

Abstract
Background and Aims
Innate immune responses are crucial for host defense against pathogens, but need to be tightly regulated to prevent chronic inflammation. Initial characterization of mice with a targeted inactivating mutation in the p110d subunit of phosphoinositide 3-kinase (PI3K p110δD910A/D910A) reveal defects in B- and T-cell signaling and chronic colitis. Here, we further characterize features of inflammatory bowel diseases (IBD) in these mice and investigate underlying innate immune defects.
Methods
Colons and macrophages from PI3K p110δD910A/D910A mice were evaluated for colonic inflammation and innate immune dysfunction. Colonic p110δ mRNA expression was examined in IL-10−/− and wild type (WT) germ free (GF) mice during transition to a conventional microbiota. To assess polygenic impact on colitis development, p110δD910A/D910A mice were backcrossed to IL-10−/− mice.
Results
A mild spontaneous colitis was demonstrated in PI3K p110δD910A/D910A mice at 8 weeks with inflammation increasing with age. An inflammatory mucosal and systemic cytokine profile was characterized by expression of IL-12/23. In PI3K p110δD910A/D910A macrophages, augmented toll-like receptor signaling and defective bactericidal activity are observed. Consistent with an important homeostatic role for PI3K p110d, WT mice raised in a GF environment markedly upregulated colonic PI3K p110δ expression with the introduction of the enteric microbiota, however colitis-prone IL-10−/− mice do not. Moreover, PI3K p110δD910A/D910A mice crossed to IL-10−/− mice developed severe colitis at an early age.
Conclusions
This study describes a novel model of experimental colitis which highlights the importance of PI3K p110δ in maintaining mucosal homeostasis and could provide insight into the pathogenesis of human IBD.
Keywords: inflammatory bowel diseases, innate immunity, PI3-kinase, enteric microbiota
INTRODUCTION
The pathogenesis of the human inflammatory bowel diseases (IBD) Crohn’s disease (CD) and ulcerative colitis (UC) is complex, with abnormal immune responses in genetically susceptible individuals eliciting uncontrolled intestinal inflammation1. Genetic variants that confer CD susceptibility highlight the importance of innate immune interactions with the enteric microbiota in controlling inflammation1. Commensal and pathogenic bacteria are recognized through conserved molecular microbial patterns by pattern-recognition receptors (PRRs), of which toll-like receptors (TLRs) form integral components2. Signaling through TLRs leads to the activation of NF-kB, culminating in the induction of inflammatory cytokines including IL-12/23 and TNF. This inflammatory response is essential for the eradication of infectious microorganisms; however, excessive and prolonged activation can be detrimental to the host. Although mechanisms by which the host distinguishes commensal from pathogenic bacteria are not well defined, under normal conditions TLR signaling initiated by the enteric microbiota is protective3.
Phosphoinositide-3 kinases (PI3Ks) have emerged as important regulators of TLR signaling4, 5. Class IA PI3Ks are a family of heterodimeric enzymes consisting of a regulatory subunit (p85, p55 or p50) and a catalytic subunit (p110α, p110β, p110δ̣)6. While p110α and p110b are expressed ubiquitously, the p110δ̣isoform is highly expressed in leukocytes7. The clearest role of PI3K in chronic inflammation is described in a mouse harboring a point mutation in the p110δ̣catalytic subunit of PI3K (p110δD910A/D910A)8. These mice demonstrate B- and T-cell defects including improper maturation, defective antigen receptor signaling and impaired humoral immune responses. Notably, these mice spontaneously develop chronic segmental colonic inflammation.
However, effects of the PI3K p110δ̣subunit on innate immune responses in mucosal inflammation remain uncharacterized. In this study, we further describe the development of chronic IBD in PI3K p110δD910A/D910A mice and investigate the role of PI3K p110δ in the regulation of TLR signaling and bactericidal pathways in macrophages.
MATERIALS AND METHODS
Mice
PI3K p110δD910A/D910A mice were on the C57BL/6 background. C57BL/6 wild type and IL-10−/− mice were obtained from Jackson Laboratories. Mice were housed in specific pathogen free conditions (SPF) in accordance with guidelines from the American Association for Laboratory Animal Care and Research. Germ free (GF) 8-week-old 129 Sv/Ev WT and IL-10−/− mice were provided by the University of North Carolina Gnotobiotic Facility. Mice were colonized with SPF enteric microbiota at 8-weeks of age with a microbiota isolated from WT mice raised in SPF conditions9. PI3K p110δD910A/D910A and IL-10−/− homozygous mice were crossed and offspring were genotyped for PI3K p110δD910A/D910A and IL-10−/− mutations. For F2 breeding, mice homozygous for one mutation and heterozygous for the other mutation were bred and mice homozygous for both PI3K p110δD910A/D910A and IL-10−/− were identified. All experimental mice were genotyped by PCR screening prior to tissue collection and littermates used as controls. The Institutional Animal Care and Use Committee of the University of Pittsburgh and the University of North Carolina approved all methods used in this study.
Reagents
Flagellin was purchased from Invivogen (San Diego, Ca). CpG DNA was obtained from Integrated DNA Technologies (Coralville, IA). Synthetic bacterial lipoprotein (sBLP) was purchased from EMC Microcollections (Tübingen, Germany). Peptidoglycan (PGN) and Lipopolysaccharide (LPS) from Salmonella enteritidis was purchased from Sigma (St. Louis, Mo). LPS was repurified by modified phenol extraction as previously described10. GM-CSF, and M-CSF were obtained from Peptotech Inc (Rocky Hill, NJ) and IFN-g was purchased from R & D Systems (Minneapolis, MN).
Cell isolation
Splenocytes and bone marrow-derived macrophages (BMMs) were cultured as described11. Lamina propria mononuclear cells (LPMCs) were isolated from mouse colons by an enzymatic method as previously described12. LPMCs were separated into CD11b+ cells using anti-CD11b microbeads (Miltenyi Biotec, Auburn, CA).
ELISAs
Murine IL-12 p40, IL-12 p70, IL-10, IFN-γ and TNF immunoassay kits (R & D Systems) and IL-23 (eBioscience) were used according to manufacturers’ instructions. IL-17, MIP1a, RANTES, KC and G-CSF levels were determined by multiplex ELISA (Luminex). Phosphorylation levels of pAKT was determined using a cell-based ELISA (Superarray Bioscience Corporation).
Western blot
Western blot analyses were performed on whole cell extracts as described11. Antibodies to phospho (p)-JNK, p-p38, p-ERK, JNK2, p38, ERK, NF-kB p65, and PI3K p110δ were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), p-p65 was obtained from Cell Signaling (Danvers, MA 01923)
Nitrite Determination
Nitrite was assayed by a standard Greiss Reaction adapted to a microplate system.
Real-time RT PCR analysis
Quantitative real-time RT-PCR was performed as described13. Primer sequences are available on request.
Colonic tissue explant cultures and histology
Colonic explant cultures were performed as described previously13. Slides were prepared for hematoxylin and eosin staining and histologic analysis was performed by a pathologist blinded to the study groups (A.R.S.) using established criteria for IL-10−/− mice (and PI3K p110δD910A/D910A/ IL-10−/− mice)13.
