

Abstract
The development of materials with biomimetic mechanical and biological properties is of great interest for regenerative medicine applications. In particular, hydrogels are a promising class of biomaterials due to their high water content, which mimic that of natural tissues. We have synthesized a hydrophilic biodegradable polymer, designated poly(glucose malate)methacrylate (PGMma), that is composed of glucose and malic acid which are commonly found in the human metabolic system. This polymer is made photocrosslinkable by the incorporation of methacrylate groups. The resulting properties of the hydrogels can be tuned by altering the reacting ratio of the starting materials, the degree of methacrylation, and the polymer concentration of the resultant hydrogel. Hydrogels exhibited compressive moduli ranging from 1.8 ± 0.4kPa to 172.7 ± 36kPa with compressive strain at failure from 37.5 ± 0.9% to 61.2 ± 1.1%, and hydration by mass ranging from 18.7 ± 0.5% to 114.1 ± 1.3%. PGMma hydrogels also showed a broad range of degradation rates and were cell-adhesive, enabling the spreading of adherent cells. Overall, this work introduces a class of cell adhesive, mechanically tunable and biodegradable glucose-based hydrogels that may be useful for various tissue engineering and cell culture applications.
1. Introduction
Synthetic biodegradable polymers are of great interest for various biomedical applications such as drug delivery and tissue engineering[1]. For many biomedical applications, it is desirable to control the mechanical and biological properties of these materials[2]. Previously, various synthetic biodegradable polymers have been made to improve the properties of biomaterials for various applications[3–10]. However, these polymers are generally hydrophobic greatly limiting the ability to encapsulate cells into the construct. Hydrogels, a class of biomaterials formed from hydrophilic polymers, are attractive for many reasons such as their biocompatibility and the fact that they contain similar water content and mechanical properties as natural tissues[11–13]. In particular, hydrogels from photocrosslinkable polymers can be injected into the body, encapsulate cells uniformly, and enable temporal and spatial control in the fabrication of complex structures[11, 12, 14–16].
Over the years, a number of synthetic hydrogels have been developed for biomedical applications. Poly(2-hydroxyethyl methacrylate) (PHEMA), poly(N-isopropylacrylamide) (PNIPAAm), poly(vinyl alcohol) (PVA) and their derivatives are vinyl monomer based synthetic polymers that have been studied for applications such as contact lenses, drug delivery, and tissue engineering[17–23]. However, these hydrogels are nondegradable and their vinyl monomers and crosslinking molecules may be toxic[11]. Poly(ethylene glycol) (PEG) is one of the most studied hydrophilic biomaterials and has been approved by the FDA for certain applications. While PEG hydrogels are inert and exhibit low toxicity, they are not biodegradable. To render PEG biodegradable, several methods have been developed such as copolymerization of PEG with biodegradable poly(α-hydroxy esters), such as poly(lactic acid) (PLA) and poly(glycolic acid) (PGA), or with peptides that are enzymatically degradable[24–27]. Recently, a new hydrophilic biodegradable polymer, poly(xylitol citrate)methacrylate (PXCma) was synthesized from non-toxic starting monomers: xylitol and citric acid[9]. While PEG based hydrogels include PEG macromers in their degradation products, PXCma hydrogels completely degrade into the original monomers, xylitol and citric acid that are endogenous to the human metabolic system. However, despite its merits, PXCma was mechanically weak and not cell adherent.
In this study, we synthesized a hydrophilic biodegradable polymer, designated poly(glucose malate)methacrylate (PGMma), which can form hydrogels that degrade into the starting monomers, glucose and malic acid. Glucose is a metabolic intermediate, which is commonly available, inexpensive and could potentially be used as an energy resource by cells when released through degradation of the polymer. Malic acid is nontoxic, an ingredient in many foods and its anion is an intermediate in the citric acid cycle[28]. The polymer form, poly(malic acid), has been demonstrated in various biomedical applications[29, 30]. As in previous reports on using polycondensation reactions with multifunctional monomer(s) to synthesize biodegradable elastomers or hydrogels, we used two hydrophilic, multifunctional monomers, glucose and malic acid, to form a randomly branched, hydrophilic, and hydrolyzable polyester, poly(glucose malate) (PGM) by polycondensation[3, 4, 9, 10]. After the polycondensation reaction, the remaining unreacted hydroxyl groups enabled further functionalization. To render the PGM photocrosslinkable, we functionalized the free hydroxyl groups of PGM with methacrylate groups by reacting PGM with methacrylic anhydride, as previously described for methacrylation of hyaluronic acid[31, 32]. Finally, we used the resulting photocrosslinkable PGMma to fabricate hydrogels through a light initiated crosslinking process. We characterized the properties of the resulting hydrogel as a function of the stoichiometric ratio of the starting monomers, the degree of methacrylation, and polymer concentration. Furthermore, cell adhesion tests showed that PGMma is cell-adhesive. Given its broad range of properties, PGMma may be useful for various tissue engineering applications or as a material in cell culture.
2. Materials and methods
2.1. Synthesis of PGMma polymers
All chemicals were obtained from Sigma-Aldrich. PGMma was synthesized as follows Fig.1.). Two batches of PGMs with varying molar ratios of starting materials were synthesized. D-(+)-glucose and DL-malic acid were mixed in a round bottom flask with molar ratio of 1:1 for PGM1:1 and 1:2 for PGM1:2. They were heated and stirred under argon gas to 135 °C. Under these conditions, malic acid melts and dissolves the glucose in the mixture. After glucose dissolved completely, vacuum was applied for five minutes and the resulting viscous intermediate material was cured at 90 °C for 2 days inside a vacuum oven. The resulting mixture was dissolved in distilled water, dialyzed by a membrane with molecular weight cutoff of 6–8 kD, and lyophilized. PGM macromers were methacrylated as previously described[32]. Briefly, methacrylic anhydride was reacted with PGM in distilled water on ice for 24 h. The pH of the solution was kept at 8 with 5 N NaOH. The solution was then dialyzed (MW cutoff 6k–8k Da) for 48 h, and lyophilized to yield PGMma. To modify the degree of methacrylation (DM), we added varying amounts of methacrylic anhydride (i.e. 1ml, 2ml, and 4ml per 1g of PGM).
Fig.1.

General synthetic scheme of PGMma hydrogel. (1) glucose, (2) malic acid, (3) PGM, (4) methacrylic anhydride, (5) PGMma. PGM and PGMma is a randomly branched polymer as R can be H, glucose, malic acid, or a polymer chain.
