6591182 on chromosome 11 (P=8.6×10−7), and rs887304 on chromosome 12 in EFCAB4B (P=7.7×10−7). SNPs associated with serum levels of alanine aminotransferase included rs2499604 on chromosome 1 (P=2.2×10−6), rs6487679 on chromosome 12 in PZP (P=1.3×10−6), rs1421201 on chromosome 18 (P=1.0×10−5), and rs2710833 on chromosome 4 (P=6.3×10−7). There were no significant associations between genotypes and steatosis, ballooning degeneration, portal inflammation, or other features of NAFLD.
Conclusions
A GWAS significantly associated genetic variants with features of hepatic histology in patients with NAFLD. These findings should be validated in larger and more diverse cohorts.
Keywords: GWAS, NASH, NAS, NASH CRN, FDFT1, AST
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a common liver condition affecting up to 35% of adults in the United States.1 It generally occurs in the context of obesity, type 2 diabetes mellitus and dyslipidemia.2 It is broadly categorized into steatosis, which is largely benign and nonalcoholic steatohepatitis (NASH), which can progress to cirrhosis and liver failure.3 The pathogenesis of NASH is not well understood. It remains unclear why some patients with NAFLD exhibit NASH, whereas patients with same set of co-morbidities and known risk factors have fatty liver only. There are data to provide evidence that genetic factors play an important role in the pathogenesis of and disease progression in NAFLD. Studies have shown that there is familial clustering of NAFLD, and in fact it is highly heritable.4,5 Schwimmer et al. have estimated that the heritability of liver fat fraction (assessed by magnetic resonance [MR] spectroscopy) as a continuous trait after controlling for age, gender, race and BMI was 39%.6 There is a striking ethnic variability in the prevalence of NAFLD, even after controlling for confounding risk factors. In the Dallas Heart Study where MR spectroscopy was performed on 2,287 multiethnic adults, Hispanics had significantly higher and Blacks had significantly lower prevalence of hepatic steatosis, in comparison to whites.1 Despite this strong suspicion and suggestive data, studies evaluating the genetic basis for NAFLD and NASH are limited. Most studies have been hypothesis-driven and have tested candidate genes in relevant pathways such as hepatic free fatty acid load, oxidative stress, response to endotoxin, release or effect of adipocytokines, and severity of hepatic fibrosis.7 Many of these studies have undoubtedly yielded interesting and important results but they have generally suffered from small sample size and insufficient statistical power.
Genome-wide association studies (GWAS) provide a broader and unbiased approach for discovery of genes involved in complex genetic traits. For example, the association between complement factor H gene and macular degeneration,8–10 TNFSF15 conferring susceptibility to Crohn’s disease,11 and association of coronary heart disease with loci near the CDKN2A and CDKN2B genes on chromosome 9 were all identified through GWAS.12 But GWAS with any pertinence to NAFLD are quite limited, and none have been performed in patients with histologically characterized NAFLD. In one study, Yuan et al. conducted a GWAS of plasma liver-enzyme levels in three populations with replication in three additional cohorts.13 The previously reported associations between the GGT1 locus and gamma glutamyl transpeptidase (GGT) levels and between the ABO locus and alkaline phosphatase (ALP) levels were confirmed. In addition, CPN1-ERLIN1-CHUK on chromosome 10 and PNPLA3-SAMM50 on chromosome 22 were found to influence plasma levels of alanineaminotransferase (ALT), HNF1A on chromosome 12 influenced GGT levels, and three loci influenced ALP levels (ALPL on chromosome 1, GPLD1 on chromosome 6, and JMJD1C-REEP3 on chromosome 10). In a more recent study, Romeo et al reported the relationship between genome-wide survey of 9,229 nonsynonymous single nucleotide polymorphisms (SNPs) and hepatic fat detected by MR spectroscopy in 1,032 African-American, 696 European-American and 383 Hispanic adults residing in Dallas County.14 This study found that variation in PNPLA3 contributes to ethnic and inter-individual differences in hepatic fat content and susceptibility to NAFLD. An allele in PNPLA3 (rs738409; I148M) was strongly associated with increased hepatic fat levels (P=5.9×10−10) and with alanine aminotransferase levels (P=3.7×10−4). But this study did not have liver histology data and furthermore it may have included a number of individuals with moderate alcohol consumption. A pilot GWAS was conducted in a cohort of patients with histologically characterized NAFLD who were enrolled into the NAFLD Database Study conducted by the NASH Clinical Research Network (CRN). While prior studies were geared towards finding genetic determinants of prevalent NAFLD, imaging parameters, and/or liver enzymes, the current study focused on genetic influences on histologic parameters in subjects with well characterized NAFLD.
Methods
Study Subjects
This GWAS was conducted on a subset of patients who were enrolled into the NAFLD Database Study (Supplemental Figure 1). The NAFLD Database Study is an ongoing multicenter, observational study conducted by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) funded NASH CRN. Initiated in 2004, this longitudinal observational study enrolled 1,215 adults and children with suspected NAFLD, definite NAFLD (confirmed by liver histology) or cryptogenic cirrhosis. Individuals enrolled into the NAFLD Database Study were evaluated for the absence of significant alcohol consumption and for competing etiologies of chronic liver disease (e.g., hepatitis B, hepatitis C, hemochromatosis) and alternate etiologies for steatosis (e.g., drugs, jejunoileal bypass). Eligibility criteria for the NAFLD Database Study are shown in Supplemental Table 1. From this cohort, non-Hispanic, Caucasian, female adults with NAFLD were selected whose liver biopsy specimens were reviewed and scored centrally by the NASH CRN Pathology Sub-committee. Features of liver histology were scored according to its published NAFLD histology scoring system.15 In brief, the severity of steatosis was graded from 0 to 3, inflammation from 0 to 2, hepatocyte ballooning degeneration from 0 to 2 and fibrosis was staged from 0 to 4. In addition, each sample was assessed for the presence or absence of NASH by pattern recognition and for the NAFLD Activity Score (NAS) which is the sum of steatosis, inflammation and hepatocyte ballooning. Additionally, all eligible subjects had liver biopsy within 6-months prior to their enrollment into the NAFLD Database Study. The NAFLD Database Study was reviewed and approved by all participating clinical centers and the data coordinating center, and all participants signed an informed consent which included specific approval for genetic studies. In addition, this GWAS ancillary study was reviewed and approved by the NASH CRN Steering Committee and by the Institutional Review Board at Cedars-Sinai Medical Center, Los Angeles, CA.