Histologic scoring of PI3K p110δD910A/D910A mice
A histological scoring system was developed to assess colonic inflammation based on the characteristic of this model. The criteria used to classify histology into grades 0 to 4 were as follows: Grade 0 was defined as (a) presence of 1 or less mitosis in the colonic crypts per 10 high power fields (HPF), (b) no epithelial hyperplasia, and (c) no neutrophils in the lamina propria. Grade 1 was established if less than two of the following criteria were found: (a) presence of epithelial hyperplasia, (b) presence of more than 2 mitosis/10 HPF in the colonic crypts, (c) any apoptotic body in the colonic crypts, (d) Infiltration by neutrophils in the lamina propria, (e) infiltration by lymphocytes and/or plasma cells in the lamina propria, (d) infiltration by lymphocytes and/or plasma cells and neutrophils in the lamina propria, (g) less than 30% of colonic crypts showing intraepithelial lymphocytes (IELs). If there were two or more of the criteria for grade 1, grade 2 was attributed. Grade 3 was defined as (a) any of the criteria for grade 2 was present and there were more than 30% of IELs involving the colonic crypts per 10 HPF, (b) or there was submucosal inflammation. Grade 4 was attributed when any of the criteria for grade 3 was identified together with the presence of crypt abscesses and/or mucosal ulcers. Histopathologic analysis of WT mice revealed no or minimal mucosal inflammation with one or less mitosis per 10 HPF, and was scored as grade 0 in the majority of mice, or 1.
Gentamicin Protection Assay
Bacterial invasion was measured by gentamicin protection assays14. Briefly, BMMs were infected with bacteria with a multiplicity of infection of 10 bacteria per cell in antibiotic free media. Cells were incubated with the bacteria for 2 hours at 37°C with 5% CO2. LPS (100 ng/mL) and IFN-g (10 ng/ml) were added 2 hours prior to bacteria where indicated. Cells were then washed twice with PBS and fresh media containing 100 mg/ml of gentamicin was added for an hour. Cells were lysed with 1% Triton X and samples were diluted and plated on LB agar plates to determine the number of colony-forming units.
Bacterial DNA Isolation
Total DNA was extracted from splenocytes and mesenteric lymph nodes as outlined in Frank et al15. Universal bacteria primer sequences were obtained from Horz, et al16. Bacterial DNA expression was determined by real-time PCR and expression was normalized to host GAPDH and represented as relative expression to control.
Statistical Analysis
Statistical significance from experiments in cells was determined using student t-test or one-way ANOVA. Statistical significance for in vivo data was assessed by the Mann-Whitney U test (SPSS, Chicago, IL, USA) with Bonferroni correction.
RESULTS
PI3K p110δD910A/D910A mice develop chronic colitis
Macroscopically, colons from 16-week-old PI3K p110δD910A/D910A mice were shorter in length and thicker than those from wild type (WT) with ten percent of mutant mice developing rectal prolapse (data not shown). To characterize histological features and progression of colitis, colonic sections were examined from 6 to 45 weeks of age (Figure 1A). A histological scoring system was developed based on features of this model to assess the severity of inflammation (see Materials and Methods). Colitis was characterized by increased colonic epithelial apoptotic bodies and a marked increase in the number of mitoses in the colonic crypts. There was an increase in lamina propria lymphocytes and neutrophils (Figure 1C, left). The colonic crypt architecture was generally well preserved with focal disruption of the tubular architecture associated with crypt abscesses (Figure 2C, middle black arrow). Histologic inflammation was detected starting at 8 weeks of age (Figure 1B). The percentage of fields demonstrating no histological inflammation (grade 0) significantly decreased and the percentage of fields with marked inflammation (grade 3 to 4) significantly increased (Figure 1A & B) with age. A reduction in goblet cells was observed in older mice (Figure 1C, right). A unique feature was the presence of numerous intraepithelial lymphocytes (IELs) in the colonic epithelium (Figure 1C, right, white arrows). Immunohistochemical analysis revealed the presence of numerous CD3+ IELs in the colonic crypts of mutant mice compared to WT mice (Supplemental Figure 1).
Figure 1. PI3K p110δD910A/D910A mice develop colitis.
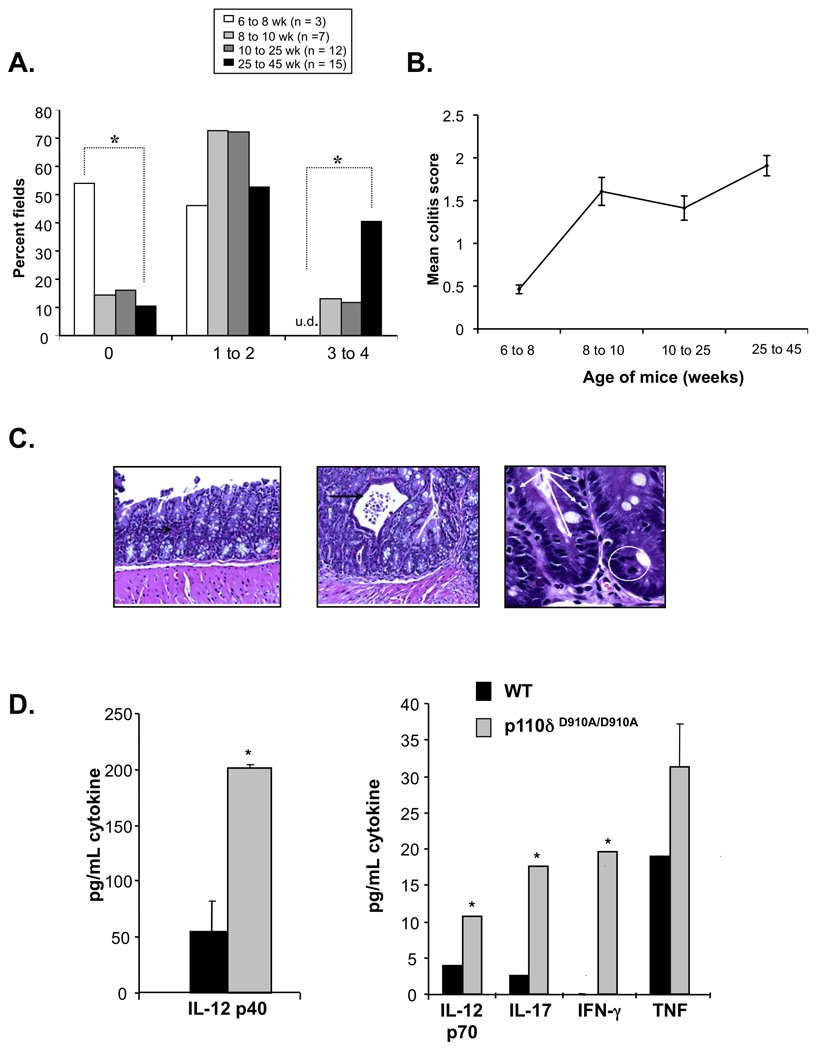
(A and B) Histological scores of colonic sections from WT and PI3K p110δD910A/D910A mice at different ages. Results are represented as percentage of microscopic fields in each age group with score 0, 1 to 2 or 3 to 4 (A); or mean colitis scores (B). (*p<0.05 v. 25–45 WT week old mice) (C) Colonic sections from 10 week old PI3K p110dD910A/D910A mice demonstrate leukocytic infiltration of the lamina propria (white circle) and intraepithelial lymphocytes (white arrows) in the crypts. Focal crypt abscesses were observed (black arrow). (D) Colonic explants from WT (black bars) and PI3K p110δD910A/D910A (grey bars) mice were assayed for spontaneous secretion of cytokines. Error bars represent mean+SEM of 3 independent experiments.
