

Abstract
Background & Aims
The identification of numerous genes that confer susceptibility to Crohn’s disease (CD) indicates that this complex disease might arise from alterations in several genes with related functions. We examined the functional interaction between the CD risk genes autophagy-related 16-like protein 1 (ATG16L1) and nucleotide-binding oligomerization domain 2 (NOD2) to identify an autophagy-dependent pathway that is altered by disease-associated variants.
Methods
We assessed Nod2 signaling and autophagy activation in response to muramyl dipeptide (MDP) by immunoblot, confocal microscopy, flow cytometry, reporter gene, and gentamycin protection assays in human epithelial cell lines and primary human macrophages and dendritic cells from healthy individuals. The requirement of Nod2 and ATG16L1 expression and the effects of CD-associated variants in MDP-stimulated autophagy and Nod2-dependent signaling were assessed in cell lines manipulated by RNA interference, inhibitors, or ATG16L1 or NOD2 variants and in primary macrophages and dendritic cells from healthy, genotyped donors.
Results
MDP stimulation of epithelial cells, macrophages, and dendritic cells activated autophagy and NF-κB and MAPK signaling; it also increased killing of Salmonella. These responses depended on ATG16L1 and Nod2 expression and were impaired by CD-associated NOD2 variants. Nod2-dependent signaling was not impaired in cells with the ATG16L1 T300A genotype, which is associated with CD. However, the ATG16L1 T300A variant blocked the increase in MDP-mediated killing of Salmonella only in epithelial cell lines and not primary macrophages or dendritic cells.
Conclusions
ATG16L1 and NOD2 are components of an autophagy-mediated, anti-bacterial pathway that is altered in a cell- and function-specific manner by CD-associated mutations.
Keywords: CARD15, inflammatory bowel disease, genetics, mucosal immunity
Introduction
Crohn’s disease (CD) is a chronic and sometimes debilitating form of inflammatory bowel disease. The underlying cause of CD is unknown; however it is clear that both environmental and genetic factors are required for its development. Substantial evidence implicates an altered immune response to microbial factors as an essential contributor to CD pathogenesis1. This is evidenced by studies demonstrating the chronic inflammation found in CD depends upon the presence of the bacterial microflora, and by clinical data of disease remission induced by fecal stream diversion or manipulation of the gut microflora2, 3.
Until recently, only one gene had been conclusively identified as a CD susceptibility gene, NOD2, which encodes an intracellular bacteria sensor of the Nod-like receptor (NLR) family4. Nod2 senses the presence of MDP, a component of the peptidoglycan cell wall from both Gram-positive and Gram-negative bacteria. Nod2 activation results in pro-inflammatory and anti-bacterial molecule production dependent on cell signaling pathways mediated by RICK/RIP2, NF-κB and MAPKs. Three major NOD2 variants are associated with CD; two missense mutations, R702W and G908R, and one frameshift mutation, L1007fsinsC (L1007fs). Human studies suggest that these NOD2 variants result in a loss of function5.
Recent genome-wide association studies have dramatically increased the number of CD-associated susceptibility genes6–9. Two of these genes, ATG16L1 and IRGM (immunity-related GTPase family, M), modulate autophagy. Autophagy is a starvation-induced cell-survival mechanism, where organelles are broken down and recycled to provide nutrients. This mechanism also plays crucial roles in the immune system for elimination of intracellular microbes, presentation of endogenous antigens via MHC class II, shaping B- and T-cell function, as well as defining central tolerance10. Importantly, knockdown of ATG16L1 or IRGM expression blocks autophagy and killing of intracellular pathogens such as Salmonella typhimurium, Listeria monocytogenes, Mycobacterium tuberculosis, Toxoplasma gondii, and adherent invasive Escherichia coli (AIEC), some of which are associated with CD pathogenesis11. The only CD-associated ATG16L1 variant, T300A, results in a specific defect in bacteria-induced autophagy7, 12. Therefore, defects in autophagy not only impact innate and adaptive immune responses to bacteria, but are implicated in CD pathogenesis.
In this report, we investigate whether NOD2 and ATG16L1 are components of a common anti-bacterial pathway that is disrupted by specific CD-associated mutations. Our data demonstrate that MDP stimulation activates autophagy as an anti-bacterial response, and implicate autophagy in Nod2 activation by MDP. Our findings also suggest that CD-associated variants of NOD2 and ATG16L1 result in defects in an autophagy-dependent, anti-bacterial pathway specifically in epithelial cells.
Materials & Methods
Reagents
MDP and MDP-LL purchased from Bachem (Torrance, CA), 3-methyl adenine and chloroquine from Sigma (St. Louis, MO), LPS from InvivoGen (San Diego, CA), and rapamycin from LC Laboratories (Woburn, MA). Cytokines obtained from Peprotech Inc (Rocky Hill, NJ).
Cell Lines
HEK293T, HCT116 (gifts of Gabriel Nuñez, Univ. of Michigan) and HT29GR cells (gift of Gerhard Rogler, Univ. of Zurich) were maintained in DMEM (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS, Lonza, Allendale, NJ) and penicillin/streptomycin (pen/strep, Invitrogen). HCT116 cells stably-expressing EGFP-LC3 (HCT116:LC3) were created by transfection with Polyfect (Qiagen, Valencia, CA), G418 selection (Invitrogen) and then FACS to select low-expressing clones. HT29 cells (gift of Carol de la Motte, Cleveland Clinic) were maintained in RPMI (Invitrogen) with 10% FBS and pen/strep. Cells were transfected by Polyfect or Amaxa nucleofection (Lonza) according to manufacturer’s instructions.