Genotyping and Quality Control (QC)
Genotyping was performed at the Medical Genetics Institute at Cedars–Sinai Medical Center using Infinium HD technology (HumanCNV370-Quadv3 BeadChips; Illumina, San Diego, CA, USA; Gunderson et al. 2005; 2006). Genotypes were determined based on clustering of the raw intensity data for the two dyes using Illumina BeadStudio software. Two samples performed in duplicate yielded 100% concordance. The average sample call rate for samples which were genotyped successfully was 99.81%.
As shown in Supplemental Figure 1, this ancillary study was approved for conducting genome-wide genotyping on 250 adult females with NAFLD and all were genotyped for 373,397 SNPs. Six samples were removed due to lack of central liver biopsy reading. Five additional samples failed genotyping, yielding 239 individuals with central histology reading and GWAS data. We also checked missing data rate for each subject and set an exclusion criterion as 0.1. No individuals were filtered out by this criterion. We found one individual with gender mismatch based on chromosome X and Y SNPs data. Two other samples were identified as outliers via principal components analysis and were therefore removed from further analysis. The remaining 236 patients constituted our study group and detailed clinical, laboratory, histological, and genotyping data were available on all these subjects. The total number of successfully genotyped SNPs was 345,111, including 334,556 autosomal SNPs. 145 SNPs with a HWE test p-value < 1e-008 were removed; 725 SNPs with missing rate > 0.1 were removed; and 9319 SNPs were removed due to minor allele frequency (MAF) < 0.02 (PLINK v1.07; Purcell et al 2007). This led to a total of 324,623 SNPs analyzed from the 22 autosomal chromosomes. Association analysis was performed on 236 samples that had their liver histology reviewed and scored centrally. Two hundred and two patients belonging to this study were also included in two other genetic studies recently accepted for publication (references 40 and 41) but neither is a GWAS.
Power calculation
We evaluated the ability to detect an association between a SNP and NAS score by power calculation implemented in QUANTO version 1.0 (3). Based on the mean and standard deviation of NAS (mean 4.27, SD 1.69) in a preliminary sample of patients enrolled into the NAFLD Database Study, we tested the power using 250 independent individuals under dominant and additive genetic models (Supplemental Table 2). For detectable effect size βG greater than 0.5, a sample size of 250 will have enough power (>0.85) to identify the association under additive model with disease allele frequency greater than 0.20.
Data Analysis
Principal components analysis (PCA) was performed using the program Eigenstrat and all GWAS SNPs to evaluate genetic homogeneity. None of the PCs explains a large proportion of the genetic variance. Outliers identified through principal component analysis (greater than 6 standard deviation) were removed from further association analysis (n=2). The scatter plot demonstrated that there was no apparent sub-cluster in the sample; therefore, all samples (after excluding the 2 outliers) were analyzed together.
End points evaluated were NAFLD Activity Score (NAS) and its individual components (steatosis, inflammation, and ballooning), fibrosis, presence of steatohepatitis and aminotransferase levels. Genotype association with trait was considered statistically significant with a p value < 1.0e-06.
The NAS was analyzed as both a quantitative trait with range from 0 to 8, and as a binary trait, defined as 0 if the activity score was less than 5, and 1 if greater than or equal to 5 (individuals with scores of 5–8 have strong evidence of NASH15). Both ALT and AST values were log transformed for association testing for normalization of the data distribution. The association between various genotypes and end-points of interests were evaluated in two different models; the first included age and BMI as covariates, and the second included age, BMI, presence of type2 diabetes, waist hip ratio, and HbA1c as covariates to adjust for potential confounders. In PLINK, logistic regression analysis was carried out for qualitative traits (NAS binary, fibrosis binary, ballooning (yes/no)), and linear regression analysis was applied for quantitative traits (NAS quantitative score, aspartate aminotransferase [AST] and alanine aminotransferase [ALT] levels, and individual histological scores for fibrosis, lobular inflammation, ballooning degeneration and steatosis). When the minor allele was rare, a dominant genetic model was assumed, and p-value and regression coefficient beta based on a dominant genetic model were obtained. For other SNPs, analysis were run for three different genetic models (additive, dominant, recessive) and the smallest p-value out of the 3 models was obtained, alone with the regression coefficient under the particular genetic model. Manhattan plots which presents the −log10(p-value) along with the 22 autosomal chromosomes were generated for the primary trait NAS and secondary end points with SNPs that reached our selection criterion of a p value < 1.0e-06.
Multiple testing is inevitable because of the large number of markers involved in this study. Given the fact that many markers are in LD, the typical Bonferroni correction is not appropriate (too conservative, resulting in loss of power). Numerous methods have been suggested for controlling the family-wise type I error rate (FWER), which is the probability of committing at least one type I error at or below a specified level in the family of hypotheses.16,17 Based on positive FDR (rate that discoveries are false, i.e. pFDR = E[V/R |R > 0]),18 q-value has been proposed as a more appropriate measure than FDR. The q-value of an individual hypothesis test is the minimum FDR at which the test may be called significant. Utilizing the software QVALUE 18 we estimated q-values corresponding to each of the p-values we reported in this study.
To obtain gene annotations for the top SNPs identified from GWAS, the software SNAP was utilized.19 The gene annotations in SNAP are from GeneCruiser, which are based on the Ensembl database. Ensembl automatically annotates and indexes data from several sources such as Entrez, RefSeq, EMBL, Uniprot/SWISS-PROT, Affymetrix, and Gene Ontology (GO). Regional plots were then generated which presents −log10(p-value) from association analysis for all SNPs in the region around the top SNP (50 kb on each side). In addition, the regional plots demonstrate the locations of all candidate genes located in the region.