2.2. Characterization of PGMma polymers
1H-NMR spectra of PGM and PGMma polymers were obtained in D2O on a Varian 300 NMR spectrometer. The chemical composition of the polymers was calculated by the signal integrals of glucose, malic acid and methacrylate group. The molar ratio of glucose and malic acid in the polymers were calculated using the peaks at 3.2–4.5 ppm from glucose compared with peaks at 2.7–3.2 ppm from malic acid. DM was calculated by the peaks at 1.8–2.0 ppm from methacrylate groups and the peaks from malic acid. It was defined as the number of methacrylate groups divided by the number of free hydroxyl groups prior to the methacrylation reaction. Since one hydroxyl group is always removed whenever either a glucose or malic acid monomer becomes attached to the PGM polymer, regardless of the location, the total number of free hydroxyl groups in the resulting PGM polymer will not vary based on the degree of branching. Therefore, the number of hydroxyl groups was counted as the number of glucose monomers multiplied by 3 plus the number of malic acid monomers because, regardless of how the polymer is branched, the number of hydroxyl groups in the resulting PGM structure will be the same as the simplified case where each glucose monomer has three remaining hydroxyl groups and each malic acid monomer has one remaining hydroxyl group. FT-IR analysis was performed on a Bruker Alpha FT-IR spectrometer. Molecular weight distribution was determined by gel permeation chromatography (GPC, Viscotek TDAmax) using PEG standard, 0.05M NaNO3 aqueous solution as eluentia and a 3× Viscotek GPMWxL column with triple detection (refractive index, light scattering, and viscometer detector). By combining the data set obtained from three kinds of detectors, the absolute molecular weight was calculated with high accuracy independent of the degree of branching. Densities of polymers were measured with an Ultrapycnometer 1000 (Quantacrome Instruments).
2.3. Photopolymerization
PGMma polymer solutions were prepared by dissolving PGMma polymers at three different concentrations (10, 15, 20 wt%) in phosphate buffered saline (PBS) containing 0.05 wt% photoinitiator (Irgacure 2959). They were subsequently molded into disks (~8mm diameter, ~1mm thickness) and cured by exposure to light (320–500nm, ~4 mW cm−2 for 10min) (EXFO OmniCure S2000).
2.4. Hydrogel Characterization
Sol content was determined by measuring the difference in mass of dried sample before (mi) and after (mf) immersion in distilled water with agitation for 1 h. It was calculated as:
| (1) |
Hydration by mass was determined by measuring the difference in mass of sol-free hydrogels in relaxed state(mr) and in swollen state(ms) after 24 h in PBS. mr was calculated by measuring the initial mass of hydrogel and subtracting sol content from it. Hydration by mass was calculated as:
| (2) |
Crosslink density (n = ρ/Mc) and molecular weight between crosslinks (Mc) were calculated by the following equation for hydrogels[12]:
| (3) |
where τ is the compression modulus of the hydrogel, R is the universal gas constant, T is temperature, ρ is the mass density, vs is the polymer volume fraction in the swollen state, vr is the polymer volume fraction in the relaxed state, and a is the elongation ratio, which is related to the polymer volume fraction in the swollen state for isotropically swollen hydrogel:
| (4) |
Compression analysis of PGMma hydrogels was performed on an Instron 5542 mechanical tester. Hydrogel disks were prepared as described above and allowed to equilibrate in PBS for 24 h and then were compressed on the tester until failure at a rate of 0.2 mm min−1. Compressive modulus was determined as the slope of the linear region in the 5–10 % strain range.
To analyze the degradation rate of the hydrogels, disks were fabricated as described above and incubated in PBS at 37 °C on an orbital shaker. PBS was replaced every 48 h. At each time point, samples were removed, lyophilized and weighed. Mass remaining was calculated by dried mass at each time point (mt) compared to initial dried mass (m0) using the following equation:
| (5) |
When the hydrogel dissociated completely, we plotted this point as zero mass remaining. Thus the final time point signified in the degradation plot (Fig. 5) occurred somewhat sooner than the true point at which mass would be actually zero.
Fig. 5.
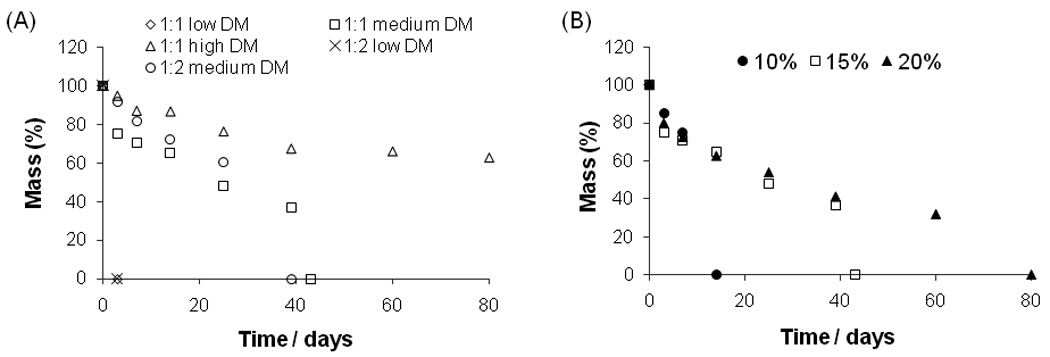
Mass-loss over time of PGMma hydrogels in PBS at 37°C (A) 15% (w/v) of each formulation, (B) PGMma1:1 medium DM.
2.5. Cell culture
NIH 3T3 fibroblasts were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen), 100 U/ml penicillin (Invitrogen), and 100 µg/ml streptomycin (Invitrogen) in a 5% CO2 atmosphere at 37 °C. Cells were passaged approximately 2 times per week and media was exchanged every 2 days.
2.6. Cell adhesion and proliferation on PGMma hydrogels
Square hydrogel pieces (1cm × 1cm × 300µm) were made by crosslinking 15% (w/v) PGMma polymer in PBS, which was previously filtered through a 0.2µm filter for sterilization, on 3-(trimethoxysilyl)propyl methacrylate coated glass slides to prevent the gels from detaching from the glass slide in growth media. After soaking the gels in growth media for six hours, they were put in 4-well plates and each well of the plates was filled with 4ml of cell suspension containing 5 × 105 cells ml−1. These plates were then incubated at 37 °C and the hydrogel surfaces were imaged at days 1, 2 and 3. To assess viability, cells were stained with live/dead viability kit (Invitrogen) and visualized under a fluorescent microscope. Confluence of the cells on the hydrogel films was determined by analyzing fluorescent images by using ImageJ software. Three formulations of PGMma – PGMma1:1 medium DM, PGMma1:1 high DM, and PGMma1:2 medium DM - were tested with poly(ethylene glycol) diacrylate 4000 (PEGDA4000, molecular weight: 4000) for comparison. In addition, to test the addition of gelatin for enhancing biological properties of the gels, hydrogels were made by mixing gelatin methacrylamide (GelMA) with PGMma1:2 medium DM (15% PGMma1:2 medium DM + 1% GelMA). Since gelatin is processed from collagen, it contains similar bioactive features as collagen[33]. GelMA was prepared according to a method previously described[34]. To better show the cell morphology, cells cultured on PEGDA4000 and PGMma1:1 medium DM were fixed and stained with Alexa Fluor 594-labelled phalloidin (Invitrogen) and DAPI to visualize F-actin filaments and cell nuclei respectively. Total cell number was quantified by counting DAPI stained nuclei.
2.6. Statistical analysis
All data are expressed as mean ± standard deviation. Statistical significance was measured by performing one-way or two-way ANOVA where appropriate (GraphPad Prism 5.02, GraphPad Software). Tukey’s multiple comparison test and Bonferroni test were used with one-way and two-way ANOVA each to determine significance between specific treatments. Differences were taken to be significant for p<0.05.