Primary Cells
Peripheral blood-derived mononuclear cells (PBMCs) were obtained from healthy donors using protocols approved by The Cleveland Clinic Institutional Review Board. Monocytes were obtained by counterflow centrifugal elutriation by the Cleveland Clinic Clinical and Translational Sciences Collaborative or purified from venous blood by Histopaque-1077 density gradient (Sigma) followed by negative selection using EasySep Monocyte Enrichment Kit Without CD16 Depletion (Stem Cell Technologies, Vancouver, BC). Monocytes were differentiated over 6–7 days through the addition of cytokines into dendritic cells (100ng/mL IL-4, 80ng/mL GM-CSF) or macrophages (50ng/mL M-CSF). Donors were screened for NOD2 and ATG16L1 CD-associated variants by TaqMan SNP genotyping (Applied Biosystems, Foster City, CA) or RFLP analysis.
Plasmids and RNAi
NLR plasmids have been previously described13. Dominant negative IKK constructs were gifts of Derek Abbott (Case Western Reserve Univ.). EGFP-tagged LC3 and kinase-dead Vps34 plasmids obtained from Tony Eissa (Baylor Univ.), myc-ATG16L1 from Origene Technologies Inc. (Rockville, MD) and NOD2 (NM_022162.1-2959s1c1), ATG16L1 (NM_030803.5-2181s1c1) and non-specific control (SHC002) MISSON shRNAs purchased from Sigma. Beclin-1 shRNA and its matching control were gifts of William Maltese (Univ. of Toledo). RICK shRNA (V2HS_17019) and pSMc2 obtained from Open Biosystems (Huntsville, AL).
Immunoblots
Immunoblots were performed on whole cell lysates with antibodies to LC3 (Novus Biologicals, Littleton, CO), tubulin (Sigma), ATG16L1 (Abgent, San Diego, CA), Beclin-1 (Novus), GFP (Santa Cruz Biotechnology Inc., Santa Cruz, CA), HA (Covance Research Products, Princeton, NJ), phosphorylated IκBα, phosphorylated p38, or GAPDH (Cell Signaling Technology, Boston, MA).
Luciferase Reporter Assays
Assays performed as previously described13. Cells were transfected in triplicate with NF-κB luciferase reporter, β-galactosidase transfection control construct, and expression plasmids. Cells were stimulated with ligands for 16h and lysates assayed using the Luciferase Reporter Assay System (Promega, Madison, WI) and for β-galactosidase activity13. Luciferase activity was normalized to β-galactosidase activity (nLuc).
Gentamycin Protection Assays
Intracellular killing of Salmonella enterica serovar typhimurium SL1344 (gift of Gabriel Nuñez) was assessed by gentamycin protection assay14. Overnight cultures in LB broth were incubated at 30°C, shaking at 180rpm, then diluted 1:7 and grown until A600=0.5. Cells were infected in triplicate at a multiplicity of infection of 1:10 for 30 minutes, washed with PBS, and further incubated in gentamycin-supplemented DMEM (50µg/ml) for 1 hour (HEK293T, HCT116, macrophages and dendritic cells) or 3 hours (HT29 and HT29GR cells). Cells lysed in 1% Triton/PBS for 10 minutes on ice and serial dilutions plated on LB agar in duplicate and grown overnight at 30°C. Colonies recovered were counted and colony forming units (cfu) per well calculated.
Flow Cytometry
HEK293T cells were plated at 105 cells/well and transfected with a total of 2µg DNA (1µg GFP-mCherry-LC3 (gift of Jaynatha Debnath, Univ. of California - San Francisco)15 and 0.1µg of NLR plasmids) using Polyfect according to manufacturer’s instructions. 48h post-transfection, cells were maintained in complete media, rapamycin-treated (25µg/mL for 1h) or starved (HBSS for 2h), followed by collection in PBS. 10,000 live events were acquired on a FACSAria II multivariable cell sorter (Becton-Dickinson, San Jose, CA). Data analyzed using FlowJo version 9.0.1 software (Tree Star, Ashland, OR). The target population was gated based on a forward- versus side-scatter plot, and transfected events determined by GFP versus mCherry density plot. The mean fluorescence intensity (MFI) of each fluorochrome was determined in the target population. Results were expressed as GFP/mCherry MFI ratio normalized to untreated sample.
Cytokine Secretion Assay
PBMCs were plated in triplicate in RPMI + 10%FBS + pen/strep at 2×105 cells/well in 48 well plates and stimulated with 0, 0.1 or 1µg/mL MDP for 18h. Cell supernatants were assayed by Quantikine Human TNFα Immunoassay (R&D Systems, Minneapolis, MN) according to manufacturer’s instructions.
Microscopy
HCT116:LC3 cells were MDP-stimulated (10µg/mL) or rapamycin-stimulated (25µg/mL), fixed in methanol and mounted on slides using Vectashield plus DAPI (Vector Laboratories, Burlingame, CA). Samples visualized by confocal microscopy using a 40× objective lens on a Leica TCS-SP spectral laser scanning confocal microscope equipped with a Q-Imaging Retiga EXi cooled CCD camera and Image ProPlus Capture and Analysis software (Media Cybernetics, Silver Spring, MD). EGFP-LC3+-vesicles were scored in z-stack overlays from at least 4 separate fields. Autophagosome analysis was performed in an automated fashion using customized visual basic Image-Pro Plus macros. Transmission electron microscopy of HCT116:LC3 cells stimulated for 1h with MDP (10µg/mL) performed by the Cleveland Clinic Imaging Core Facility.