Results
Table 1 shows selected clinical and laboratory characteristics of 236 patients included in the genotype-phenotype association analyses. All individuals were non-Hispanic, Caucasian, adult females. The age and body mass index (BMI) presented as median (interquartile range, IQR) were 53 years (46–60) and 36 kg/m2 (31–40). Median (IQR) AST and ALT values were 42 U/L (28–60) and 51 U/L (31–76). Forty two percent of patients had type 2 diabetes and their median (IQR) fasting glucose at enrollment was 98 mg/dL (88–119).
Table 1.
Baseline characteristics of the population (white, non-Hispanic, adult females, N=236)
| Patient demographic and clinical characteristics | |
| Age at enrollment, median years (IQR) | 53 (46–60) |
| Diabetes mellitus, n (%) | 99 (42%) |
| BMI, median kg/m2 (IQR) | 36 (31–40) |
| Waist to hip ratio, median (IQR) | 0.91 (0.86–0.95) |
| Laboratory measures, median (IQR) | |
| ALT, U/L | 51 (31–76) |
| AST, U/L | 42 (28–60) |
| HbA1c, % | 5.9 (5.5–6.6) |
| Triglycerides, mg/dL | 152 (112–202) |
| Total cholesterol, mg/dL | 196 (172–224) |
| HDL cholesterol, mg/dL | 46 (38–53) |
| LDL cholesterol, mg/dL | 122 (96–145) |
| Serum glucose, mg/dL | 98 (88–119) |
| Serum insulin, μU/mL | 17 (11–26) |
| Histologic characteristics | |
| Steatosis, n (%) | |
| <5% | 11 (5%) |
| 5–33% | 101 (43%) |
| 34–66% | 76 (32%) |
| >66% | 48 (20%) |
| Lobular inflammation, n (%) | |
| 0 | 1 (<1%) |
| <2 under 20x mag | 125 (53%) |
| 2–4 under 20x mag | 78 (33%) |
| >4 under 20x mag | 32 (14%) |
| Ballooning, n (%) | |
| None | 83 (35%) |
| Few | 44 (19%) |
| Many | 109 (46%) |
| Fibrosis, n (%) | |
| None | 57 (24%) |
| Mild, zone 3, perisinusoidal | 22 (9%) |
| Moderate, zone 3, perisinusoidal | 20 (9%) |
| Zone 3 and periportal, any combination | 43 (18%) |
| Bridging | 51 (22%) |
| Cirrhosis | 34 (15%) |
| NASH diagnosis, n (%) | |
| No NASH | 54 (23%) |
| Suspicious/borderline/indeterminate: Zone 3 or Zone 1 | 52 (22%) |
| Yes, definite | 129 (55%) |
| NAS (range 0–8), median (IQR)† | 4 (3–6) |
NAS is the NAFLD Activity Score, a sum of scores for steatosis, lobular inflammation, and ballooning.
All individuals had liver biopsy performed within 6-months prior to their enrollment and liver histology was reviewed centrally. The histological characteristics are shown in Table 1. Fifty-five percent of patients were deemed to have definite NASH by pattern recognition and 22% had biopsy findings that were suspicious of NASH./ The median (IQR) NAS was 4 (3–6) (Table 1). There was no fibrosis in 24%, mild zone 3 and/or periportal fibrosis in 39%, bridging fibrosis in 22% and cirrhosis in 15% of patients.
Primary outcome: Novel association between NAS (NASH activity score) and FDFT1 (Farnesyl diphosphate farnesyl transferase 1) gene
The SNPs with the most significant evidence for association (p<1.0e-06) with nonalcoholic fatty liver disease-related measures are summarized in Table 2. The quantitative histologic activity score (NAS) was the primary outcome of interest. The most significant association with NAS was with rs2645424 on chromosome 8 in the FDFT1 gene (p = 8.0e-07 when adjusting for age and BMI only; and 7.0e-07 when further adjusting for diabetic status (yes/no), waist-hip ratio, and HbA1c) (Figure 1a). Upon further examination of SNPs in the FDFT1 gene, we found one additional SNP had p-value less than 1.0e-04, and several more with p-value <0.01 (Figure 2a). This relationship between FDFT1 gene variants and NAS generally persisted even when NAS was treated as a binary variable, although none of the associations reached our cutoff p-value 1.0e-06. A strong association was observed between SNP rs2645424 and NAS as a binary variable with a p-value of 1.03e-05 after adjusting for age and BMI only and a p-value of 8.39e-06 after further adjusting for diabetes, waist-hip ratio, and HbA1c. The details of the association analysis results for binary NAS score and SNPs in the FDFT1 gene, including genotype frequencies in cases and controls, odd ratio, 95%CI, and corresponding p-values for each of the two models are given in Supplementary Table 2.
Table 2.
Top SNPs from GWAS for primary and secondary end points
| Trait | SNP | CHR | POSITION | Genes | Minor | MAF1 | P12 | Q-value1 | β13 | P24 | Q-value2 | β2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary end point | ||||||||||||
| NAS | rs2645424A | 8 | 11721872 | FDFT1 | A | 0.398 | 7.81E-07 | 1.05E-06 | −0.76 | 6.75E-07 | 1.30E-06 | −0.77 |
| Secondary end points | ||||||||||||
| Fibrosis | rs343064R | 7 | 35521313 | - | A | 0.4 | 4.73E-08 | 4.26E-07 | 1.29 | 2.72E-08 | 2.45E-07 | 1.3 |
| Lobular | rs1227756R | 10 | 71258510 | COL13A1 | G | 0.461 | 1.14E-07 | 5.13E-07 | 0.58 | 2.01E-07 | 9.05E-07 | 0. 57 |
| rs6591182R | 11 | 65106332 | LTBP3 | A | 0.467 | 6.69E-07 | 1.05E-06 | 0.54 | 8.66E-07 | 1.30E-06 | 0.54 | |
| rs887304A | 12 | 3627809 | EFCAB4B | A | 0.312 | 4.00E-07 | 1.05E-06 | 0.37 | 7.66E-07 | 1.30E-06 | 0.36 | |
| AST | rs2499604D | 1 | 236170124 | ZP4 | A | 0.437 | 9.34E-07 | 1.05E-06 | 0.41 | 2.23E-06 | 2.51E-06 | −0.38 |
| rs6487679D | 12 | 9262599 | PZP | G | 0.164 | 8.74E-07 | 1.05E-06 | 0.41 | 1.26E-06 | 1.62E-06 | 0.39 | |
| rs1421201R | 18 | 41226287 | - | G | 0.492 | 7.34E-07 | 1.05E-06 | 0.44 | 1.09E-05 | 1.09E-05 | 0.39 | |
| rs2710833D | 4 | 169646533 | DDX60L PALLD | A | 0.168 | 2.35E-06 | 2.35E-06 | 0.39 | 6.27E-07 | 1.30E-06 | 0.41 | |
Minor allele frequency;
β1 and β2 are the estimates of regression coefficient beta for the first and second covariate models, respectively;
P1 is the best p-value among three genetic models (additive, dominant, recessive) for the first covariate model: with age and BMI as covariates;
P2 is the best p-value among three genetic models for the 2nd covariate model: with age, BMI, diabetic status, waist-hip-ratio, and hba1c;
Recessive model;
Additive model;
Dominant model. Q-value1 is the Q-value corresponding to P1; Q-value2 is the Q-value corresponding to P2.