3. Results
3.1. PGMma polymer synthesis and characterization
To generate a hydrogel, we first synthesized the PGM polymer through bulk polycondensation reaction of D-(+)-glucose and DL-malic acid. Since every hydroxyl group of glucose and malic acid can react, the PGM polymer is randomly branched rather than linear (Fig. 1. (3)). Two stoichiometric ratios of glucose to malic acid were made: PGM1:1 and PGM1:2. Fig. 2. (A) shows a representative 1H NMR spectrum of PGM1:1. The chemical composition of these two PGMs were determined by 1H NMR to be 1 : 1.01 and 1 : 1.97 (Table 1) as determined by the signal integral of glucose and malic acid. These chemical compositions were similar to the molar ratio of the starting materials. An FT-IR spectrum of PGM (Fig. 2. (C)) confirmed ester bond formation with a peak at 1732cm−1. A broad band centered at 3388cm−1 was also detectable, most likely due to hydrogen bonded hydroxyl groups, while a small band centered at 2934cm−1 was detectable due to sp3-hybridized C-H bonds. The number average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity index (PDI) determined by GPC with three types of detectors (refractive index, light scattering, and viscometer detector) are summarized in Table 1.
Fig.2.
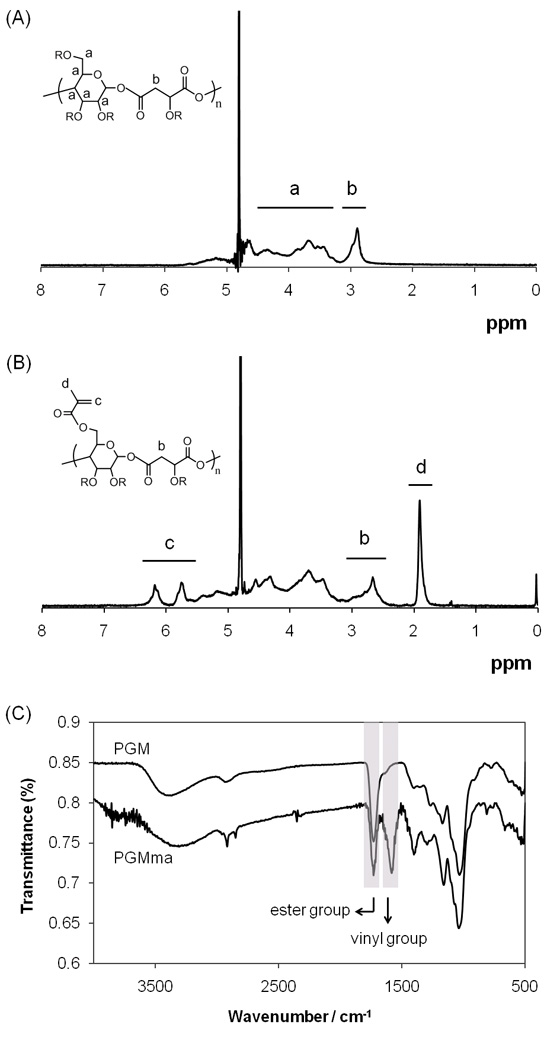
(A) Representative 1H NMR spectrum of PGM. (B) Representative 1H NMR spectrum of PGMma. (C) Representative FT-IR spectra of PGM and PGMma.
Table 1.
Composition by 1H NMR and molecular weight distribution of PGM.
| Polymer | Composition by 1H NMR |
Mn (× 104) / g mol−1 |
Mw (× 104) / g mol−1 |
PDI |
|---|---|---|---|---|
| PGM1:1 | 1 : 1.01 | 1.51 | 9.77 | 6.5 |
| PGM1:2 | 1 : 1.97 | 1.18 | 6.41 | 5.4 |
Following polymerization of PGM, methacrylation was performed as described above to obtain PGMma. To achieve differences in DM, we systematically varied the amount of methacrylic anhydride added to the reaction. The amount of methacrylic anhydride added and DM of each PGMma polymer determined by 1H NMR spectra (representative image in Fig. 2. (B)) are summarized in Table 2. Low (16%), medium (28%), and high (44%) DM were obtained for PGMma1:1, and low (23%) and medium (28%) DM were obtained for PGMma1:2. A representative FT-IR spectrum of PGMma also confirmed methacrylation with a peak due to vinyl groups at 1596cm−1. Average molecular weight and distribution determined using GPC and density measured with a pycnometer are summarized in Table 2.
Table 2.
The amount of methacrylic anhydride added to PGM, the resulting DM of PGMma polymers, and physical properties of PGMma polymers and hydrogels. Crosslink density and molecular weight between crosslinks were calculated for 15% hydrogels.
| Polymer | Amount of methacrylic anhydride added per 1g of PGM / ml |
DM (%) |
Mn (× 104) / g mol−1 |
Mw (× 104) / g mol−1 |
PDI | Density of polymer / g cm−3 |
Crosslink density / mol m−3 |
Molecular weight between crosslinks (× 103) / g mol−1 |
|---|---|---|---|---|---|---|---|---|
| PGMma1:1 low DM | 1 | 16 | 1.01 | 2.58 | 2.5 | 1.98±0.02 | 391 | 5.06 |
| PGMma1:1 medium DM | 2 | 28 | 0.94 | 2.05 | 2.2 | 1.96±0.06 | 423 | 4.64 |
| PGMma1:1 high DM | 4 | 44 | 0.83 | 1.87 | 2.2 | 1.98±0.08 | 496 | 3.99 |
| PGMma1:2 low DM | 2 | 23 | 1.14 | 2.27 | 2.0 | 1.85±0.01 | 324 | 5.71 |
| PGMma1:2 medium DM | 4 | 28 | 0.96 | 1.99 | 2.1 | 2.01±0.04 | 435 | 4.61 |
3.2. Characterization of PGMma hydrogels
Physical properties were measured to characterize the resulting PGMma hydrogels. To measure the amount of the soluble fraction, including microgels that could diffuse out of the gel following crosslinking, the sol content of 15% (w/v) hydrogel of all formulations of PGMma polymers and PEGDA4000 was determined as described above. For PGMma1:1 medium DM, the sol content of 10% and 20% (w/v) hydrogels was measured as well. As seen in Fig. 3. (A), as DM increased, sol content decreased. PGMma1:1 low DM exhibited the highest sol content, 29.6 ± 0.5%, while PGMma1:1 high DM exhibited the lowest sol content, 5.8 ± 1.1%. Sol content of the lowest DM hydrogel (PGMma1:1 low DM) was similar (p>0.05) to that of PEGDA4000. However, the polymer concentration in the hydrogel did not affect the sol content. The difference in sol content for different polymer concentrations in the hydrogel was not significant (p>0.05) (Fig. 3. (B)). Hydration by mass of sol-free PGMma hydrogels are summarized in Fig. 3. (C) and (D). Hydration was also measured for 15% (w/v) hydrogel of every formulation of PGMma, in addition to 10% and 20% (w/v) for PGMma1:1 medium DM. As expected, as DM increased, hydration decreased. Among all the formulations, PGMma1:2 medium DM exhibited the highest hydration, 114.1 ± 1.3%, while PGMma1:2 low DM exhibited the lowest hydration, 18.7 ± 0.5%. With respect to the concentration, the 20% hydrogel exhibited higher hydration than the other two concentrations, however, the difference between 15% and 20% hydrogels was not significant (p>0.05).