Statistical Analyses
Statistical significance between groups was determined by unpaired two-tailed Student t test. For multiple group comparisons, data were analyzed by the Quantitative Health Sciences Department (Cleveland Clinic) using natural logarithm transformation to achieve normality, and results summarized using mean and standard deviation or confidence limits for the mean. Repeated measure analysis of variance models were fit on the log-transformed responses, using ATG16L1 genotype, MDP dose, and their interaction as predictors. An autoregressive correlation structure was used to model the association within each subject across MDP dose, as was a genotype-specific compound symmetry correlation structure. Statistical analysis was performed using SAS software (version 9.1, Cary NC). Differences were considered significant when P<.05.
Results
The bacterial cell wall component, MDP, stimulates autophagy
We examined the role of autophagy in Nod2-mediated responses in both dendritic and epithelial cells, both of which play significant roles in the pathogenesis of CD1. Specifically, we explored whether the Nod2 ligand, MDP stimulates autophagy in primary human dendritic cells and the human colon epithelial cell line, HCT116. The recruitment of microtubule-associated protein 1 light chain 3 (LC3) from the cytoplasm to vesicle membranes has been defined as a specific marker of autophagosomes10. This process involves the modification of a soluble form of LC3 (LC3-I) to a lipidated form (LC3-II), which inserts into the forming autophagosomal membrane. This can be detected by a shift in molecular weight in an immunoblot or the localization of LC3 to vesicles by microscopy. We assessed autophagy induction by immunoblot and found that in both HCT116 and dendritic cells, MDP stimulation increased LC3-II levels within 30 minutes to 1 hour (Fig. 1A&B). This result was confirmed by the microscopic examination of HCT116 cells stably expressing EGFP-LC3 (HCT116:LC3 cells), which showed a modest, but reproducible increase (~2 fold) in the number of LC3-positive cells after MDP stimulation (Fig. 1C&D). Together, the results of these two assays show LC3-II accumulation preceding the formation of LC3-positive puncta. The formation of autophagosomes was further confirmed by electron microscopy in MDP-stimulated HCT116:LC3 cells (Fig 1E). This increase in autophagy was specific to the stimulatory form of MDP, as treatment of HCT116 cells with a non-stimulatory MDP isomer, MDP-LL, did not increase LC3-II levels (Fig. 1F).
Figure 1.

MDP stimulates autophagy in epithelial and dendritic cells. (A) Immunoblot of HCT116 cells treated with 10µg/mL MDP. LC3-II accumulation quantified by LC3-II/GAPDH ratio. p-p38, phosphorylated p38. (B) Assay performed as in A. with primary human dendritic cells. (C) Confocal micrographs of HCT116 expressing EGFP-LC3 (green) in media or MDP-stimulated (20µg/mL) for 1h. (D) Quantification of cells with punctuate EGFP-LC3, mean ±SD, n≥4 fields with >50 cells/field. (E) Electron micrograph of MDP-stimulated HCT116:LC3 cells (10µg/mL for 1h). Arrows indicate autophagosomes (F) Immunoblot of MDP-LL-stimulated (10µg/mL) HCT116 cells.
We assessed the effect of MDP stimulation on the killing of an intracellular pathogen, Salmonella typhimurium to measure autophagy in the context of an immune response. Salmonella is targeted by autophagy and has become a model organism to study the autophagic process16. We confirmed that Salmonella infection induces an autophagic response by increasing LC3-II accumulation in immunoblots of HCT116 cell lysates (Supplemental Fig. 1A). In addition, rapamycin stimulation of autophagy increased Salmonella killing in gentamycin protection assays, while reduction of ATG16L1 expression by RNA interference (RNAi) decreased bacterial clearance (Supplemental Figs. 1B, 1C & 2D). Using this system, we asked whether MDP-stimulated autophagy enhanced the clearance of intracellular Salmonella infection. MDP stimulation of HCT116 cells during infection significantly increased Salmonella killing (Fig. 2A). This anti-bacterial effect was specific to the stimulatory form of MDP, as treatment with MDP-LL had no effect. The MDP-enhancement of bacterial killing was autophagy-dependent, since RNAi knockdown of ATG16L1 expression abrogated the MDP response (Fig 2B). Similar results were observed in both primary human macrophages and dendritic cells infected with Salmonella (Fig. 2C). These results demonstrate that MDP stimulation of autophagy results in increased anti-bacterial activity in both primary immune cells, as well as in epithelial cells.
Figure 2.

MDP enhances Salmonella killing via an ATG16L1-dependent mechanism in gentamycin protection assays. (A) Infected HCT116 cells treated with media, MDP (10µg/mL), or MDP-LL (10µg/mL), mean ±SD. (B) HCT116 cells transfected with control or ATG16L1 RNAi constructs and infected ±MDP (10µg/mL), mean ±SD. (C) Assay performed as in A. using primary human macrophages or dendritic cells, mean ±SD.
Nod2 is necessary and sufficient for MDP-stimulated autophagic responses
With the generation of Nod2-deficient mice, it has been demonstrated that Nod2 mediates most of the cellular effects of MDP17. However, additional NLRs are activated by MDP, such as NLRP3, to activate caspase-1 and cause secretion of IL-1β and IL-1818. Therefore, we investigated the requirement of Nod2 in autophagic, anti-bacterial killing in response to MDP stimulation. We found MDP-enhanced Salmonella killing was blocked in HCT116 cells transfected with Nod2 RNAi (Fig. 3A), indicating that Nod2 expression is required for MDP-enhanced anti-bacterial activity.
Figure 3.