Figure 2. PI3K p110δD910A/D910A mice display enhanced expression of IL-12 p40.
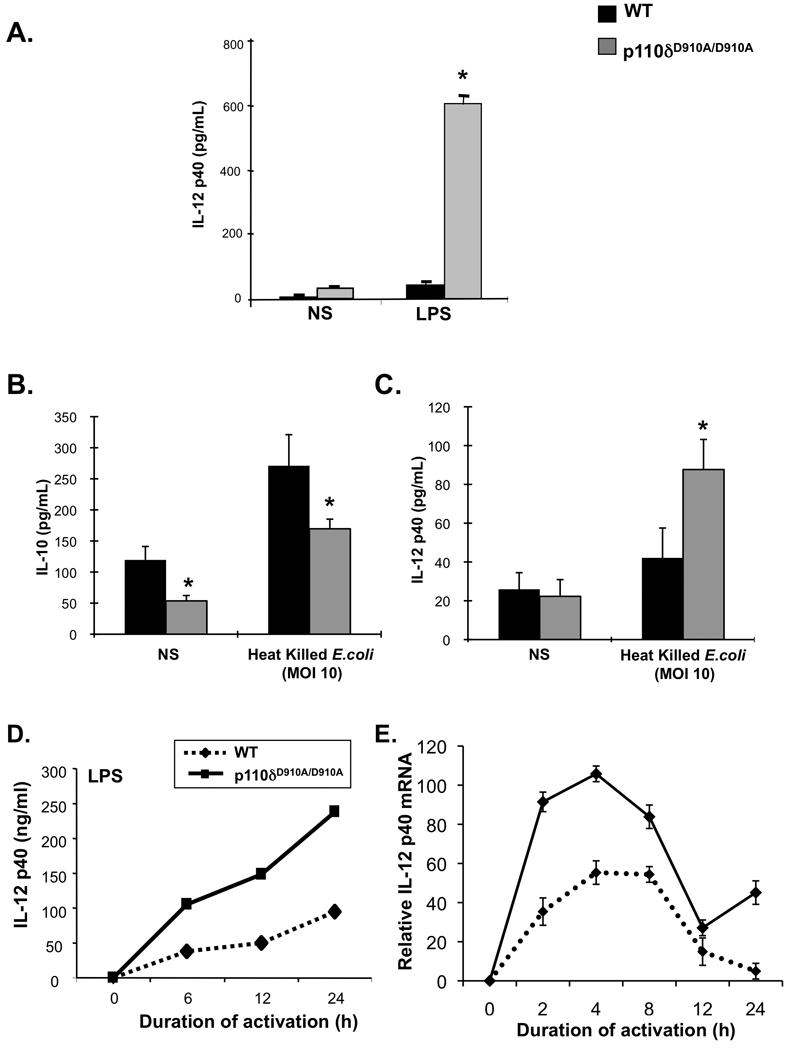
(A) Splenocytes from WT (black bars) and PI3K p110δD910A/D910A (grey bars) mice were untreated (un) or stimulated with LPS (1 µg/ml) alone or LPS and IFN-γ (10 ng/ml) for 24 hours. IL-12 p40 was measured by ELISA. (B and C) Colonic macrophages from WT or PI3K p110dD910A/D910A mice were stimulated with heat killed E. coli (mode of multiplicity 10) for 24 hours. ELISAs were performed to assess IL-12 p40 (B) and IL-10 (C) levels. (D and E) Bone-marrow derived macrophages (BMMs) from WT (black bars) and PI3K p110δD910A/D910A (grey bars) mice were unstimulated (Un.) or stimulated with LPS (1 µg/ml) and supernatants analyzed for IL-12 p40 (D). PI3K p110dD910A/D910A BMMs were harvested at each time point and IL-12 p40 (Il12b) mRNA (E) levels were assessed by real-time RT-PCR. Results are expressed as fold induction normalized to β-actin. Error bars represent mean+SEM of 3 independent experiments (*p<0.05 v. WT cells).
PI3K p110δD910A/D910A mice display an exaggerated mucosal and systemic Th1/ Th17 cytokine profile
Colonic explants from p110δD910A/D910A mice secreted significantly elevated amounts of inflammatory cytokines IL-12 p40, IL-12 p70, TNF, IFN-g and IL-17 (Figure 1D), as well as the growth factors and chemokines G-CSF, MIP1a, RANTES and KC (Supplemental Figure 2). LPS-stimulated PI3K p110δD910A/D910A splenocytes secreted elevated levels of IL-12 p40 (Figure 2A) and TNF (Supplemental Figure 3) compared to WT splenocytes.
CD11b+ lamina propria mononuclear cells (LPMC), comprising macrophages and dendritic cells, were isolated from colons of p110δD910A/D910A and WT mice. PI3K p110δD910A/D910A CD11b+ colonic LPMCs produced lower basal levels of IL-10 relative to WT LPMCs (Figure 2B). Moreover, p110δD910A/D910A CD11b+ LPMCs activated with heat killed E. coli expressed increased levels of IL-12 p40 and decreased levels of IL-10 compared to wild type CD11b+ cells (Figure 2 B&C). Furthermore, CD11b+ LPMC from PI3K p110δD910A/D910A mice demonstrated upregulation of numerous activation markers, TLR4, and CD14 (Supplemental Figure 4B) compared to WT CD11b+ LPMC, consistent with in vivo activation and/or recruitment of macrophages during the development of colitis.
PI3K p110δD910A/D910A macrophages are hyperresponsive to TLR signaling
The role of PI3K p110δ̣in the regulation of IL-12 p40 gene expression was next studied as a biologically relevant target of TLR signaling in macrophages. BMMs from PI3K p110δD910A/D910A mice secreted significantly greater amounts of IL-12 p40 protein with LPS stimulation compared to WT BMMs (Figure 2D). Although the kinetics of IL-12 p40 induction were similar between WT and p110δD910A/D910A BMMs, the magnitude of induction at each time point was significantly greater in the latter. PI3K p110δD910A/D910A and WT BMMs revealed similar kinetics of IL-12 p40 (Il12b) mRNA expression that peaked 4 hours post-stimulation and significantly attenuated by 12 hours. However, there was increased magnitude of expression at all time points until 12 hours in p110δD910A/D910A BMMs (Figure 2E).
Next, BMMs from WT and p110δD910A/D910A mice were stimulated with TLR9 (CpG), TLR2 (synthetic bacterial lipoprotein-sBLP) or TLR5 (flagellin) ligands. IL-12 p40, IL-12 p70, IL-23 and nitric oxide (NO) production were assessed. PI3K p110δD910A/D910A BMMs produced enhanced amounts of inflammatory cytokines and nitric oxide in response to multiple TLR ligands (Figure 3). There were no differences in cell surface phenotypic or activation marker expression between PI3K p110δD910A/D910A and WT BMMs, including TLR4 and CD14 (Supplemental Figure 4A), suggesting that augmented TLR signaling in PI3K p110δD910A/D910A BMMs is secondary to intrinsic defects in TLR signaling pathways and not a result of a heightened activation state or increased expression of TLRs or TLR co-receptors.