Fig. 3.
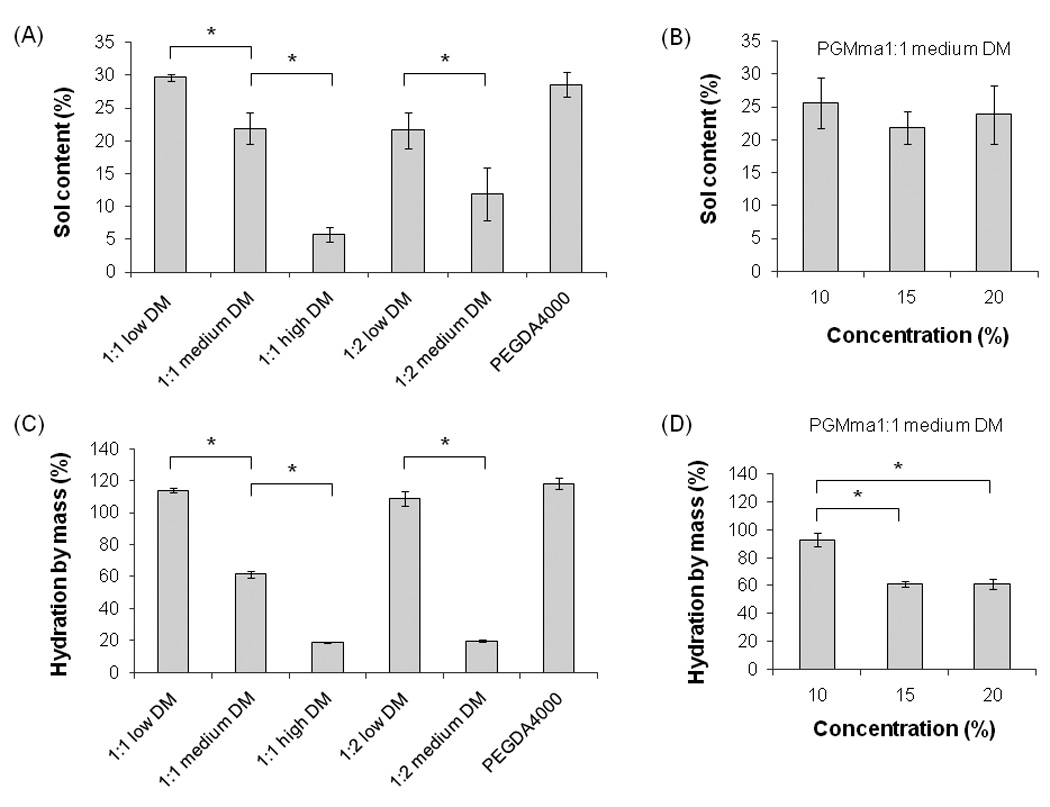
Sol content of (A) 15% (w/v) hydrogels of each PGMma formulation compared with PEGDA4000, and (B) PGMma1:1 medium DM hydrogels with different concentrations. Hydration by mass of (C) 15% (w/v) hydrogels of each PGMma formulation compared with PEGDA4000, and (D) PGMma1:1 medium DM hydrogels with different concentrations. (*) indicates significant difference (p<0.05).
To characterize the mechanical properties of PGMma hydrogels, compression analysis was performed using unconfined, uniaxial compression. Fig. 4. (A) shows representative stress-strain curves of PGMma1:1 15% hydrogels with varying DM (low, medium, high), while panel (B) shows representative stress-strain curves of PGMma1:1 medium DM hydrogel with varying polymer concentration (10%, 15%, 20% (w/v)). Compressive modulus and compressive strain at failure of all formulations of PGMma hydrogels measured are shown in Fig. 4. (C) and (D) together with those of 15% polymer concentration PEGDA4000 hydrogel for comparison. For both PGMma1:1 and PGMma1:2 with low DM, hydrogels could not be made from 10% polymer solution. Overall, the compressive modulus increased as DM increased or polymer concentration increased. For example, 15% hydrogels of PGMma1:2 low DM exhibited a compressive modulus of 1.8 ± 0.4kPa, while 20% hydrogels of PGMma1:2 medium DM exhibited a modulus of 172.7 ± 36kPa. The compressive modulus of both PGMma1:1 high DM and PGMma1:2 medium DM was comparable to that of PEGDA4000. The compressive strain at failure generally decreased as DM increased or polymer concentration increased. However, the difference was not as great as that seen with the compressive modulus. For example, the compressive strain at failure of 15% PGMma1:1 high DM was not significantly different from that of the 20% hydrogel of the same polymer (p>0.05). The compressive strain at failure of PGMma hydrogels ranged from 37.5 ± 0.9% to 61.2 ± 1.1%, which shows PGMma was not as compressible as PEGDA4000, which exhibited compressive strain at failure values ranging from 65.7 ± 6.7% to 79.6 ± 1.6%.
Fig. 4.
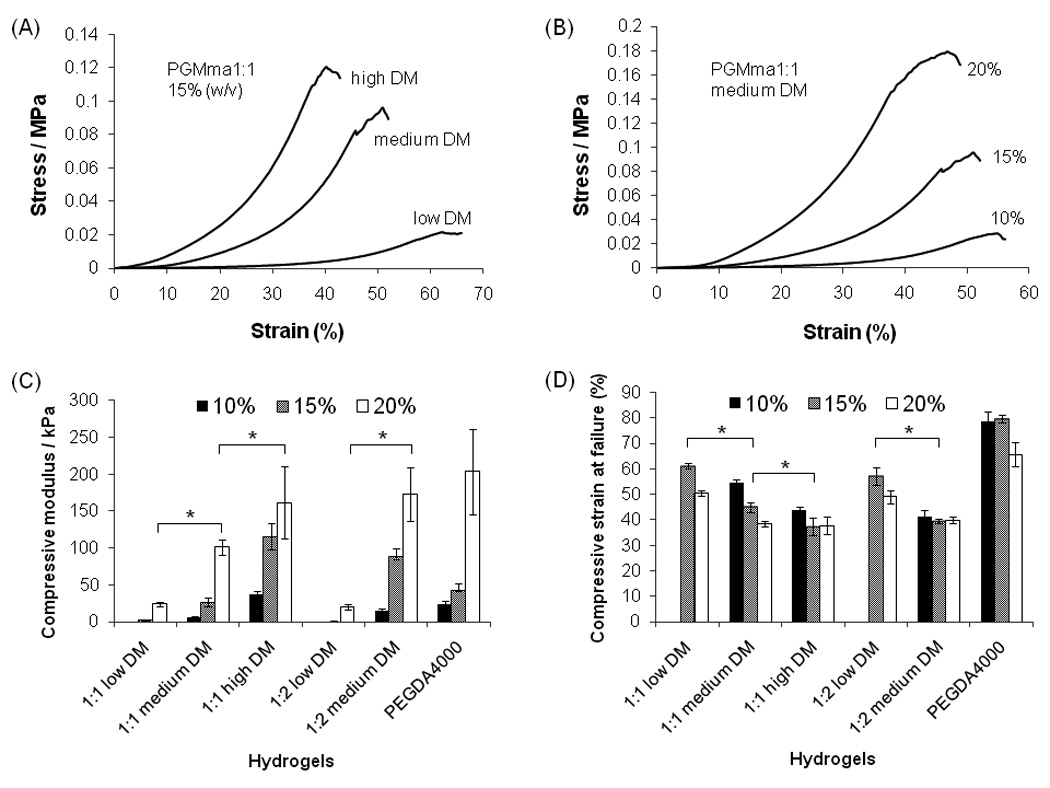
Stress-strain curve of PGMma1:1 hydrogels (A) 15%(w/v) of each DM, (B) different polymer concentrations (10%, 15%, 20% (w/v)) of medium DM. (C) Compressive modulus of PGMma1:1 and PGMma1:2 hydrogels compared with that of PEGDA4000 hydrogel. (D) Compressive strain at failure of PGMma1:1 and PGMma1:2 hydrogels compared with that of PEGDA4000 hydrogel. (*) indicates significant difference (p<0.05).