Nod2 is necessary and sufficient for MDP-enhanced Salmonella killing by autophagy. (A) HCT116 cells transfected with control or Nod2 RNAi and infected ±MDP (10µg/mL), mean ±SD. (B) Gentamycin protection assay performed with HEK293T cells transfected with indicated amounts of Nod2 plasmid, mean ±SD. (C) Control or RICK RNAi transfected HCT116 cells and infected ±MDP (10µg/mL), mean ±SD. (D) Gentamycin protection assay performed with HEK293T cells transfected with vector, Nod2 or NLRP3 expression plasmid and control or ATG16L1 RNAi construct, mean ±SD. (E) Autophagosomal maturation assessed by flow cytometric analysis of GFP-mCherry-LC3 in HEK293T cells transfected with vector, Nod2 or NLRP3 expression vectors. Histogram of GFP MFI of the mCherry-positive population (left). Quantification of GFP/mCherry MFI ratio normalized to vector transfected sample, mean ±SD (right, n=4).
Next, we investigated whether Nod2 activity is sufficient for an anti-bacterial response mediated by autophagy. For these experiments, we used HEK293T cells transiently transfected with NLR expression constructs. HEK293T cells do not express detectable levels of NLR or Toll-like receptor (TLR) proteins19 and therefore are commonly used to assess components of these microbial sensor pathways in isolation. In addition, NLR overexpression causes ligand-independent activation of these proteins19. We found that Nod2 overexpression significantly increased Salmonella killing in a dose-dependent manner (Fig. 3B). This Nod2-dependent increase was dependent on Nod2 signal transduction, as RNAi knockdown of the signaling adaptor RICK blocked Nod2-dependent anti-bacterial activity (Fig. 3C). A similar increase was not observed when NLRP3 was expressed, suggesting that, of the NLRs activated by MDP, this effect is specific to Nod2 (Fig. 3D). Autophagy is also required for this effect, since RNAi knockdown of ATG16L1 expression decreased the basal level of Salmonella killing, as well as blocked Nod2-enhanced killing (Fig. 3D).
These findings were further confirmed as an autophagy-dependent response by a separate autophagy assay which detects the maturation of autophagosomes. The LC3-positive autophagosome matures through fusion with lysosomes, providing an acidic environment promoting degradation of autophagosome contents. Autophagosomal maturation can be tracked by a GFP-mCherry-LC3 tandem tagged construct, as the acidic environment in the mature autophagolysome quenches the fluorescent signal of GFP, but not the mCherry fluorophore20. Traditionally, the maturation of dual-positive autophagosomes to single mCherry-positive autophagolysosomes is quantified by fluorescent confocal microscopy. We adapted this method for analysis by flow cytometry to increase the speed of analysis and decrease investigator bias. In response to starvation or rapamycin treatment, HEK293T cells expressing GFP-mCherry-LC3 demonstrated a significant decrease in GFP mean fluorescent intensity (MFI) without a similar change in mCherry fluorescence (Supplemental Fig. 3C–F). This change is reflected in a statistically significant decreased GFP/mCherry ratio over a series of independent experiments (Supplemental Fig. 3B). Using this system, we transfected HEK293T cells with GFP-mCherry-LC3 and either vector, Nod2 or NLRP3 expression constructs and detected a significant increase in autophagosomal maturation in the Nod2 expressing population (Fig. 3E). Expression of NLRP3 did not change the GFP/mCherry ratio (Fig. 3E). These results demonstrate that MDP-stimulated anti-bacterial responses are mediated by Nod2 signaling and autophagy. In addition, Nod2 is sufficient to induce these effects when activated in a ligand-independent manner.
CD-associated NOD2 variants are impaired in both pro-inflammatory signaling and autophagy induction
Next, we assessed the activation of autophagic responses by CD-associated NOD2 variants. The responses of HEK293T cells transfected with expression plasmids for the three major CD-associated Nod2 variants were compared to cells transfected with wild-type Nod2 or empty vector. Consistent with previous reports,5 the Nod2 variants R702W and G908R activated significantly less NF-κB activity than wild-type Nod2 in a luciferase reporter assay, while L1007fs was unable to activate the reporter (Fig. 4A). When these Nod2 mutants were tested for enhancement of Salmonella killing, a similar profile was seen. The anti-bacterial killing by Nod2 was significantly impaired by the R702W and G908R mutations and completely lost by the L1007fs mutant (Fig. 4B). A recent report described a role for the NF-κB activating kinases IKKα and IKKβ in autophagy activation21; therefore, we investigated whether these IKKs are required for Nod2 enhancement of bacterial killing. Dominant-negative IKKα and IKKβ were expressed in HEK293T cells to test their effect on Nod2-dependent functions. While both dominant negative IKKs blocked NF-κB activity stimulated by Nod2 overexpression (Fig. 4C), Nod2-enhanced bacterial killing was unaffected (Fig. 4D). These results indicate that CD-associated Nod2 mutations decrease pro-inflammatory NF-κB signaling, as well as NF-κB-independent anti-bacterial Nod2 function.
Figure 4.

CD-associated NOD2 mutants are impaired in signaling, anti-bacterial killing, and autophagy. (A) NF-κB luciferase reporter assay in HEK293T cells transfected with Nod2 plasmids and stimulated with MDP (1µg/mL) for 18h. Luciferase values normalized to β-gal transfection control (nLuc), mean ±SD. (B) Gentamycin protection assay of HEK293T cells transfected with Nod2 plasmids, mean ±SD. (C) NF-κB luciferase reporter assay in HEK293T cells transfected with Nod2 and IKK dominant negative (DN) constructs, mean ±SD. (D) Gentamycin protection assay of HEK293T cells transfected with Nod2 and IKK DN plasmids, mean ±SD. (E) Autophagosomal maturation assessed by flow cytometric analysis of GFP-mCherry-LC3 in HEK293T cells transfected with vector, Nod2 or Nod2 L1007fs plasmids. Histogram of GFP MFI of the mCherry-positive population (left). Quantification of GFP/mCherry MFI ratio normalized to vector transfected sample, mean ±SD (right, n=7). (F) LC3 immunoblots of HEK293T cells transfected with Nod2 or Nod2 L1007fs plasmids treated with MDP (1µg/mL) or rapamycin (25µg/mL) for the indicated times (hours). Quantification of LC3-II amount indicated by LC3-II/tubulin ratio (right).