Figure 3. PI3K p110δD910A/D910A macrophages demonstrate heightened sensitivity to TLR stimulation.
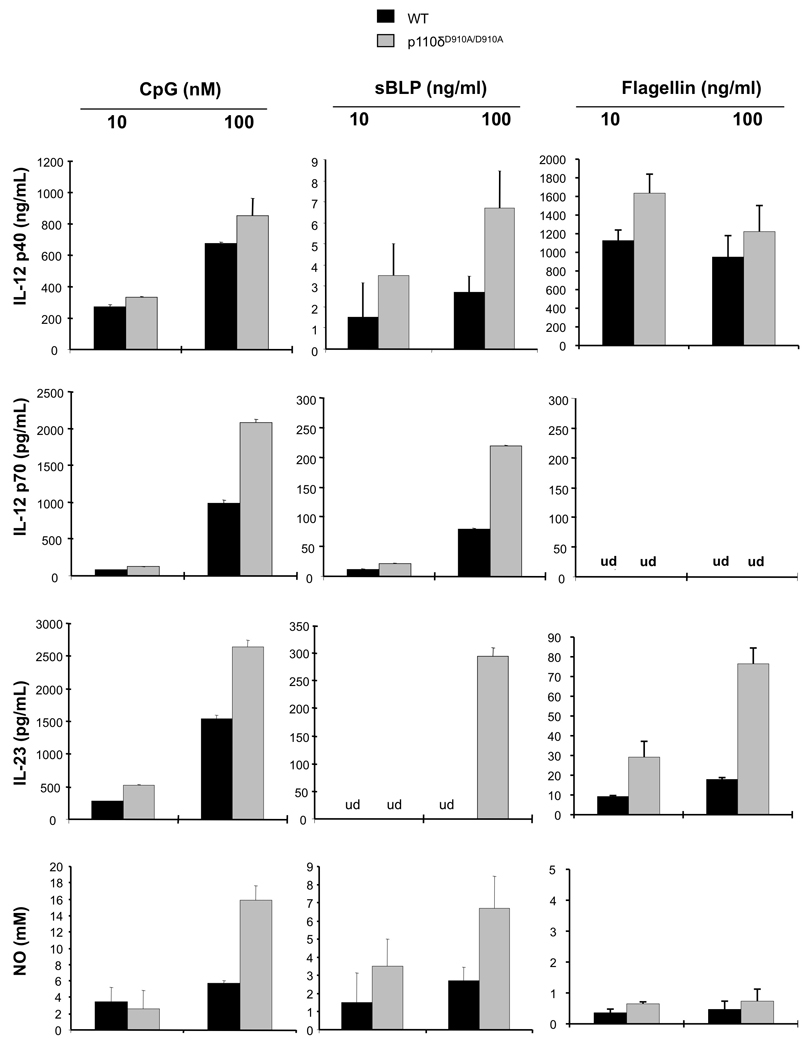
BMMs from WT (black bars) and PI3K p110δD910A/D910A (grey bars) mice were stimulated with TLR9 (CpG), TLR2 (sBLP) or TLR5 (Flagellin) ligands for 24 hours. Supernatants were analyzed for IL-12 p40, IL-12 p70 or IL-23 secretion by ELISA and nitric oxide secretion by Greiss reaction. Error bars represent mean+SEM of 3 independent experiments.
PI3K p110δD910A/D910A macrophages display enhanced MAPK activation
PI3K signaling is significantly diminished in PI3K p110δD910A/D910A BMMs as demonstrated by decreased phosphorylation of the PI3K downstream target Akt in LPS or sBLP activated PI3K p110δD910A/D910A BMMs compared to WT BMMs (Figure 4A). TLR signaling in macrophages is positively regulated by the MAP kinases p38 and JNK17, 18; and negatively regulated by the ERK MAP kinase pathway18. LPS-activated p110δD910A/D910A BMMs displayed a different kinetic pattern of JNK and p38 MAP kinase activation compared to WT macrophages (Figure 4B), with earlier activation and enhanced phosphorylation of p38 MAP kinase. There were no significant differences in ERK activation between p110δD910A/D910A and WT BMMs. Likewise, there was no difference in magnitude or kinetics of NF-kB p65 phosphorylation in LPS-stimulated WT and PI3K p110δD910A/D910A BMMs (Supplemental Figure 5A).
Figure 4. PI3K p110δD910A/D910A macrophages demonstrate altered kinetics and magnitude of MAPK activation.

(A) BMMs from WT or PI3K p110δD910A/D910A mice were stimulated with (right) LPS (100 ng/ml) or (left) sBLP (100 ng/ml) for the indicated periods of time and phosphorylation of Akt (p-Akt) was assayed by ELISA. Results are presented as a ratio of p-Akt to total Akt. (B) BMMs from WT and PI3K p110δD910A/D910A mice were stimulated with LPS (1 µg/ml) for the indicated times. Whole cell extracts were analyzed for phosphorylation of MAPK (JNK, ERK, p38) by Western blot. Results represent mean+SEM of 3 independent experiments (*p<0.05 vs WT).
PI3K p110δD910A/D910A macrophages demonstrate decreased bactericidal activity
To determine whether PI3K p110dD910A/D910A macrophages are defective in eradicating intracellular bacteria, gentamicin protection assays were performed with the commensal enteric bacteria K12 E. coli, NC101 E. coli, and the invasive enteric organism S. typhimurium. NC101 E. coli is a colitogenic bacterial strain isolated from IL-10−/− mice19. PI3K p110δD910A/D910A BMMs display decreased bactericidal activity when infected with K12 E. coli, NC101 E. coli, and S. typhimurium (Figure 5A, right). Moreover, bacterial colonies recovered 1 hour following infection were not significantly different from WT BMMs (Figure 5A, left). Additionally, WT and PI3K p110δD910A/D910A BMMs were infected for 1 hour with K12 E. coli, washed and permeabilized, and then immunostained with anti-E. coli LPS antibodies. No immunoreactivity was demonstrated in non-permeabilized cells, and wild type and PI3K p110δD910A/D910A BMMs demonstrated similar numbers of intracellular bacteria, demonstrating that uptake/phagocytosis is not defective in PI3K p110δD910A/D910A BMMs (Supplemental Figure 6). Culture supernatants showed a marked increase in IL-12 p40 protein in PI3K p110δD910A/D910A BMMs that inversely correlated with bactericidal activity (Figure 5B).
Figure 5. PI3K p110δD910A/D910A BMMs demonstrate defective bactericidal activity.