Figure 1.
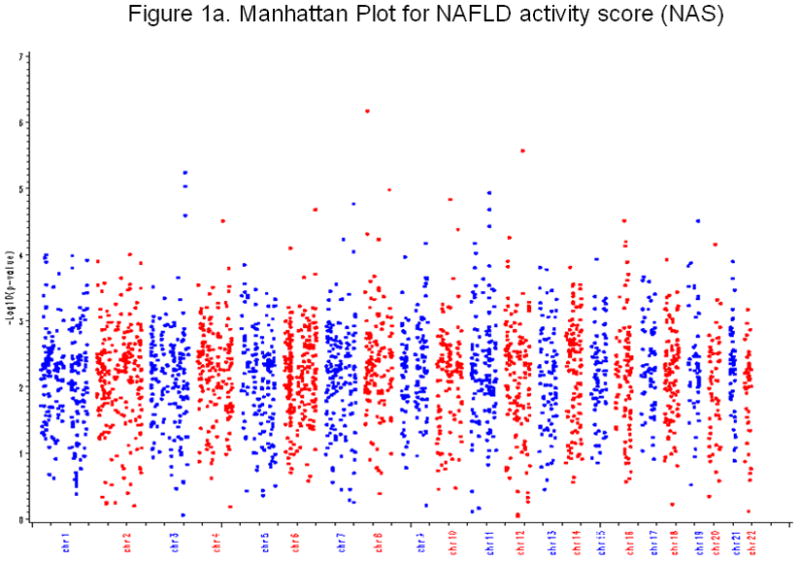
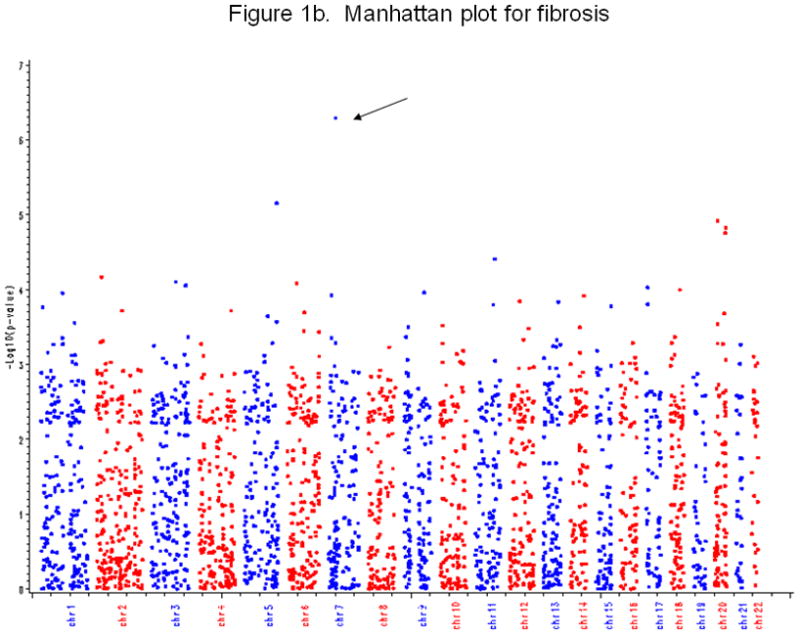
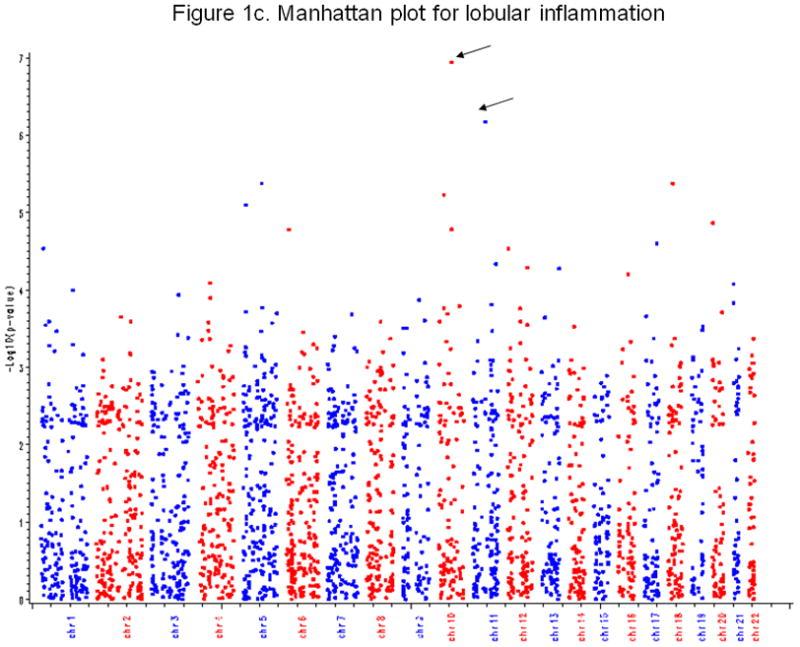
Figure 1a. Manhattan Plot NASH activity score (NAS)
Figure 2a shows the Manhattan plot for NAS. X-axis is chromosome number, and Y-axis is −log10(p-value) from GWAS. Each point represents a profiled SNP.