Next, we examined whether the defect in Salmonella killing by CD-associated Nod2 mutants is due to an impairment of autophagy. We assessed maturation of autophagosomes by two separate assays. Overexpression of Nod2 in HEK293T cells, but not the L1007fs mutant, induced autophagosome maturation, as reflected by a significant decrease in the GFP/mCherry ratio (Fig. 4E). In addition, MDP-stimulated LC3-II accumulation in HEK293T cells transfected with low amounts of wild-type or L1007fs mutant Nod2 was assessed by immunoblot. Significant changes in LC3-II levels were detected in cells expressing wild-type Nod2 but not the L1007fs mutant (Fig. 4F). This was not due to a global autophagy defect in these cells, as rapamycin treatment activated autophagy in both conditions. These results demonstrate that MDP-stimulated autophagy is a Nod2-dependent anti-bacterial response which may be altered in individuals carrying specific NOD2 mutations.
Autophagy inhibition impairs activation of Nod2 signaling
MDP must be delivered to the cell cytosol to activate Nod2. MDP enters cells by a vesicular pathway and trafficks to an acidic compartment to stimulate Nod2 signaling17, 22, 23. Many of the vesicular characteristics of MDP-containing vesicles are shared with autophagosomes, leading us to investigate whether autophagy is involved in the intracellular trafficking of MDP and activation of Nod2 signaling.
We tested the contribution of autophagy to Nod2 signaling activation using NF-κB luciferase reporter assays in HCT116 cells. MDP activates both Nod2 and NLRP3; however, MDP stimulation of NF-κB signaling is a Nod2-dependent activity as demonstrated by previous studies17 and RNAi knockdown of Nod2 expression (Fig 5D). The first stage of autophagy is dependent on a PI3K complex containing Vps34 and Beclin-1, which causes the formation of autophagic isolation membranes (Fig. 5A). When autophagy initiation was inhibited by treatment with a PI3K inhibitor, 3-methyl adenine (3-MA) or expression of a kinase-dead Vps34 mutant, Nod2 signaling was dose-dependently inhibited (Fig. 5B & Supplemental Fig. 4). Similarly, Nod2-dependent NF-κB activation was decreased with RNAi knockdown of Beclin-1 expression (Fig. 5C). ATG16L1 assists in the recruitment of LC3 to autophagosomal membranes mediating the elongation and closure of autophagosomes. RNAi-mediated knockdown of ATG16L1 impaired MDP-stimulated NF-κB signaling (Fig. 5D). These findings demonstrate a novel, functional interaction between two CD risk genes, whereby ATG16L1 expression affects Nod2 function. In addition, when lysosomal fusion was inhibited by chloroquine treatment, a dose-dependent reduction in Nod2 signaling was observed (Fig. 5E). This result concurs with published data from mouse macrophages22, where bafilomycin A1 treatment inhibits Nod2 signaling. Our results demonstrate that inhibition of autophagy, at several distinct steps of the autophagy pathway, significantly decreases the activation of Nod2 signaling.
Figure 5.
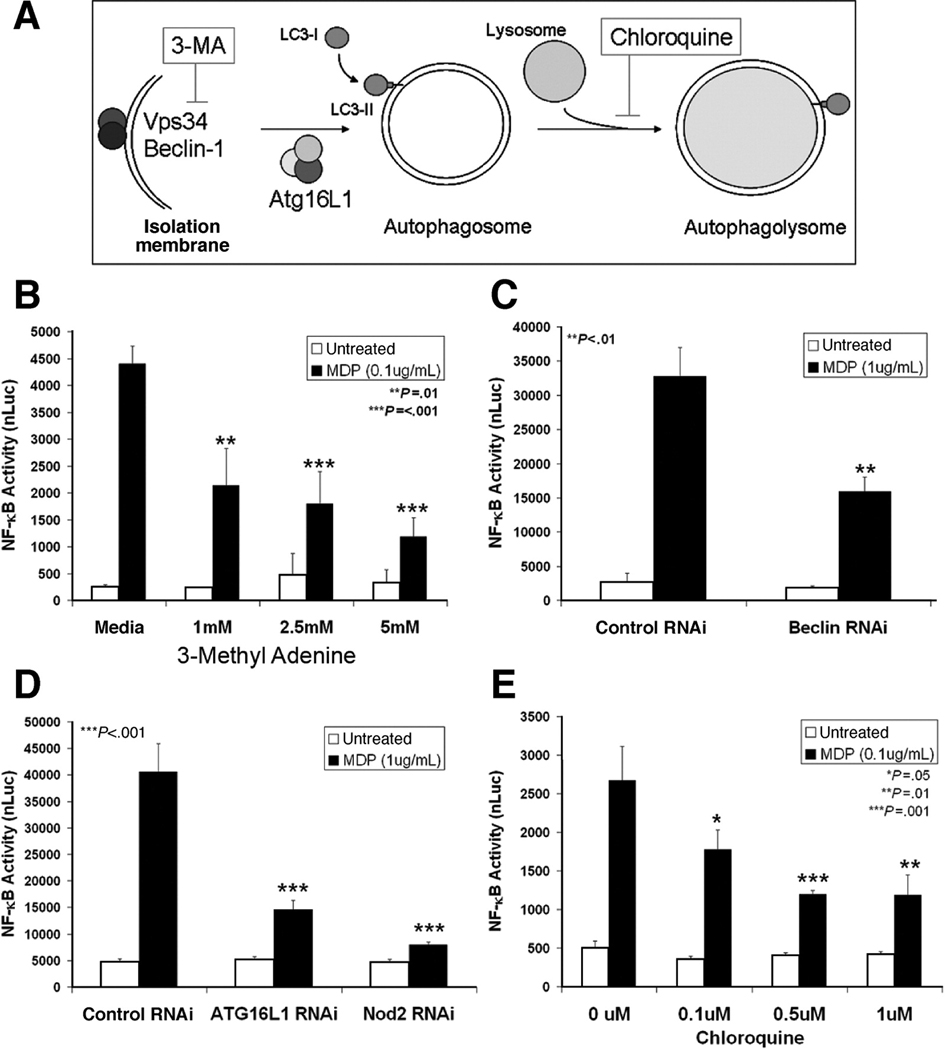
Inhibition of autophagy impairs activation of Nod2 signaling. NF-κB luciferase reporter assays were performed in HCT116 cells in the presence of autophagy inhibitors. Luciferase values normalized to β-gal transfection control (nLuc), mean ±SD. (A) Steps of autophagosome formation and maturation with key molecules and action of inhibitors indicated. (B) Cells stimulated with MDP and indicated amounts of 3-methyl adenine (3-MA). (C) Cells transfected with either control or Beclin-1 RNAi constructs. (D) Cells transfected with control, ATG16L1, or Nod2 RNAi constructs. (E) Cells stimulated with MDP and indicated amounts of chloroquine.