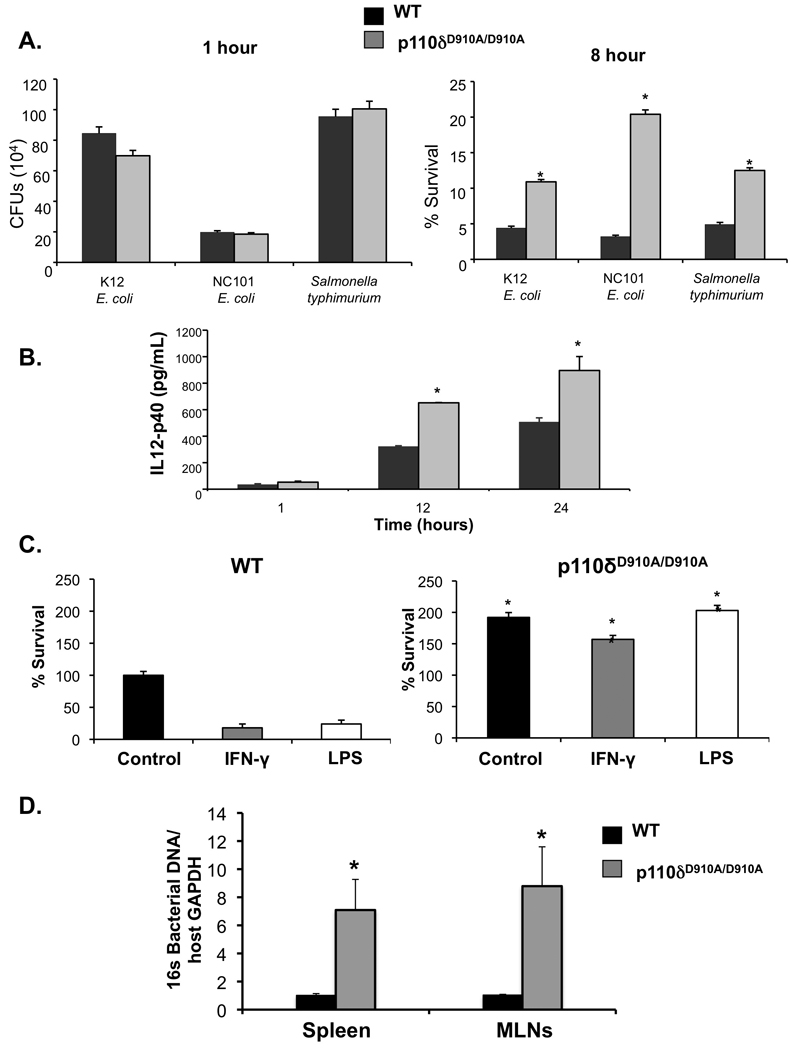
(A) WT and p110dD910A/D910A BMMs were cultured with K12 E. coli. NC101 E. coli or S. typhimurium. No significant bacterial recoveries were seen 1-hour post-bacterial infection (left panel). P13K p110δD910A/D910A BMMs demonstrate decreased bactericidal activity relative to wild type BMMs at 8 hours post-infection (right panel) (*p< 0.05 v WT BMMs). (B) IL-12 p40 production by ELISA was assessed in P13K p110dD910A/D910A BMMs infected with K12 E. coli. (C) BMMs were treated with LPS (100 ng/mL) or IFN-γ (10 ng/mL) prior to bacterial infection, and bacteria recovered from lysed cells 8 hours post-infection (*p< 0.05 vs WT BMMs). (D) Total bacterial DNA in spleen and mesenteric lymph nodes were detected by real-time PCR using primers for total 16S rRNA genes Primers for GAPDH were used to show loading control of host genomic DNA. Error bars represent mean+SEM of 3 independent experiments (*p < 0.05 vs. WT).
Bacterial products such as LPS and inflammatory cytokines like IFN-g activate macrophages and augment bactericidal responses20, 21. Bactericidal activity was significantly enhanced in WT BMMs treated with LPS and IFN-γ̃ This augmentation was completely absent in PI3K p110δD910A/D910A BMMs (Figure 5C).
To obtain in vivo evidence of defective enteric bacterial clearance in PI3K p110δD910A/D910A mice, the presence of bacterial DNA in WT and p110δD910A/D910A spleens and mesenteric lymph nodes (MLNs) was determined using universal bacterial 16S ribosomal RNA (rRNA) gene primers. Markedly increased bacterial rDNA was detected in spleens and MLNs of PI3K p110δD910A/D910A mice compared to WT mice, consistent with defective bacterial clearance and/or increased bacterial translocation (Figure 5D).
The enteric microbiota induce colonic PI3K p110δ expression in WT but not in colitisprone IL-10−/− mice
Colonic expression of PI3K p110δ was studied in WT and colitis-prone IL-10−/− mice raised in a GF environment and then colonized with a specific pathogen free microbiota. PI3K p110δ mRNA (Figure 6A) expression increased in WT mice beginning 7 days following colonization and was most strongly upregulated 14 days following transition. This increase was specific for the p110d isoform, as colonic mRNA expression for the p55a and p85 subunits were not significantly altered (Figure 6B). This robust increase in colonic p110MRNA was not observed in GF IL-10−/− mice transitioned to a conventional microbiota (Figure 6A). These results, in a well-established model of experimental colitis, support the hypothesis that PI3K p110 is an important homeostatic pathway limiting the extent and duration of intestinal inflammation.
Figure 6. The enteric microbiota induces colonic PI3K p110δ expression in WT but not colitis-prone IL-10−/− mice.
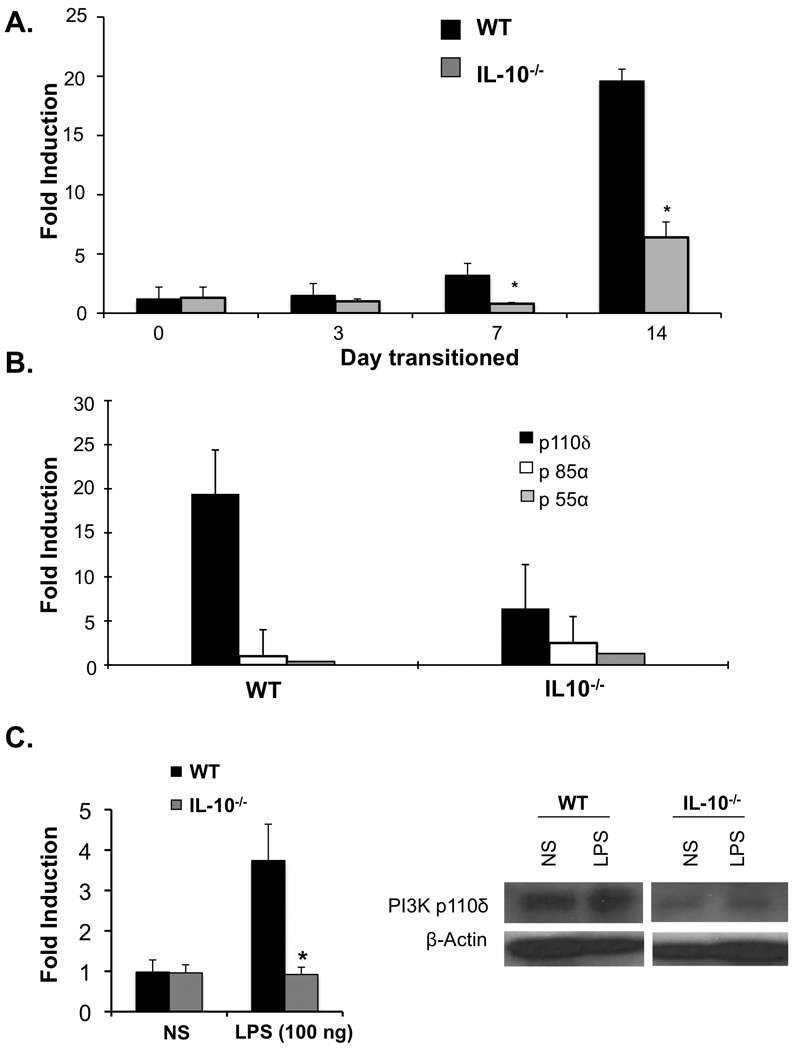
Germ-free (GF) WT and IL-10−/− mice were transitioned to a specific pathogen free (SPF) microbiota. Colonic mRNA was isolated and expression of PI3K p110δ, p85a, and p55a mRNA was assessed by real-time RT-PCR. (A) Colonic PI3K p110d was determined in WT and IL-10−/− mice at 0, 3, 7, and 14 days post colonization of GF mice with SPF microbiota (*p < 0.05 vs WT). (B) Colonic expression of PI3K p110δ, p85a, and p55a mRNA was examined 14 days post-transition of GF mice to SPF microbiota. Results are expressed as fold induction normalized to β-actin. Error bars represent mean+SEM of 3 independent experiments. (C) BMMs from WT and IL-10−/− mice were stimulated with LPS (100 ng) for the indicated times. PI3K p110δ mRNA levels were assessed by real-time RT-PCR (left panel). Results are expressed as fold induction normalized to β-actin and represent mean+SEM of 3 independent experiments (*p<0.05 vs WT). BMMs from WT and IL-10−/− mice were stimulated with LPS (100 ng) for 16 hours. Whole cell extracts were analyzed for PI3K p110δ by Western blot (right panel). Results are representative of three independent experiments.