Figure 1b. Manhattan plot for fibrosis
Figure 2b shows the Manhattan plot for fibrosis. X-axis is chromosome number, and Y-axis is −log10(p-value) from GWAS. Each point represents a profiled SNP. The arrow indicates the SNP with a p-value less than 1.0e-06.
Figure 1c. Manhattan plot for lobular
Figure 2c shows the Manhattan plot for lobular. X-axis is chromosome number, and Y-axis is −log10(p-value) from the GWAS. Each point represents a profiled SNP. The arrow indicates the SNP with a p-value less than 1.0e-06.
Figure 2.
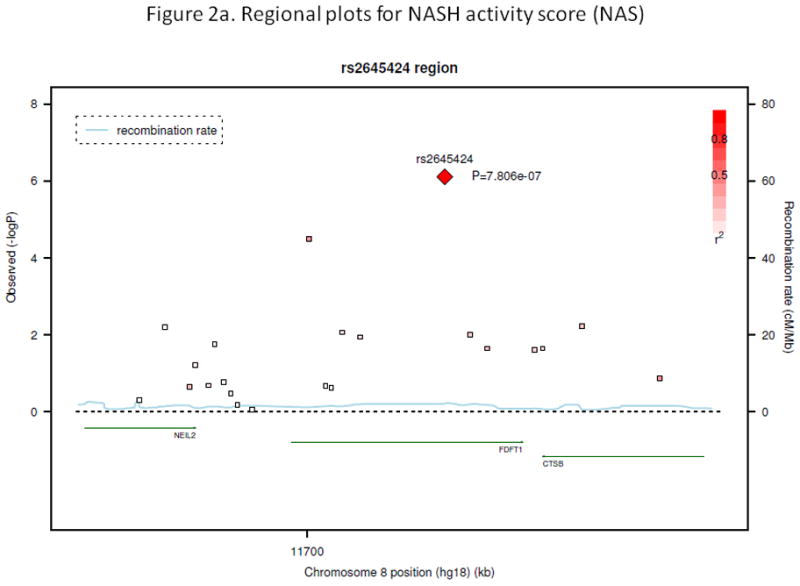
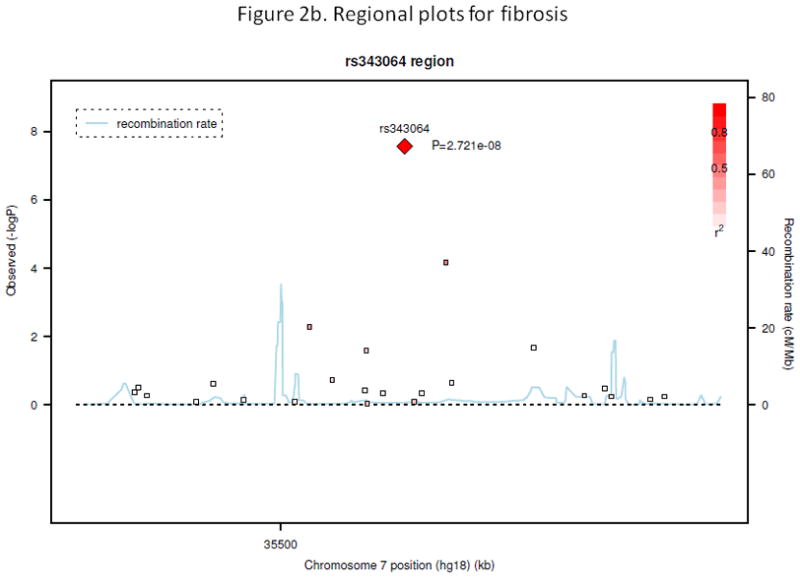
Figure 2a. Regional plot for NASH activity score (NAS)
Figure 3a shows −log10(p-value) from association analysis for all SNPs in the region around the top SNP rs2645424 for NAS (50 kb on each side). X-axis shows position of the SNPs along chromosome 8; Y-axis in the left gives −log10(p-value). P-values were obtained from association analysis when including age and BMI as covariates and assuming an additive genetic model. Y-axis in the right shows recombination rate (cM/Mb). r2 shows measure of the LD between this SNP and target SNP. The genes (NEIL2, FDFT1, and CTSB) are located in the region as indicated at the bottom portion of the figure.
Figure 2b. Regional plot for fibrosis
Figure 3b shows −log10(p-value) from association analysis for all SNPs in the region around the top SNP rs343064 for fibrosis (50 kb on each side). X-axis shows position of the SNPs along chromosome 7; Y-axis gives −log10(p-value). P-values were obtained from association analysis which included age, BMI, diabetic status, waist-hip-ratio and HbA1c as covariates and assuming a recessive genetic model. Y-axis in the right shows recombination rate (cM/Mb). r2 shows measure of the LD between this SNP and target SNP.
Secondary outcomes
There were several genetic variants associated with secondary outcomes. The most significant was between SNP rs343062 on chromosome 7 and the degree of fibrosis (Figure 1b. p=5.0e-08 when adjusting for age and BMI; 3.0e-08 after further adjusting for diabetic status, waist hip ratio, and HbA1c). As shown in the regional plot for rs343062 (Figure 2b), an additional SNP in the region, rs867177, also had modest association with fibrosis (p<0.0001). Three SNPs were associated with the lobular inflammation phenotype: SNP rs1227756 on chromosome 10 in the COL13A1 gene, rs6591182 on chromosome 11, and rs887304 on chromosome 12 in the EFCAB4B gene (p≤9.0e-07, Figure 1c). Three more SNPs in the COL13A1 gene (rs7077164, rs2763341, rs1227771) had p-values less than 0.0001 (regional plot, Supplemental figure 2). Four SNPs met the selection criterion for an association with AST levels: rs2499604 on chromosome 1 (p1 = 9.0e-07, p2 = 2.0e-06), rs6487679 on chromosome 12 in the PZP gene (p1 = 9.0e-07, p2 = 1.0e-06), rs1421201 on chromosome 18 (p1 = 7.0e-07, p2 = 1.0e-05), and rs2710833 on chromosome 4 (p1 = 2.0e-06, p2 = 6.0e-07) (Supplemental Figure 3), where p1 and p2 represent p-values calculated from two different covariate models previously defined. Several additional SNPs in the PZP gene also showed modest evidence of association with AST, with one having a p-value of less than 1.0e-04 (Supplemental Figure 4).