The CD-associated ATG16L1 T300A variant specifically impairs Nod2-dependent anti-bacterial functions in epithelial cells
The CD-associated variant of ATG16L1 T300A has specific reductions in bacteria-stimulated autophagy and not other forms of autophagy12, 24. As loss of ATG16L1 expression had dramatic effects on Nod2 function, we examined whether the CD-associated variant of ATG16L1 affected Nod2 function in both monocytic and epithelial cells.
We obtained monocytic cells from healthy donors genotyped for the ATG16L1 T300A variant to test the effect of this variant on Nod2-dependent responses. First, we assessed activation of Nod2 signaling by measuring TNFα secretion (a cytokine central to CD pathogenesis and a target for CD therapy1) and MDP-stimulated phosphorylation of signaling proteins. Studies suggest the T300A variant is a loss of function allele;12, 24 therefore, we combined individuals heterozygous for the T300A variant and wild-type individuals and compared their responses to T300A homozygous individuals. We found no statistically significant differences in TNFα secretion by MDP-stimulated PBMCs in the two groups (Fig. 6A). In addition, the timing and amplitude of Nod2 signaling in dendritic cells from these donors was not drastically different, as assessed by phosphorylation of p38 MAPK in immunoblots (Fig 6B). Autophagic responses were not significantly altered either, as there were no consistent differences in the timing or amount of LC3-II accumulated in MDP-stimulated dendritic cells from wild-type, heterozygous, or homozygous donors (Fig. 6B). Macrophages from all donor groups also had comparable mean basal Salmonella killing and MDP-stimulated bacterial killing was increased by a similar amount (30–40%, Fig. 6C). These results suggest that the CD-associated ATG16L1 T300A variant does not significantly affect Nod2 function in monocytic cells.
Figure 6.

ATG16L1 T300A impairs MDP-enhanced bacterial killing in a cell type specific manner. (A) TNFα secreted by MDP-stimulated PBMCs (0, 0.1 or 1µg/mL for 18h) with the indicated genotypes, median values of the group (n=6) indicated by bars. WT, wild-type ATG16L1; HT, heterozygous ATG16L1; T300A, homozygous ATG16L1 T300A. (B) Immunoblot analysis of MDP-stimulated, (10µg/mL), genotyped, dendritic cells. Autophagy induction quantified by LC3-II accumulation indicated by LC3-II/GAPDH ratio, with similar results seen by alternative quantification of LC3-I/LC3-II ratios. p-p38, phosphorylated p38. (C) Gentamycin protection assay of genotyped macrophages performed in media ± MDP (10µg/mL). Bars indicate the geometric mean values (WT, n=7; HT, n=7; T300A, n=14) with the 95% confidence limits shown by the boxes. The range of response is indicated by the vertical bars. (D) Gentamycin protection assay of HT29 or HT29GR cells performed in media ± MDP (20µg/mL), mean ±SD. (E) NF-κB luciferase reporter assay (left) and gentamycin protection assay (right) in HT29GR cells transfected with vector or Nod2 plasmid and treated with MDP (10µg/mL), mean ±SD. (F) Gentamycin protection assay of Nod2-expressing HT29GR cells transfected with vector, ATG16L1 or ATG16L1 T300A plasmids and control or ATG16L1 RNAi performed in media ± MDP (20µg/mL), mean ±SD.