Additionally, this result suggested that IL-10 might be an important cofactor for expression of p110δ by enteric bacteria. Therefore, induction of p110δ was assessed in LPS activated WT and IL-10−/− BMMs. LPS-activated IL-10−/− BMMs demonstrated decreased PI3K p110δD910A/D910A mRNA (Figure 6C, left) and protein expression compared to WT BMMs (Figure 6C, right). Moreover, PI3K p110δ mRNA induction in LPS-activated BMMs is through a MyD88 dependent pathways as significantly less p110δ mRNA and protein was observed in MyD88−/− BMMS compared to WT BMMS (Supplemental Figure 5B), confirming that PI3K p110δ induction is through the canonical TLR signaling pathway.
IL -10/PI3K p110δD910A/D910A mice exhibit severe colitis at an early age
The phenotype of murine and human IBDs are influenced by polygenic contributions. In PI3K p110dD910A/D910A and IL-10−/− mice on the C57BL/6 background, the phenotype of colitis is relatively mild. Moreover, partial but not complete abrogation of colonic PI3K p110δ expression was observed in GF IL-10−/− mice transitioned to a microbiota (Figure 6A). Therefore, to address whether a combined genetic defect in p110δ and IL-10 alters the phenotype of colitis, IL-10−/− mice were crossed with PI3K p110δD910A/D910A to create IL-10−/−/PI3K p110δD910A/D910A mice. While 4 week old PI3K p110δD910A/D910A and IL-10−/− mice do not demonstrate colonic inflammation, IL-10−/−/PI3K p110δD910A/D910A mice developed severe colitis and were notably smaller in size. Gross colonic appearance showed 100% disease penetrance with all mice developing colitis by 4 weeks of age and over 50% displaying severe inflammatory changes (Supplemental Figure 7). Colitis scores from IL-10−/−/PI3K p110δD910A/D910A mice were significantly higher than age-matched PI3K p110δD910A/D910A, IL-10−/−, and WT mice (Figure 7A). Intestinal explant cultures demonstrated increased secretion of IL-12 p40, IL-12 p70 and IL-23 in from IL-10−/−/PI3K p110δD910A/D910A mice compared to age-matched IL-10−/− and PI3K p110δD910A/D910A mice (Figure 7B–D).
Figure 7. IL -10−/− PI3K p110δD910A/D910A mice exhibit severe colitis at an early age.
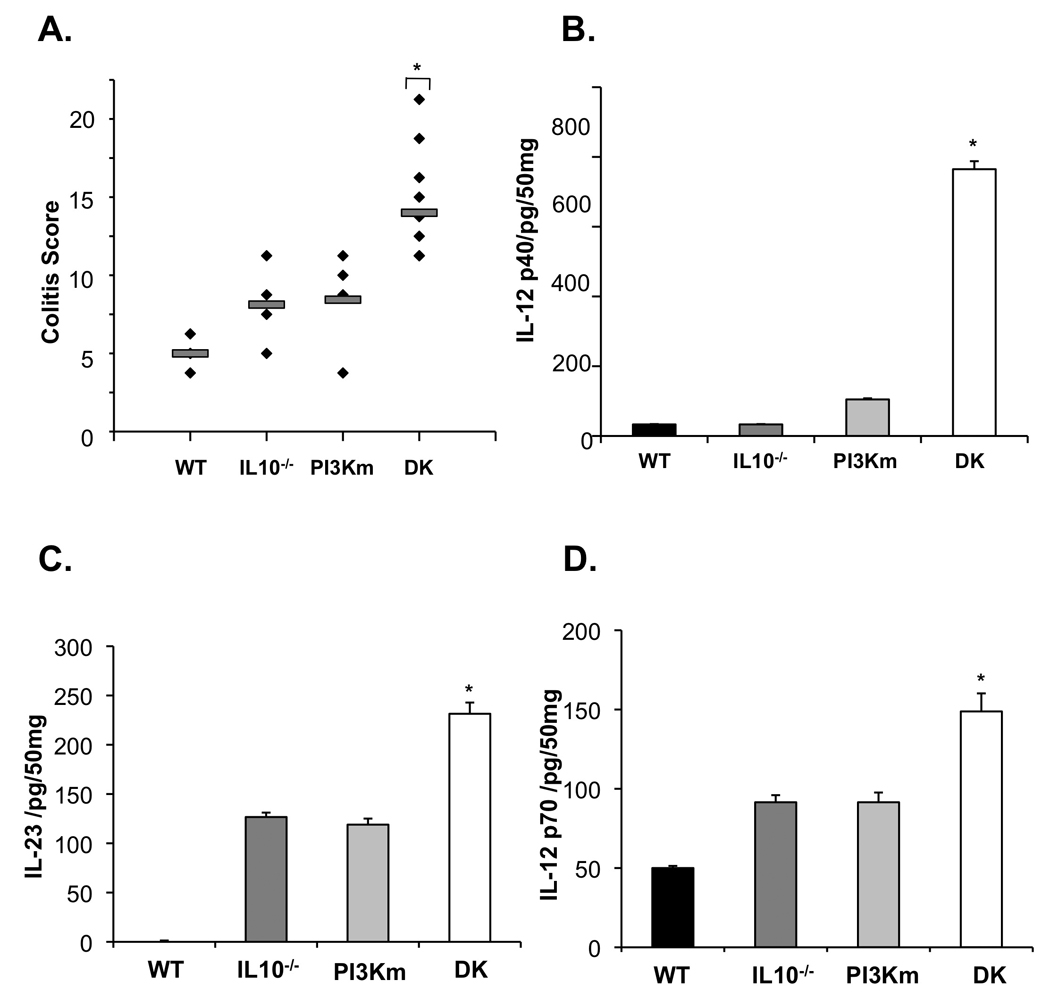
(A) Colitis scores were determined for 4-week-old IL-10−/−/PI3K p110δD910A/D910A (DK), WT, IL-10−/−, and PI3K p110δD910A/D910A (PI3Km) mice using criteria established for IL-10−/− mice13 by a pathologist blinded to genotype (*p < 0.05 vs WT). IL-12 p40 (C), IL-12 p70 (E) and IL-23 (D) protein in supernatants from colon explant cultures from IL-10−/−/PI3K p110δD910A/D910A (DK), WT, IL-10−/−, and PI3K p11δD910A/D910A (PI3Km) mice were analyzed by ELISA. Error bars represent mean+SEM of 3 independent experiments (*p< 0.05 vs. WT).