No SNPs were identified at the pre-specified significance level of 1.0e-06 for degree of steatosis or ballooning degeneration, presence or absence of NASH, and for ALT levels. A number of SNPs manifested a modest level of evidence of association (0.05 < p value < 1.0e-06) for each of the histologically assessed nonalcoholic fatty liver disease measures, as summarized in Supplementary Tables 3 and 4: 46 SNPs for NAS, 38 for fibrosis, 60 for lobular, 72 for AST, 43 SNPs for steatosis, 56 for ALT, 27 SNPs for balloon, 23 SNPs for fibrosis as a qualitative trait, 38 SNPs for NAFLD when the full covariate adjustment model was applied.
Fifteen SNPs in the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene were evaluated in this GWAS. SNP (rs738409) was not captured by the GWAS so genotypic association could not be assessed. However, five out of the 15 genotyped SNPs in the PNPLA3 gene were in complete linkage disequilibrium (D′ = 1) with rs738409, but none of the genotypes showed significant association with any of the outcome variables.
QQ plots for NAS, fibrosis, lobular inflammation, and AST are shown as Supplementary Figures 5a–5d.
Discussion
In this pilot GWAS in non-Hispanic, Caucasian, women with liver biopsy proven NAFLD, a novel association was found between NAS and SNPs within the FDFT1 gene locus on chromosome 8. The FDFT1 gene is a key regulator of cholesterol biosynthesis.20,21 These findings indicate potential role of lipid metabolism and possibly lipotoxicity in the pathogenesis of NASH.22 A significant association was also found between various SNPs and fibrosis, lobular inflammation and AST levels. To our knowledge, this is the first genome wide association study of histologic features of NAFLD and NASH in a well-characterized cohort derived from geographically diverse eight centers within United States.
In this study, fibrosis was associated with SNP rs343062 on chromosome 7. Although the exact function of this SNP is unknown, it is in close proximity of the platelet-derived growth factor-alpha (PDGFA) gene that appears to play a role in regulation of liver inflammation and fibrosis in animal models.23 Lobular inflammation was found to be associated with LTBP3 (latent transforming growth factor-beta-protein 3), EF-hand calcium binding domain 4B (EFCAB4B) and collagen, type XIII, alpha 1 (COL13A1) gene. LTBP3 secretion is known to be dependent upon co-expression of TGF-beta24 which is a key pro-fibrogenic cytokine that is activated in chronic inflammation and leads to collagen deposition.25,26 COL13A1 has been previously linked with modifying inflammatory response genes in intestine.27 A recent GWAS study showed that EFCAB4B is associated with hypertension in Chinese population.28 This pleiotropy may explain genetic bases of co-occurrence of hypertension among individuals with NAFLD.
AST levels were associated with SNPs in the pregnancy zone protein (PZP) gene on chromosome 12. PZP is a proteinase that is strongly related to alpha-2-macroglobulin and is involved in clearance of TGF-β clearance from human plasma, which plays an important role in hepatic fibrogenesis.29
Biological rationale and proposed mechanism
In this GWAS, a novel association was found between the NAS (NASH activity score) and the FDFT1 gene, which is involved in lipid metabolism and cholesterol biosynthesis. FDFT1 encodes squalene synthase (FDFT1) that catalyzes the production of squalene from farnesyl pyrophosphate and is the first step in the sterol biosynthesis pathway. Inhibitors of squalene synthase can decrease plasma cholesterol levels.30 The nuclear hormone receptor, liver X receptor (LXR) α, regulates cholesterol biosynthesis by silencing the expression of squalene synthase via negative response elements (nLXREs) located in each of the FDFT1 genes. LXR is activated by dietary sugars and then induces fatty acid synthesis.31 LXR agonists are being considered in the therapy of dyslipidemia and NAFLD.32
Furthermore, an association was identified between the PDGFA gene and the degree of fibrosis in NAFLD. A recent study showed that transgenic mice over-expressing PDGF develop significant hepatic fibrosis spontaneously.33 Previous studies have shown that PDGFA mRNA expression is increased in mononuclear and ductular cells and that 23 PDGF expression correlates with ductular proliferation.23 Recent human studies have confirmed that ductular reaction and proliferation is linked to advanced fibrosis in patients with NASH.34 Although type 1, 3 and 4 collagen production has been linked to hepatic fibrogenesis,35 the association between collagen type 13 and liver disease has not been previously reported. The COL13A1 gene is linked to intestinal inflammatory response and it may play a role in liver inflammation that has yet to be appreciated. Serum AST was linked to the PZP gene. PZP is structurally closely related to alpha-2-macroglobulin, which is a non-invasive biomarker for predicting hepatic fibrosis.36 PZP binds to TGF-beta 1 and 2 in a manner that is similar to binding of the TGF-beta and alpha 2-macroglobulin. PZP binds to TGF-beta 1 preferentially over TGF-beta-2 and regulates it’s plasma clearance.37 TGF-beta signaling is strongly linked to hepatic fibrogenesis and carcinogenesis and is involved in the pathogenesis of NASH fibrosis. These multiple associations require further studies to confirm the linkage and to elucidate the biological basis for the associations.
Strengths and limitations of the study
The strengths of this study include that patients were derived from diverse geographic areas within United States, well-characterized liver histology, prospectively collected and measured laboratory and recorded demographic variables, and biological plausibility of findings. A significant strength of the study includes well-characterization of liver histology using a validated scoring system derived from this dataset. Additionally, the study was able to exclude individuals who may have alcoholic liver disease because of stringent entry criteria. However, limitations of the study include that the study population was exclusively White (non-Hispanic) women residing in the United States. Thus, these results need to be validated in larger cohorts that include men, and participants of other ethnicities and nationalities.