These findings are in contrast to the results obtained from epithelial cells. We tested Nod2 function in human colon epithelial cell lines screened for ATG16L1 and NOD2 risk alleles. We compared two different derivatives of the HT29 cell line, one which has wild-type ATG16L1 and expresses Nod2 (“HT29”) and one with ATG16L1 T300A and does not express Nod2 (“HT29GR”). Initially, we compared the MDP-induced functions of these cells. As expected, in the absence of Nod2 expression, MDP treatment of HT29GR cells did not enhance Salmonella killing (Fig. 6D) or activation of a NF-κB luciferase reporter (Fig. 6E). When exogenous Nod2 is expressed by transfection in HT29GR cells, we saw a Nod2-dependent increase in basal NF-κB luciferase reporter expression and an MDP-stimulated enhancement of its expression (Fig. 6E). However, when Nod2-dependent anti-bacterial killing was measured in the same cells, we found no differences in the basal levels of Salmonella killing and no MDP-enhanced killing (Fig. 6E). To determine if the ATG16L1 T300A risk variant was responsible for this defect in bacterial killing, we knocked down expression of endogenous ATG16L1 T300A using RNAi targeting the 3’UTR and expressed wild-type ATG16L1 or mutant ATG16L1 T300A in its place by transfection of cDNA expression plasmids. We saw a restoration of MDP-stimulated anti-bacterial killing in cells which now expressed wild-type ATG16L1, while expression of the mutant ATG16L1 T300A was unable to complement the defect (Fig. 6F). These results indicate that the CD-associated ATG16L1 T300A risk variant causes a specific loss of Nod2 anti-bacterial function in colon epithelial cells.
Discussion
Great advances have been made in understanding the genetics of CD through the use of genome-wide association studies6–9. However, significant challenges remain to identify the functional consequences of disease-associated variants and how they contribute to disease pathogenesis. Instead of focusing on the effects of a single gene, we examined the functional interplay of CD risk genes to identify common pathways altered by specific genetic variants. Although it is of great interest to study these pathways in the context of the disease, the inherent variability of patient-derived cells (i.e. current inflammatory status, medications, disease subtype, etc.) and limited amounts of sample, make interpretation of results challenging in small sample sizes. Therefore, we examined the interplay of two CD risk genes in healthy individuals to reduce the complexity of our system and provide a basis for our future studies in patient-derived cells. Our findings demonstrate that ATG16L1 and NOD2 functionally interact in an autophagy-dependent, anti-bacterial pathway, which is altered by CD-associated variants in a cell-type specific manner.
Multiple links are evident between autophagy and microbial sensors. Autophagy has been demonstrated to be activated by TLR signaling, playing a positive role in the control of mycobacteria25–28. TLR-stimulated autophagy was shown to require signaling adaptor proteins, such as MyD88 and TRIF25, 27, 28. Related studies proposed the requirement for a TLR-independent pathway in autophagic killing of Salmonella29. While our studies were underway, it was demonstrated by two other groups that Nod2 activates autophagy to enhance intracellular bacterial killing30, 31. Cooney et al., found MDP-activated autophagy enhanced bacterial killing and MHC class II-dependent antigen presentation in primary human dendritic cells and that this process was dependent on ATG16L1, Nod2 and the signaling adaptor RICK, but not NLRP330. Another group demonstrated that Nod1 and Nod2 stimulate autophagy in a RICK and NF-κB-independent manner, possibly through ATG16L1 recruitment bacterial entry sites31. Our findings confirm these results and clarify discrepancies between these reports; supporting a role for Nod2-dependent signaling mediated by RICK, but not NF-κB in this process.
Autophagy is also implicated in the delivery of microbial ligands to intracellular compartments containing TLRs to stimulate an anti-microbial response. Autophagy is essential for the stimulation of TLR7 and TLR8 by viral ssRNA25, 32, as well as the delivery of co-stimulatory ligands to TLR933. Our results implicate autophagy in intracellular trafficking of MDP essential for Nod2-dependent signaling. However, it should be noted that autophagy inhibition did not completely ablate MDP-stimulated Nod2 signaling, suggesting a redundant mechanism in this process. This agrees with reports demonstrating multiple entry routes for MDP into cells, as well as cell-type specific mechanisms for uptake4. For example, a recent study demonstrates no difference in MDP-stimulated cytokine secretion between wild-type or ATG16L1 null mouse macrophages29, while our data in human epithelial cell lines show a dramatic reduction in MDP-stimulated NF-κB activity when ATG16L1 expression is knocked-down. Our results suggest autophagy is an additional, novel mechanism for MDP intracellular trafficking, which may contribute to a positive feedback loop during intracellular bacterial infection.
The ATG16L1 T300A risk variant is highly prevalent in healthy individuals (0.53)7. In our study of cells from healthy donors, the ATG16L1 T300A variant specifically impaired Nod2-dependent anti-bacterial function in colon epithelial cells, but not in monocytic cells. This is in contrast to recent reports demonstrating MDP-stimulated autophagy impairment in dendritic cells and lymphoblastoid cells from CD patients with the ATG16L1 T300A variant30, 31. This raises the intriguing possibility of other pathway modulators (environmental, microbial, genetic, etc.) contributing to pathway impairment and disease development. In addition, severe Paneth cell abnormalities were found in CD patients carrying the ATG16L1 T300A variant34 or NOD2 risk variants35. In mice, Nod2 deficiency results in increased intestinal bacterial load and greater susceptibility to pathogenic bacterial colonization36. This finding could result from either Paneth cell dysfunction, defects in intestinal autophagic responses, or more likely, a combination of the two. Our results suggest that in addition to Paneth cell defects, ATG16L1 and NOD2 risk variants alter intestinal epithelial cell anti-microbial responses, potentially altering the amount or composition of gut bacteria and increasing CD susceptibility.
Finally, we demonstrated that both ATG16L1 and NOD2 contribute to an autophagy-dependent, anti-bacterial pathway, which is altered in a cell-type specific manner by CD-associated mutations. Two additional CD risk genes (IRGM and NCF4) have obvious links to autophagy, making them candidates for inclusion in this pathway as well37. Future studies will need to examine the contribution of these and other CD risk genes to this autophagic, anti-microbial pathway. Examining the interplay of risk genes may lead to additional, more effective and individually tailored treatments for CD based on underlying disease mechanisms.