DISCUSSION
This study describes an important role for the activity of the PI3K p110δ isoform in the regulation of TLR signaling and bactericidal pathways in macrophages. Notably, PI3K p110δ̣plays a critical role in intestinal homeostasis in experimental colitis models.
Class IA PI3Ks are heterodimers consisting of a catalytic subunit (p110α, p110β, p110δ) which complexes with one of five regulatory units (p85 isoforms). While p110α and p110β are ubiquitously expressed, p110δ expression is low or absent in most cell types but is abundantly expressed in leukocytes22. PI3K p110δD910A/D910A mice have significant defects in B cell antigen receptor signaling, substantial declines in immunoglobulin levels, as well as diminished numbers of immature and mature B cells. Interestingly, in B cells, p110δ regulates TLR-induced proliferation23. PI3K p110δ also suppresses TLR9-induced IL-12 production in B cells, inhibiting a Th1-skewed response24. T cell functional abnormalities have been described in PI3K p110δD910A/D910A mice, including defects in T cell signaling through the T cell receptor, and defective T CD4+/CD25+/Foxp3+ T regulatory cell function, recently demonstrated in an adoptive transfer model of colitis25.
We provide the first detailed characterization of spontaneously occurring colitis in PI3K p110δD910A/D910A mice. Immunologically, an exuberant inflammatory Th1 and Th17 cytokine profile was observed in the colon and systemically. Several characteristics of colitis are reminiscent of features of human IBD, including leukocytic and neutrophilic infiltrates, intestinal epithelial cell damage, and goblet cell depletion. Although older mice had more significant histologic inflammation, the majority of mice demonstrated colonic inflammatory changes that were not severe in nature. However, genetic background is an important modifier of phenotype in murine experimental colitis and human IBD. For instance, IL-10−/− mice on the C57BL/6 background have a relatively mild colitis phenotype. In fact, in our comparative studies, the incidence and severity of histological inflammation and intestinal inflammatory cytokine secretion were similar in PI3K p110δD910A/D910A mice and age-matched IL-10−/− mice on the C57BL/6 background. As polygenic contributions and genetic background can alter phenotype, IL-10−/− mice were backcrossed to PI3K p110δD910A/D910A mice. Using a scoring system devised for IL-10−/− mice, IL-10−/−/PI3K p110δD910A/D910A mice develop severe colitis and an exuberant mucosal inflammatory cytokine response at an early age compared to each of the founder strains. This finding implicates IL-10 and PI3K p110δ as two important, non-redundant, homeostatic pathways that function in normal physiology to suppress intestinal inflammation directed against the enteric microbiota.
The importance of the enteric microbiota in the initiation of IBD is illustrated by IL-10−/− mice, where the development of spontaneous colitis is dependent on the presence of the microbiota1. We show dramatically increased levels of PI3K p110δ mRNA in the colon of WT GF mice transitioned to a microbiota. Importantly, augmented expression was not seen in transitioned colitis-prone IL-10−/− mice, which correlated with the development of intestinal inflammation. Strikingly similar findings were observed in LPS-activated BMMs from IL-10−/− and WT mice. These findings suggest that p110δ regulation may be an important homeostatic pathway in other models of intestinal inflammation. Based on our results, PI3K induced through TLR signaling is an event that limits the extent and duration of TLR-activated of pro-inflammatory responses. In IL-10 deficiency, one mechanism for exuberant and prolonged inflammatory responses may be loss of induction of p110δ (See Supplemental Figure 8 for model).
A prominent histological feature in PI3K p110δD910A/D910A mice that is not characteristic of human Crohn’s disease or ulcerative colitis is the presence of numerous intraepithelial lymphocytes. However, intraepithelial lymphocytosis is characteristic of three forms of human IBD; celiac disease, lymphocytic colitis and collagenous colitis26, 27. Although the purpose of this study was to correlate the development of colitis with defects in innate immunity in PI3K p110dD910A/D910A mice, a goal of future research will be to characterize the role of intraepithelial lymphocytes in this model.
PI3K p110δD910A/D910A macrophages demonstrate heightened sensitivity to stimulation by TLR ligands. This finding underscores the importance of PI3K p110δ̣in dampening TLR signaling and also suggests that aberrant regulation of innate immune responses could contribute to the development of colitis in PI3K p110δD910A/D910A mice. Accumulating evidence has established the role of PI3K in the attenuation of TLR signaling 28–31. For instance, mice with genetic deletion of the PI3K p85α subunit display altered balance of Th1-Th2 responses 28, and dendritic cells produce enhanced levels of IL-12 in response to TLR2 (PGN), TLR4 (LPS) and TLR9 (CpG) ligands. However, these mice do not develop chronic colonic inflammation. It is possible that mice deficient in PI3K subunits demonstrate compensatory changes in expression and availability of regulatory subunits, which could affect phenotypic and functional analyses8. As we demonstrate in PI3K p110δD910A/D910A mice, TLR activation in macrophages elicits an exuberant inflammatory response. However, TLR signaling in intestinal epithelial cells is protective against inflammation2. We speculate that the genetic defects in PI3K p110δD910A/D910A mice that lead to the development of colitis are limited to the hematopoietic compartment as p110δ is not highly expressed in epithelial cells32
Genetic variants linked to Crohn’s disease include genes that mediate autophagy and phagosomal function1. Recent studies indicate the importance of PI3K signaling in phagosomal maturation and acidification, essential for optimal bacterial killing33,34. Here, we show that PI3K p110δD910A/D910A macrophages are less efficient than WT macrophages at eliminating enteric commensal and pathogenic enteric bacteria. Our results indicate that PI3K p110δ̣is not necessary for phagocytosis of bacteria into the cell as there is no difference in bacterial survival following one hour of incubation with bacteria between wild-type and PI3K p110δD910A/D910A macrophages. We also demonstrated that defective bactericidal activity in PI3K p110δD910A/D910A macrophages is associated with increased inflammatory cytokine production. Additionally, PI3K p110δD910A/D910A mice are defective at clearing enteric bacteria in vivo, suggesting that the inability of PI3K p110δD910A/D910A macrophages to efficiently kill and clear microbes may contribute to prolonged inflammatory responses. However, macrophage function is not globally compromised in PI3K p110δD910A/D910A mice. Indeed, the ability of macrophages and dendritic cells from PI3K p110δD910A/D910A mice to produce NO and destroy intracellular Leishmania parasites was recently reported to be similar to WT mice2.
In summary, the PI3K p110δD910A/D910A mouse is an interesting model for understanding the pathogenesis of human IBD as it provides an example of how a genetic defect in a specific intracellular signaling molecule can lead to global defects in innate and adaptive homeostatic pathways in the intestine. Furthermore, polygenic contributions alter the phenotype of colitis as IL-10−/−/PI3K p110δD910A/D910A mice develop severe colitis at a young age compared to the parent strains. This study describes aberrant innate immunity including exuberant TLR signaling and defective bactericidal activity in macrophages that contribute to the pathogenesis of colitis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by NIH grants RO1 DK054452 (SEP), UNC Center for Gastrointestinal Biology and Disease P30 DK34987 (Histology Core, Gnotobiotic Core, and Immunotechnologies Core). SPIRE fellowship K12GM000678 (JKU) NRSA F32 DK083186 (SZS), and a Crohn’s and Colitis Foundation of America Research Fellowship Award (KM).