It is noteworthy that this study did not find significant association between PNPLA3 genetic variants and liver histology in patients with NAFLD. Since the seminal observation by Romeo et al.,14 that showed a very strong relationship between rs738409 C→G polymorphism of PNPLA3 and hepatic steatosis, several studies have examined the relationship between genetic variability in PNPLA3 and liver histology in patients with NAFLD.38–41 In a study that combined 321 NAFLD patients from UK and 253 NAFLD patients from Italy, Valenti et al., have shown that PNPLA3 genotype was associated with severity of steatosis, presence of steatohepatitis, and severity of fibrosis. However, these associations could not be reproduced entirely in the subset of NAFLD patients from Italy, presumably due to small sample size. More recently, Rotman et al., reported the association between genetic variability in PNPLA3 and liver histology in 1197 individuals with NAFLD (96% from the NASH CRN studies, 19% children). This study showed a statistically significant relationship between PNPLA3 genotype and degree of steatosis, inflammation, NAS, and fibrosis (but not steatohepatitis). Interestingly, in a subgroup analysis, investigators did not find any significant relationship between PNPLA3 genotype and liver histology in pediatric patients with NAFLD. In another study that consisted of 678 NAFLD patients (also from the NASH CRN), PNPLA3 genotype was significantly associated with the degree of inflammation and fibrosis, but not with steatosis severity, ballooning or the presence of steatohepatitis. It is unclear why we did not find any relationship between PNPLA3 genetic variability and liver histology in this study, but this may be a reflection of our modest sample size and highly select group of patients (i.e., adult Caucasian women).
Conclusions
In this GWAS, novel associations were found between genes involved in lipid metabolism, growth factors and collagen deposition and with histologic and biochemical features of NAFLD in a well-characterized cohort of non-Hispanic, Caucasian women participants of the NASH CRN. These findings suggest that several complex genes are involved and interlinked in the pathogenesis of NAFLD and NASH. The specific histologic phenotype may be regulated by genes involved in diverse metabolic pathways. This pilot study provides preliminary data that GWAS approach may lead to identification of novel genes involved in the pathogenesis of NAFLD and NASH. In future, large, international, collaborative studies including geographically and ethnically diverse population with well-characterized liver histologic features of NAFLD are needed to confirm and validate these findings.
Supplementary Material
Acknowledgments
Funding: This study was supported by U01DK061737 and K24DK069290 (N.C.), NCRR grant M01-RR00425 to the Cedars-Sinai General Research Center Genotyping core, P30DK063491 to J.R., and R01DK079888 to M.O.G. There are no relevant financial conflicts of interest to declare. This work is supported in part by the American Gastroenterological Association (AGA) Foundation – Sucampo – ASP Designated Research Award in Geriatric Gastroenterology and by a T. Franklin Williams Scholarship Award; Funding provided by: Atlantic Philanthropies, Inc, the John A. Hartford Foundation, the Association of Specialty Professors, and the American Gastroenterological Association to RL. This research was funded in part with the support of the UCSD Digestive Diseases Research Development Center, U.S. PHS grant #DK080506.
Abbreviations
- NAFLD
Nonalcoholic Fatty Liver Disease
- NASH
Nonalcoholic steatohepatitis
- NASH CRN
Nonalcoholic Steatohepatitis Clinical Research Network
- NAS
NAFLD Activity Score
- SNP
Single nucleotide polymorphism
- AST
Aspartate aminotransferase
- ALT
Alanine aminotransferase
Footnotes
Authors contributions: Drs. Chalasani and Guo: Study supervision, concept/design, acquisition of samples, interpretation of data, draft and revision of the manuscript
Drs. Loomba and Goodarzi: Concept/design, interpretation of data, draft and revision of the manuscript
Dr. Rotter: Study supervision, concept/design, acquisition of funding, interpretation of data, draft and revision of the manuscript
Drs. Taylor, Chen, Haritunians, and Cui: Sample analyses, data analyses, data interpretation, draft and revision of the manuscript. Wilson and Cummings: Concept/design, cohort assembly, cohort characterization, interpretation of data, critical review of the manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–95. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–62. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 3.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–9. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 4.Abdelmalek MF, Liu C, Shuster J, Nelson DR, Asal NR. Familial aggregation of insulin resistance in first-degree relatives of patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:1162–9. doi: 10.1016/j.cgh.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Loomba R, Hwang SJ, O’Donnell CJ, Ellison RC, Vasan RS, D’Agostino RB, Sr, Liang TJ, Fox CS. Parental obesity and offspring serum alanine and aspartate aminotransferase levels: the Framingham heart study. Gastroenterology. 2008;134:953–9. doi: 10.1053/j.gastro.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, Shiehmorteza M, Yokoo T, Chavez A, Middleton MS, Sirlin CB. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1585–92. doi: 10.1053/j.gastro.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilfred de Alwis NM, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin Liver Dis. 2007;27:44–54. doi: 10.1055/s-2006-960170. [DOI] [PubMed] [Google Scholar]
- 8.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 9.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 10.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, Cardon L, Takazoe M, Tanaka T, Ichimori T, Saito S, Sekine A, Iida A, Takahashi A, Tsunoda T, Lathrop M, Nakamura Y. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14:3499–506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 12.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–53. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, Zhang W, Vollenweider P, Stirnadel H, Johnson T, Bergmann S, Beckmann ND, Li Y, Ferrucci L, Melzer D, Hernandez D, Singleton A, Scott J, Elliott P, Waeber G, Cardon L, Frayling TM, Kooner JS, Mooser V. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet. 2008;83:520–8. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–5. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 16.Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- 17.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 18.Storey JD. A direct approach to false discovery rates. J R Statist Soc B. 2002;64:479–498. [Google Scholar]