Supplementary Material
Acknowledgements
We thank all the members of the Cleveland Clinic’s Inflammatory Bowel Disease group, Laura Nagy and Derek Abbott for their constructive comments, Judy Drazba and Amit Vasanji for their imaging assistance, James Bena for help with statistical analysis, Keyonna Smith for recruiting subjects for this study, and all the individuals who donated blood. Thanks to Brian Rubin, Gabriel Nuñez, Gerhard Rogler, Derek Abbott, Tony Eissa, and William Maltese for reagents and Luigi Franchi for advice on Salmonella infection assays.
Grant support:
This work was supported by a Career Development Award from the Crohn’s & Colitis Foundation of America (C.M.) and National Institutes of Health research grants R01DK082437 (C.M.) and K23DK068112 (J.P.A.). These studies were also supported in part by the generosity of Gerald and Nancy Goldberg, Kenneth and Jennifer Rainin, and the National Institutes of Health National Center for Research Resources, CTSA 1UL1RR024989, Cleveland, Ohio.
Abbreviations
- AIEC
adherent invasive Escherichia coli
- ATG16L1
autophagy related 16-like protein 1
- CD
Crohn’s disease
- DC
dendritic cell
- IRGM
immunity-related GTPase family, M
- LC3
microtubule-associated protein 1 light chain 3
- 3-MA
3-methyl adenine
- MDP
muramyl dipeptide
- NLR
Nod-like receptor
- Nod2
nucleotide-binding, oligomerization domain 2
- RNAi
RNA interference
- TLR
Toll-like receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors disclose no conflicts of interest.
Author contributions:
CRH was involved in acquisition of data, analysis and interpretation of data, and critical revision of the manuscript. ALR and NAR were involved in acquisition of data, technical and material support. JPA provided technical and material support, critical revision of the manuscript, as well as obtained funding. CM was responsible for the study concept and design, acquisition, analysis and interpretation of data, drafting of the manuscript and obtained funding for these studies.
References
- 1.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 2.Rutgeerts P, Goboes K, Peeters M, et al. Effect of faecal stream diversion on recurrence of Crohn's disease in the neoterminal ileum. Lancet. 1991;338:771–774. doi: 10.1016/0140-6736(91)90663-a. [DOI] [PubMed] [Google Scholar]
- 3.Sartor RB. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics. Gastroenterology. 2004;126:1620–1633. doi: 10.1053/j.gastro.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 4.Inohara N, Chamaillard M, McDonald C, et al. NOD-LRR proteins: role in microbial-host interactions and inflammatory disease. Ann Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 5.Abraham C, Cho JH. Functional consequences of NOD2 (CARD15) mutations. Inflamm Bowel Dis. 2006;12:641–650. doi: 10.1097/01.MIB.0000225332.83861.5f. [DOI] [PubMed] [Google Scholar]
- 6.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 8.Massey DC, Parkes M. Common pathways in inflammatory diseases revealed by genomics. Gut. 2007 doi: 10.1136/gut.2007.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pineton de Chambrun G, Colombel JF, et al. Pathogenic agents in inflammatory bowel diseases. Curr Opin Gastroenterol. 2008;24:440–447. doi: 10.1097/MOG.0b013e3283023be5. [DOI] [PubMed] [Google Scholar]
- 12.Kuballa P, Huett A, Rioux JD, et al. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS One. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald C, Chen FF, Ollendorff V, et al. A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. JBC. 2005;280:40301–40309. doi: 10.1074/jbc.M508538200. [DOI] [PubMed] [Google Scholar]
- 14.Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 15.Debnath J. Detachment-induced autophagy in three-dimensional epithelial cell cultures. Methods Enzymol. 2009;452:423–439. doi: 10.1016/S0076-6879(08)03625-2. [DOI] [PubMed] [Google Scholar]
- 16.Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy. 2006;2:156–158. doi: 10.4161/auto.2825. [DOI] [PubMed] [Google Scholar]
- 17.Marina-Garcia N, Franchi L, Kim YG, et al. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via Cryopyrin/NLRP3 independently of Nod2. J Immunol. 2008;180:4050–4057. doi: 10.4049/jimmunol.180.6.4050. [DOI] [PubMed] [Google Scholar]
- 18.Martinon F, Agostini L, Meylan E, et al. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 19.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. JBC. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 20.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Criollo A, Senovilla L, Authier H, et al. The IKK complex contributes to the induction of autophagy. EMBO. 29:619–631. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marina-Garcia N, Franchi L, Kim YG, et al. Clathrin- and dynamin-dependent endocytic pathway regulates muramyl dipeptide internalization and NOD2 activation. J Immunol. 2009;182:4321–4327. doi: 10.4049/jimmunol.0802197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herskovits AA, Auerbuch V, Portnoy DA. Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS Pathog. 2007;3:e51. doi: 10.1371/journal.ppat.0030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lapaquette P, Glasser AL, Huett A, et al. Crohn's disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2009 doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delgado MA, Elmaoued RA, Davis AS, et al. Toll-like receptors control autophagy. EMBO. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 27.Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. JBC. 2008;283:33175–33182. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Y, Jagannath C, Liu XD, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 30.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 31.Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 32.Lee HK, Lund JM, Ramanathan B, et al. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 33.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petnicki-Ocwieja T, Hrncir T, Liu YJ, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. PNAS. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts RL, Hollis-Moffatt JE, Gearry RB, et al. Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population-based cohort. Genes Immun. 2008;9:561–565. doi: 10.1038/gene.2008.49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.