Abbreviations
- BMM
bone-marrow derived macrophages
- GF
germ-free
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IL
interleukin
- LPMC
lamina propria mononuclear cells
- LPS
lipopolysaccharide
- M-CSF
macrophage colony-stimulating factor
- NO
nitric oxide
- NF-κB
nuclear factor kappa B
- PRRs
pattern-recognition receptors
- PGN
peptidoglycan
- PI3K
phosphoinositide 3-kinase
- SPF
specific pathogen free
- sBLP
Synthetic bacterial lipoprotein
- TNF
tumor necrosis factor alpha
- TLR
toll-like receptor
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions of Authors: Study concept and design (JKU, KNR, SEP); Acquisition of data (JKU, KNR, KM, SZS, FL, ECS, ARS, SEP, TK); Analysis and interpretation of data (JKU, KNR, KM, SZS, FL, ARS, SEP, TK); Drafting of the manuscript (JKU, KNR, ECS, BV, SEP); Critical revision of the manuscript for important intellectual content (JKU, KNR, BV, SEP); statistical analysis (JKU, SEP); Obtained funding (JKU, SZS, KM, RBS, SEP); Technical or material support (FL, BV, RBS); study supervision (RBS, SEP)
Conflict of Interest: None of the authors listed above have any conflicts of interest to disclose.
REFERENCES
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 3.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 5.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 7.Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, Higashi K, Volinia S, Downward J, Waterfield MD. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A. 1997;94:4330–4335. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJ, Vanhaesebroeck B. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297:1031–1034. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 9.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 11.Xiong H, Zhu C, Li F, Hegazi R, He K, Babyatsky M, Bauer AJ, Plevy SE. Inhibition of interleukin-12 p40 transcription and NF-kappaB activation by nitric oxide in murine macrophages and dendritic cells. J Biol Chem. 2004;279:10776–10783. doi: 10.1074/jbc.M313416200. [DOI] [PubMed] [Google Scholar]
- 12.Kamada N, Hisamatsu T, Okamoto S, Sato T, Matsuoka K, Arai K, Nakai T, Hasegawa A, Inoue N, Watanabe N, Akagawa KS, Hibi T. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J Immunol. 2005;175:6900–6908. doi: 10.4049/jimmunol.175.10.6900. [DOI] [PubMed] [Google Scholar]
- 13.Hegazi RA, Rao KN, Mayle A, Sepulveda AR, Otterbein LE, Plevy SE. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J Exp Med. 2005;202:1703–1713. doi: 10.1084/jem.20051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 15.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horz HP, Vianna ME, Gomes BP, Conrads G. Evaluation of universal probes and primer sets for assessing total bacterial load in clinical samples: general implications and practical use in endodontic antimicrobial therapy. J Clin Microbiol. 2005;43:5332–5337. doi: 10.1128/JCM.43.10.5332-5337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu C, Gagnidze K, Gemberling JH, Plevy SE. Characterization of an activation protein-1-binding site in the murine interleukin-12 p40 promoter. Demonstration of novel functional elements by a reductionist approach. J Biol Chem. 2001;276:18519–18528. doi: 10.1074/jbc.M100440200. [DOI] [PubMed] [Google Scholar]
- 18.Feng GJ, Goodridge HS, Harnett MM, Wei XQ, Nikolaev AV, Higson AP, Liew FY. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J Immunol. 1999;163:6403–6412. [PubMed] [Google Scholar]
- 19.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol. 2008;15:164–187. doi: 10.1159/000136357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yap GS, Ling Y, Zhao Y. Autophagic elimination of intracellular parasites: convergent induction by IFN-gamma and CD40 ligation? Autophagy. 2007;3:163–165. doi: 10.4161/auto.3655. [DOI] [PubMed] [Google Scholar]
- 22.Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34:115–127. doi: 10.1016/j.tibs.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Al-Alwan MM, Okkenhaug K, Vanhaesebroeck B, Hayflick JS, Marshall AJ. Requirement for phosphoinositide 3-kinase p110delta signaling in B cell antigen receptor-mediated antigen presentation. J Immunol. 2007;178:2328–2335. doi: 10.4049/jimmunol.178.4.2328. [DOI] [PubMed] [Google Scholar]
- 24.Dil N, Marshall AJ. Role of phosphoinositide 3-kinase p110 delta in TLR4- and TLR9- mediated B cell cytokine production and differentiation. Mol Immunol. 2009;46:1970–1978. doi: 10.1016/j.molimm.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, Rowan WC, Sancho S, Walker LS, Vanhaesebroeck B, Okkenhaug K. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177:6598–6602. doi: 10.4049/jimmunol.177.10.6598. [DOI] [PubMed] [Google Scholar]
- 26.Moayyedi P, O'Mahony S, Jackson P, Lynch DA, Dixon MF, Axon AT. Small intestine in lymphocytic and collagenous colitis: mucosal morphology, permeability, and secretory immunity to gliadin. J Clin Pathol. 1997;50:527–529. doi: 10.1136/jcp.50.6.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sollid LM. Intraepithelial lymphocytes in celiac disease: license to kill revealed. Immunity. 2004;21:303–304. doi: 10.1016/j.immuni.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 29.Kuo CC, Lin WT, Liang CM, Liang SM. Class I and III phosphatidylinositol 3'-kinase play distinct roles in TLR signaling pathway. J Immunol. 2006;176:5943–5949. doi: 10.4049/jimmunol.176.10.5943. [DOI] [PubMed] [Google Scholar]
- 30.Yu Y, Nagai S, Wu H, Neish AS, Koyasu S, Gewirtz AT. TLR5-mediated phosphoinositide 3-kinase activation negatively regulates flagellin-induced proinflammatory gene expression. J Immunol. 2006;176:6194–6201. doi: 10.4049/jimmunol.176.10.6194. [DOI] [PubMed] [Google Scholar]
- 31.Aksoy E, Vanden Berghe W, Detienne S, Amraoui Z, Fitzgerald KA, Haegeman G, Goldman M, Willems F. Inhibition of phosphoinositide 3-kinase enhances TRIF-dependent NF-kappa B activation and IFN-beta synthesis downstream of Toll-like receptor 3 and 4. Eur J Immunol. 2005;35:2200–2209. doi: 10.1002/eji.200425801. [DOI] [PubMed] [Google Scholar]
- 32.Papakonstanti EA, Zwaenepoel O, Bilancio A, Burns E, Nock GE, Houseman B, Shokat K, Ridley AJ, Vanhaesebroeck B. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J Cell Sci. 2008;121:4124–4133. doi: 10.1242/jcs.032763. [DOI] [PubMed] [Google Scholar]
- 33.Booth JW, Telio D, Liao EH, McCaw SE, Matsuo T, Grinstein S, Gray-Owen SD. Phosphatidylinositol 3-kinases in carcinoembryonic antigen-related cellular adhesion molecule-mediated internalization of Neisseria gonorrhoeae. J Biol Chem. 2003;278:14037–14045. doi: 10.1074/jbc.M211879200. [DOI] [PubMed] [Google Scholar]
- 34.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.