- 19.Johnson C, Drgon T, Liu QR, Zhang PW, Walther D, Li CY, Anthony JC, Ding Y, Eaton WW, Uhl GR. Genome wide association for substance dependence: convergent results from epidemiologic and research volunteer samples. BMC Med Genet. 2008;9:113. doi: 10.1186/1471-2350-9-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schechter I, Conrad DG, Hart I, Berger RC, McKenzie TL, Bleskan J, Patterson D. Localization of the squalene synthase gene (FDFT1) to human chromosome 8p22-p23.1. Genomics. 1994;20:116–8. doi: 10.1006/geno.1994.1135. [DOI] [PubMed] [Google Scholar]
- 21.Ness GC, Zhao Z, Keller RK. Effect of squalene synthase inhibition on the expression of hepatic cholesterol biosynthetic enzymes, LDL receptor, and cholesterol 7 alpha hydroxylase. Arch Biochem Biophys. 1994;311:277–85. doi: 10.1006/abbi.1994.1238. [DOI] [PubMed] [Google Scholar]
- 22.Ness GC, Eales S, Lopez D, Zhao Z. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase gene expression by sterols and nonsterols in rat liver. Arch Biochem Biophys. 1994;308:420–5. doi: 10.1006/abbi.1994.1059. [DOI] [PubMed] [Google Scholar]
- 23.Malizia G, Brunt EM, Peters MG, Rizzo A, Broekelmann TJ, McDonald JA. Growth factor and procollagen type I gene expression in human liver disease. Gastroenterology. 1995;108:145–56. doi: 10.1016/0016-5085(95)90019-5. [DOI] [PubMed] [Google Scholar]
- 24.Taipale J, Miyazono K, Heldin CH, Keski-Oja J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J Cell Biol. 1994;124:171–81. doi: 10.1083/jcb.124.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czaja MJ, Weiner FR, Flanders KC, Giambrone MA, Wind R, Biempica L, Zern MA. In vitro and in vivo association of transforming growth factor-beta 1 with hepatic fibrosis. J Cell Biol. 1989;108:2477–82. doi: 10.1083/jcb.108.6.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshinaga K, Obata H, Jurukovski V, Mazzieri R, Chen Y, Zilberberg L, Huso D, Melamed J, Prijatelj P, Todorovic V, Dabovic B, Rifkin DB. Perturbation of transforming growth factor (TGF)-beta1 association with latent TGF-beta binding protein yields inflammation and tumors. Proc Natl Acad Sci U S A. 2008;105:18758–63. doi: 10.1073/pnas.0805411105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuomisto A, Sund M, Tahkola J, Latvanlehto A, Savolainen ER, Autio-Harmainen H, Liakka A, Sormunen R, Vuoristo J, West A, Lahesmaa R, Morse HC, 3rd, Pihlajaniemi T. A mutant collagen XIII alters intestinal expression of immune response genes and predisposes transgenic mice to develop B-cell lymphomas. Cancer Res. 2008;68:10324–32. doi: 10.1158/0008-5472.CAN-08-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang YGY, Zhang X, Huang W, Zhu D. A Genome-wide Association Study of Hypertension and Blood Pressure in Chinese Population. Circulation. 2009;120:S555, 1781. [Google Scholar]
- 29.Ling TY, Huang YH, Lai MC, Huang SS, Huang JS. Fatty acids modulate transforming growth factor-beta activity and plasma clearance. FASEB J. 2003;17:1559–61. doi: 10.1096/fj.02-1063fje. [DOI] [PubMed] [Google Scholar]
- 30.Hiyoshi H, Yanagimachi M, Ito M, Yasuda N, Okada T, Ikuta H, Shinmyo D, Tanaka K, Kurusu N, Yoshida I, Abe S, Saeki T, Tanaka H. Squalene synthase inhibitors suppress triglyceride biosynthesis through the farnesol pathway in rat hepatocytes. J Lipid Res. 2003;44:128–35. doi: 10.1194/jlr.m200316-jlr200. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Rogers PM, Su C, Varga G, Stayrook KR, Burris TP. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J Biol Chem. 2008;283:26332–9. doi: 10.1074/jbc.M804808200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lund EG, Menke JG, Sparrow CP. Liver X receptor agonists as potential therapeutic agents for dyslipidemia and atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1169–77. doi: 10.1161/01.ATV.0000056743.42348.59. [DOI] [PubMed] [Google Scholar]
- 33.Thieringer F, Maass T, Czochra P, Klopcic B, Conrad I, Friebe D, Schirmacher P, Lohse AW, Blessing M, Galle PR, Teufel A, Kanzler S. Spontaneous hepatic fibrosis in transgenic mice overexpressing PDGF-A. Gene. 2008;423:23–8. doi: 10.1016/j.gene.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 34.Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, Weltman MD, Tilg H, Moschen AR, Purdie DM, Demetris AJ, Clouston AD. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology. 2007;133:80–90. doi: 10.1053/j.gastro.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 35.Ala-Kokko L, Pihlajaniemi T, Myers JC, Kivirikko KI, Savolainen ER. Gene expression of type I, III and IV collagens in hepatic fibrosis induced by dimethylnitrosamine in the rat. Biochem J. 1987;244:75–9. doi: 10.1042/bj2440075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naveau S, Poynard T, Benattar C, Bedossa P, Chaput JC. Alpha-2-macroglobulin and hepatic fibrosis. Diagnostic interest. Dig Dis Sci. 1994;39:2426–32. doi: 10.1007/BF02087661. [DOI] [PubMed] [Google Scholar]
- 37.Philip A, Bostedt L, Stigbrand T, O’Connor-McCourt MD. Binding of transforming growth factor-beta (TGF-beta) to pregnancy zone protein (PZP). Comparison to the TGF-beta-alpha 2-macroglobulin interaction. Eur J Biochem. 1994;221:687–93. doi: 10.1111/j.1432-1033.1994.tb18781.x. [DOI] [PubMed] [Google Scholar]
- 38.Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Research. 2009;50:2111–2116. doi: 10.1194/jlr.P900013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, Mozzi E, Roviaro E, Bugianesi E, Maggioni M, Fracanzani AL, Fargion S, Day CP. Homozygosity for the patalin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–1217. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 40.Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ the NASH CRN. The association of genetic variability in PNPLA3 with histological severity of nonalcoholic fatty liver disease. Hepatology. 2010 doi: 10.1002/hep.23759. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Speliotes EK, Butler JL, Palmer C, Voight BF, Hirschhorn JN the GIANT Consortium, the MIGen Consortium, the NASH CRN. PNPLA3 variants specifically confer increased risk for histologic NAFLD but not metabolic disease. Hepatology. 2010 doi: 10.1002/hep.23768. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.