3.3. In vitro degradation of PGMma hydrogels
PGMma hydrogels were hydrolytically degradable in PBS (Fig. 5). As shown in Fig. 5. (A), PGMma polymers exhibited a broad range of degradation rates. For example, hydrogels with a low DM dissociated in 3 days, whereas those with high DM showed a much slower degradation rate (at least 80 days). The degradation rates of PGMma1:1 medium DM hydrogels with varying polymer concentrations (10%, 15%, 20% (w/v)) are shown in (B). Interestingly, the initial degradation rates were similar for all concentrations, however, at a critical point, the hydrogels dissociated abruptly. This critical time occurred sooner for hydrogels with lower polymer concentrations. For example, 10% hydrogels dissociated by day 14, while 15% hydrogels dissociated by day 43, and 20% hydrogels did not dissociate throughout the 80 day study.
3.4. Cell adhesion and proliferation on PGMma hydrogels
To determine the adhesivity of the PGMma hydrogels, the initial attachment and subsequent proliferation of 3T3-fibroblasts on three formulations of PGMma hydrogels was compared to PEGDA4000 (Fig. 6). Consistent with previously published reports, cells did not significantly adhere to PEGDA4000[35, 36]. Interestingly however, cells readily attached to and spread on all PGMma hydrogels. The levels of initial cell attachment as measured by the extent of confluence at day 1 were similar for all glucose-based hydrogels tested (p>0.05). To analyze cell proliferation, cell-seeded hydrogels were cultured for 3 days. We observed that after 2 days, cells proliferated on all PGMma hydrogels and there was no significant difference in the level of confluence among PGMma hydrogels. However, after 3 days, differences in proliferation were seen among PGMma hydrogels. At this time, the confluence on PGMma1:1 medium DM was the highest while that on PGMma1:2 medium DM was the lowest (p<0.05) among the three formulations of PGMma. To assess the effect of the addition of GelMA on enhancing biological properties of PGMma hydrogels, the same tests were performed on GelMA-added PGMma1:2 medium DM and the data was compared with that of pure PGMma1:2 medium DM. In these studies, the extent of cell attachment at day 1 and the confluence at day 2 was not significantly different. However at day 3, GelMA-added PGMma1:2 medium DM exhibited significantly higher confluence than the same hydrogels without GelMA (p<0.05). To better demonstrate cell morphology and changes in the total cell number over time, we stained the cells cultured on PEGDA4000 and PGMma1:1 medium DM for F-actin (phalloidin) and cell nuclei (DAPI). The cells on PGMma1:1 medium DM hydrogels clearly exhibited spread morphology, and the cell density increased significantly by day 3 (p<0.05).
Fig. 6.
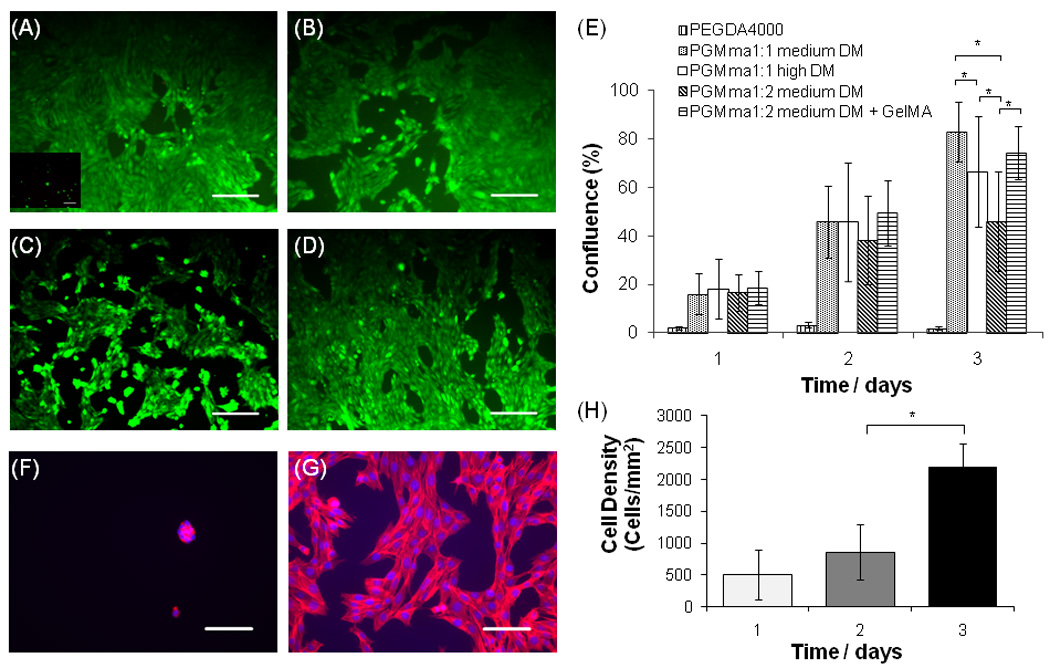
Fluorescence images (10×) of live/dead stained 3T3 fibroblasts after 3 days of culture on PEGDA4000 (inset in (A)), (A) PGMma1:1 medium DM, (B) PGMma1:1 high DM, (C) PGMma1:2 medium DM, and (D) PGMma1:2 medium DM + GelMA. The polymer concentration of hydrogels is 15% (w/v). Scale bars represent 200µm. (E) Attachment and proliferation of 3T3 fibroblasts on different hydrogels. (*) indicates significant difference (p<0.05) to the other hydrogel(s) for that time point. Phalloidin/DAPI staining for F-actin/cell nuclei on day 2 of culture on (F) PEGDA4000 and (G) PGMma1:1 medium DM. Scale bars represent 100µm. (H) Determination of cell density, defined as the number of DAPI stained nuclei per PGMma1:1 medium DM hydrogel area. (*) indicates significant difference (p<0.05).
4. Discussion
The melting point of D-(+)-glucose and DL-malic acid are 150–152°C and 131–133°C, respectively. Once the glucose begins to melt at temperatures above 150°C, it starts to caramelize by a browning reaction due to oxidation. Therefore, the first polycondensation step to synthesize PGM, which involves heating and curing, was done at 135 °C and 90 °C to minimize this effect. To do this, glucose was dissolved into liquid malic acid, instead of melting the glucose and malic acid mixture above 150 °C. Despite this reduced reaction temperature, glucose and malic acid reacted thoroughly to yield a polyester material, PGM, which was confirmed by GPC, FT-IR and 1H-NMR analysis. Furthermore, the chemical composition of PGM, as determined by 1H-NMR, correlated well to the initial molar ratios of glucose and malic acid. Even though the multiple hydroxyl groups in glucose and one in malic acid make PGM polymers structurally heterogeneous, the remaining unreacted hydroxyl groups following polycondensation enable PGM to be water-soluble, which is one of the critical reasons that glucose was chosen as a starting material. Glucose can exist either in the ring form with no aldehyde groups, or in the open-chain form containing an aldehyde group[37]. Since the 1H NMR spectrum of PGM does not contain a peak indicative of the aldehyde groups, we conclude that the majority of the glucose in PGM remained in the ring form.
There were differences in the reaction characteristics for methacrylation of the different PGM polymers. For example, it was more difficult to methacrylate PGM1:2 than it was for PGM1:1 (Table 2). When 2 ml of methacrylic anhydride per 1 g of PGM was added, the DM of PGMma1:1 was 28%, whereas the DM of PGMma1:2 was 23%. By doubling the amount of methacrylic anhydride, the DM of PGMma1:1 was increased to 44%, whereas the DM was only increased to 28% for PGMma1:2. This is likely due to the presence of more unreacted hydroxyl groups which are available for methacrylation in PGMma1:1 as compared to the PGMma1:2 polymers. Another possible explanation is that PGM1:2 is more branched than PGM1:1 so methacrylate groups cannot access the free hydroxyl groups in PGM1:2 as easily as is possible in PGM1:1. It appeared from Table 1 and 2 that methacrylation resulted in a decrease in both the molecular weight and PDI, and as DM increased, the molecular weight decreased. The ester bonds of PGM can be hydrolyzed in aqueous solution, particularly faster in either acidic or basic conditions. Since the methacrylation is done in a weak basic aqueous solution, some hydrolysis likely occurred to decrease the molecular weight of the resultant PGMma. If the molecular weight is larger, the chance of hydrolysis in the macromer is higher, and small macromers were removed by dialysis. These are likely to be the reasons that PDI decreased. The reason that as DM increased, molecular weight decreased is likely to be that increased methacrylic acid derived from methacrylic anhydride and sodium hydroxide which was added to keep the pH constant, led to increased hydrolysis of ester bonds. In addition, more methacrylic anhydride remained after the 24h reaction leading to some continued hydrolysis during the dialysis step.
The control or repeatability of the synthesis is important because if the structure, such as degree of branching, of the material varies, the properties of hydrogels are likely to be different. Therefore, tight adherence to the protocols would be required to avoid significant batch to batch variation. As the number of remaining hydroxyl groups after the polycondensation reaction does not vary according to the degree of branching, unless there is a big difference in the degree of branching and thus a big difference in the accessibility of the remaining hydroxyl groups, DM would not be much different from batch to batch and neither would be the mechanical properties of the hydrogels. If the methacrylation process were automated to input NaOH as needed according to the instantaneous pH, the material could be synthesized much more consistently for potential applications.
As expected, the sol content decreased as DM increased in PGMma hydrogels (Fig. 3. (A)). This is because as DM increases and crosslinking increases, the amount of soluble polymer chains remaining in the gel decreases. Interestingly, there was no significant change in sol content as a function of polymer concentration changes in the hydrogels (Fig. 3. (B)). This indicates that at least in this concentration range, the concentration increase does not result in more propagation of crosslinking in the hydrogels. A state of equilibrium hydration is reached in which two opposing driving forces of an elastic retractive force by polymer networks and a diluting force related to the enthalpy and entropy of mixing are balanced[38]. Although the PGMma1:2 polymer exhibited a narrower range of DM (23% (low) and 28% (medium)) compared to that of PGMma1:1 (16% (low), 28% (medium), 44% (high)), they displayed a comparable range of hydration by mass (Fig. 3. (C)) to that of PGMma1:1. This could be explained by the difference in the hydrophilicity of glucose and malic acid, the accessibility to vinyl groups during crosslinking, and the network structure of PGMma1:1 and PGMma1:2. As shown in Fig. 3. (D), hydration did not decrease substantially when the polymer concentration increased from 15% to 20%. In this case, the concentration change did not lead to a significant change in the balance of the diluting force and the retractive force.
For potential tissue engineering applications, matching the mechanical properties of the polymer scaffold with those of the natural tissues is desirable[39]. The compressive modulus of PGMma hydrogels varied from 1.8 ± 0.4kPa to 172.7 ± 36kPa (Fig. 4.) which is in the range of moduli for many natural tissues. For example, human relaxed (6kPa) and contracted smooth muscle (10kPa), human carotid artery (84 ± 22kPa), rat skeletal muscle (100kPa), human spinal cord (89kPa), mouse cardiac muscle (20–150kPa), human thyroid (9kPa), and guinea pig lung (5–6kPa) have moduli in this range[39, 40]. As shown in equation (3), the molecular weight (Mn) of the polymer has a great influence on the resulting moduli of hydrogel. Thus, increasing the average molecular weight of PGMma could be a potential method to increase the stiffness of PGMma hydrogels. In addition, PGMma is a branched polymer before it forms a hydrogel, so the degree of entanglement in the polymer solution and in the hydrogel would be reduced. This could explain why PEGDA4000 hydrogels were as stiff as PGMma gels with a high DM. Thus, developing a synthesis method to make linear PGMma-like polymers could be useful in creating hydrogels with greater stiffness. Given the range of DM values obtained and the molecular weight between crosslinks (Table 2) calculated by using equation (3), it is likely that a major fraction of the acrylic moieties react with each other to make only local links so they do not greatly affect the modulus of the hydrogels. Only a minor fraction of the acrylic moieties are being reticulated.
Within the PGMma hydrogels, ester bonds can undergo hydrolysis to induce bulk degradation, where the material degrades throughout its entire volume at the same time[41]. PGMma gels degraded more quickly than hydrophobic polyester biopolymers many of which undergo surface-erosion degradation, where degradation occurs only at the surface[4, 9, 10]. As expected, the DM, which is related to the crosslinking density, was responsible for altering the degradation rate, so as the DM increased, the degradation rate decreased. However, the effect of polymer concentration on the degradation rate behaved differently. Until the point at which hydrogels dissociated, hydrogels of different polymer concentrations displayed similar degradation rates. The time at which hydrogels dissociated completely came earlier for hydrogels at lower polymer concentrations. This indicates that the crosslinking density did not vary significantly with the concentration change. However, there is an absolute quantity of polymer that is necessary to preserve hydrogel shape, causing a decrease in time for lower concentration hydrogels to dissociate completely due to bulk degradation. This degradation data was obtained without washing the gels with distilled water before freeze drying because at times the gels were too fragile to withstand washing. Thus, salts in the PBS may have contributed somewhat to the dry weight of the gels causing the remaining mass to be slightly overestimated. However, although the absolute value of actual remaining mass could be slightly different from the presented data, the trend of lower DM PGMma degrading faster than higher DM PGMma will be preserved. In the case of different concentrations in Fig. 5. (B), since the hydration by mass of 15% and 20% gels is similar, the trend of these two having similar degradation rates until the point of dissociation is not likely to change. In addition, the time needed for dissociation is not affected by the effect of salts.
Since PGMma is hydrophilic and does not contain known cell-adhesive motifs, we anticipated that cells would not attach onto PGMma hydrogels. However, we found that after soaking the gels in cell culture media for 6 hrs, PGMma gels were cell adhesive (Fig. 6). These hydrogels displayed significantly greater cell attachment as compared to PEGDA4000 which was treated in the same manner. This may be due to adsorption of a layer of adhesive proteins from the serum onto PGMma hydrogels, however this was not confirmed. This protein adsorption is possible because PGMma has a greater quantity of hydrophobic methacrylate groups than PEGDA, which only has acrylate groups at the ends of each PEG molecule. In addition, by comparing PGMma1:2 medium DM with and without co-polymerized GelMA, we concluded that GelMA appeared to improve cell proliferation on the PGMma hydrogel surface. Through simple mixing of GelMA with PGMma, the biological properties of PGMma hydrogels could be easily tuned. This demonstrates that it is possible to improve the biological properties of PGMma hydrogels through incorporation of bioactive materials such as RGD motifs. Since the amount of GelMA mixed into hydrogels was small (1%) compared to that of PGMma (15%), and GelMA hydrogels are less stiff than PGMma hydrogels [42], the inclusion of GelMA into PGMma hydrogels is not likely to greatly affect the resultant mechanical properties of the composite hydrogel. However, if greater quantities of GelMA were used, this would have to be considered.
PGMma hydrogels degrade into glucose, malic acid, and a group of molecules derived from reacted methacrylate groups. Glucose could be used as an energy source for cells, which is likely to be useful in tissue engineering applications although the overall scaffold size should be carefully considered to avoid unsafe glucose levels in certain patients, such as those with diabetes. However, high concentrations of malic acid released from the gels could be harmful to cells due to its acidity if the hydrogels degrade rapidly. Thus, addition of other materials to offset the acidity of the malic acid, or use of slow degrading formulations could be useful. In addition, as shown in the 1H NMR spectrum of PGMma (Fig. 2. (B)), the ratio of the amount of methacrylate groups in PGMma is large as compared to other methacrylated natural polymers for biomedical applications [31, 43–45]. Since the potential effect of the molecules derived from methacrylate groups are not well characterized, it is desirable to decrease the needed quantity of methacrylate groups to be able to form hydrogels. This could be achieved by synthesizing polymers with higher molecular weight. Alternatively, chemical modification of the PGM polymer to produce other degradation products could also render PGM applicable to wider range of biomedical applications.
5. Conclusions
In this study, we synthesized a hydrophilic, biodegradable polymer, PGMma, from biologically relevant molecules, glucose and malic acid, without the need for organic solvents. This polymer is photocrosslinkable by the incorporation of methacrylate groups that initiate the crosslinking of polymer chains upon exposure to light. We demonstrated that by altering the chemical composition, the degree of methacrylation, and the polymer concentration in the hydrogels, the properties of PGMma polymers and hydrogels could be tuned. PGMma hydrogels were degradable and cell-adhesive. Given their wide range of properties, PGMma hydrogels could be potentially useful for a number of biomedical applications such as scaffolds for tissue engineering or tissue culture.
Acknowledgements
Hyeongho Shin acknowledges financial support from the Samsung Scholarship. Jason Nichol was supported by the U.S. Army Construction Engineering Research Laboratory, Engineering Research and Development Center (USACERL/ERDC). This work was supported by the National Institutes of Health (EB007249; DE019024; HL092836), the NSF CAREER award, the Institute for Soldier Nanotechnology and the US Army Corps of Engineers. The authors would like to thank Prof. Robert Langer for access to FT-IR, GPC and Instron testing devices, Prof. Patrice Hildgen and Dr. Shilpa Sant for access to Ultrapycnometer measurements, and Dr. Che Hutson for assistance on statistical evaluation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peppas N, Langer R. New challenges in biomaterials. Science. 1994;263:1715–1720. doi: 10.1126/science.8134835. [DOI] [PubMed] [Google Scholar]
- 2.Langer R, Vacanti J. Tissue Eng. Science. 1993;260(5110):920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Ameer G, Sheppard B, Langer R. A tough biodegradable elastomer. Nature Biotechnology. 2002;20(6):602–606. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, Webb A, Ameer G. Novel citric acid-based biodegradable elastomers for tissue engineering. Advanced Materials. 2004;16(6):511–516. [Google Scholar]
- 5.Bettinger C, Bruggeman J, Borenstein J, Langer R. Amino alcohol-based degradable poly (ester amide) elastomers. Biomaterials. 2008;29(15):2315–2325. doi: 10.1016/j.biomaterials.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Younes H, Bravo-Grimaldo E, Amsden B. Synthesis, characterization and in vitro degradation of a biodegradable elastomer. Biomaterials. 2004;25(22):5261–5269. doi: 10.1016/j.biomaterials.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 7.Guan J, Wagner W. Synthesis, characterization and cytocompatibility of polyurethaneurea elastomers with designed elastase sensitivity. Biomacromolecules. 2005;6(5):2833–2842. doi: 10.1021/bm0503322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dey J, Xu H, Shen J, Thevenot P, Gondi S, Nguyen K, et al. Development of biodegradable crosslinked urethane-doped polyester elastomers. Biomaterials. 2008;29(35):4637–4649. doi: 10.1016/j.biomaterials.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruggeman J, Bettinger C, Nijst C, Kohane D, Langer R, Stichting J, et al. Biodegradable xylitol-based polymers. Advanced Materials. 2008;20(10):1922–1927. [Google Scholar]
- 10.Bruggeman J, de Bruin B, Bettinger C, Langer R. Biodegradable poly (polyol sebacate) polymers. Biomaterials. 2008;29(36):4726–4735. doi: 10.1016/j.biomaterials.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee K, Mooney D. Hydrogels for tissue engineering. Chemical Reviews. 2001;101(7):1869. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 12.Peppas N, Hilt J, Khademhosseini A, Langer R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Advanced Materials. 2006;18(11):1345. [Google Scholar]
- 13.Khademhosseini A, Vacanti JP, Langer R. Progress in tissue engineering. Scientific American. 2009 May;300(5):64–71. doi: 10.1038/scientificamerican0509-64. [DOI] [PubMed] [Google Scholar]
- 14.Ifkovits J, Burdick J. Review: photopolymerizable and degradable biomaterials for tissue engineering applications. Tissue Engineering. 2007;13(10):2369–2385. doi: 10.1089/ten.2007.0093. [DOI] [PubMed] [Google Scholar]
- 15.Khademhosseini A, Langer R, Borenstein J, Vacanti JP. Microscale technologies for tissue engineering and biology. Proceedings of the National Academy of Sciences of the United States of America. 2006 Feb;103(8):2480–2487. doi: 10.1073/pnas.0507681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khademhosseini A, Langer R. Microengineered hydrogels for tissue engineering. Biomaterials. 2007 Dec;28(34):5087–5092. doi: 10.1016/j.biomaterials.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 17.Lu SX, Anseth KS. Photopolymerization of multilaminated poly(HEMA) hydrogels for controlled release. Journal of Controlled Release. 1999 Feb;57(3):291–300. doi: 10.1016/s0168-3659(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 18.Kidane A, Szabocsik JM, Park K. Accelerated study on lysozyme deposition on poly(HEMA) contact lenses. Biomaterials. 1998 Nov;19(22):2051–2055. doi: 10.1016/s0142-9612(98)00111-2. [DOI] [PubMed] [Google Scholar]
- 19.Stile RA, Burghardt WR, Healy KE. Synthesis and characterization of injectable poly(N-isopropylacrylamide)-based hydrogels that support tissue formation in vitro. Macromolecules. 1999 Nov;32(22):7370–7379. [Google Scholar]
- 20.Brazel CS, Peppas NA. Pulsatile local delivery of thrombolytic and antithrombotic agents using poly(N-isopropylacrylamide-co-methacrylic acid) hydrogels. Journal of Controlled Release. 1996 Mar;39(1):57–64. [Google Scholar]
- 21.Schmedlen R, Masters K, West J. Photocrosslinkable polyvinyl alcohol hydrogels that can be modified with cell adhesion peptides for use in tissue engineering. Biomaterials. 2002;23(22):4325–4332. doi: 10.1016/s0142-9612(02)00177-1. [DOI] [PubMed] [Google Scholar]
- 22.Nuttelman C, Henry S, Anseth K. Synthesis and characterization of photocrosslinkable, degradable poly (vinyl alcohol)-based tissue engineering scaffolds. Biomaterials. 2002;23(17):3617–3626. doi: 10.1016/s0142-9612(02)00093-5. [DOI] [PubMed] [Google Scholar]
- 23.Anseth K, Metters A, Bryant S, Martens P, Elisseeff J, Bowman C. In situ forming degradable networks and their application in tissue engineering and drug delivery. Journal of Controlled Release. 2002;78(1–3):199–209. doi: 10.1016/s0168-3659(01)00500-4. [DOI] [PubMed] [Google Scholar]
- 24.Sawhney A, Pathak C, Hubbell J. Bioerodible Hydrogels Based on Photopolymerized Poly (ethylene glycol)-co-poly (. alpha.-hydroxy acid) Diacrylate Macromers. Macromolecules. 1993;26(4):581–587. [Google Scholar]
- 25.Metters A, Anseth K, Bowman C. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer. 2000;41(11):3993–4004. [Google Scholar]
- 26.Rice M, Anseth K. Encapsulating chondrocytes in copolymer gels: bimodal degradation kinetics influence cell phenotype and extracellular matrix development. Journal of Biomedical Materials Research Part A. 2004;70(4):560–568. doi: 10.1002/jbm.a.30106. [DOI] [PubMed] [Google Scholar]
- 27.West J, Hubbell J. Polymeric biomaterials with degradation sites for proteases involved in cell migration. Macromolecules. 1999;32:241–244. [Google Scholar]
- 28.Poon Y, Cao Y, Zhu Y, Judeh Z, Chan-Park M. Addition of -Malic Acid-Containing Poly (ethylene glycol) Dimethacrylate To Form Biodegradable and Biocompatible Hydrogels. Biomacromolecules. 2009;10(8):2043–2052. doi: 10.1021/bm801367n. [DOI] [PubMed] [Google Scholar]
- 29.He B, Wan E, Chan-Park M. Synthesis and Degradation of Biodegradable Photo-Cross-Linked Poly ([alpha],[beta]-malic acid)-Based Hydrogel. Chem Mater. 2006;18(17):3946–3955. [Google Scholar]
- 30.Lee B, Fujita M, Khazenzon N, Wawrowsky K, Wachsmann-Hogiu S, Farkas D, et al. Polycefin, a New Prototype of a Multifunctional Nanoconjugate Based on Poly ([beta]-l-malic acid) for Drug Delivery. Bioconjugate Chem. 2006;17(2):317–326. doi: 10.1021/bc0502457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burdick J, Chung C, Jia X, Randolph M, Langer R. Controlled degradation and mechanical behavior of photopolymerized hyaluronic acid networks. Biomacromolecules. 2005;6:386. doi: 10.1021/bm049508a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smeds K, Grinstaff M. Photocrosslinkable polysaccharides for in situ hydrogel formation. Journal of Biomedical Materials Research. 2001;54(1):115–121. doi: 10.1002/1097-4636(200101)54:1<115::aid-jbm14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 33.Benton J, DeForest C, Vivekanandan V, Anseth K. Photocrosslinking of gelatin macromers to synthesize porous hydrogels that promote valvular interstitial cell function. Tissue Engineering Part A. 2009;15(11):3221–3230. doi: 10.1089/ten.tea.2008.0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Den Bulcke A, Bogdanov B, De Rooze N, Schacht E, Cornelissen M, Berghmanss H. Structural and rheological properties of methacrylamide modified gelatin hydrogels. Biomacromolecules. 2000;1(1):31–38. doi: 10.1021/bm990017d. [DOI] [PubMed] [Google Scholar]
- 35.Hern DL, Hubbell JA. Incorporation of adhesion peptides into nonadhesive hydrogels useful for tissue resurfacing. Journal of Biomedical Materials Research. 1998 Feb;39(2):266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 36.Drumheller PD, Hubbell JA. Polymer networks with grafted cell-adhesion peptides for highly biospecific cell adhesive substrates. Analytical Biochemistry. 1994 Nov;222(2):380–388. doi: 10.1006/abio.1994.1506. [DOI] [PubMed] [Google Scholar]
- 37.Berg J, Tymoczko J, Stryer L. Biochemistry. International Edition. New York: WH Freeman and Company; 2002. [Google Scholar]
- 38.Flory P. Principles of polymer chemistry. Cornell Univ Pr; 1953. [Google Scholar]
- 39.Amsden B. Curable, biodegradable elastomers: emerging biomaterials for drug delivery and tissue engineering. Soft Matter. 2007;3:1335–1348. doi: 10.1039/b707472g. [DOI] [PubMed] [Google Scholar]
- 40.Levental I, Georges P, Janmey P. Soft biological materials and their impact on cell function. Soft Matter. 2007;3(3):299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- 41.Nair L, Laurencin C. Polymers as biomaterials for tissue engineering and controlled drug delivery. Advances in Biochemical Engineering Biotechnology. 2006;102:47–90. doi: 10.1007/b137240. [DOI] [PubMed] [Google Scholar]
- 42.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials. 31(21):5536–5544. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia X, Burdick J, Kobler J, Clifton R, Rosowski J, Zeitels S, et al. Synthesis and characterization of in situ cross-linkable hyaluronic acid-based hydrogels with potential application for vocal fold regeneration. Macromolecules. 2004;37(9):3239–3248. [Google Scholar]
- 44.Bencherif S, Srinivasan A, Horkay F, Hollinger J, Matyjaszewski K, Washburn N. Influence of the degree of methacrylation on hyaluronic acid hydrogels properties. Biomaterials. 2008;29(12):1739–1749. doi: 10.1016/j.biomaterials.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 45.Amsden B, Sukarto A, Knight D, Shapka S. Methacrylated glycol chitosan as a photopolymerizable biomaterial. Biomacromolecules. 2007;8(12):3758–3766. doi: 10.1021/bm700691e. [DOI] [PubMed] [Google Scholar]