

Abstract
The age dependence of the oval cell response and bile duct carcinomas of male F344 rats exposed to 2 weeks on-1 week off cyclic CDE diet supports the concept of loss of potential of liver stem cells to form cancers with aging. Livers of rats exposed at 3 weeks of age demonstrated a robust and wide-spread oval cell proliferation followed by cholangiofibrosis and bile duct metaplasia with extensive mucinous cysts throughout all lobes, and induction of cholangiocarcinomas (CCAs) in 7/8 rats. Livers of rats exposed beginning at 8 weeks of age had much less oval cell response and cholangiofibrosis with only 1/15 rats developing a CCA. Livers in old (10–12 months when started) rats remained virtually unaffected, with minimal oval cell proliferation, only occasional and small foci of ductular dysplasia and 0/16 CCAs. In contrast to most published studies using uninterrupted CD plus a carcinogen, hepatocellular carcinoma was not observed under the conditions of this study. The week off during cyclic CDE may allow putative liver stem cells to avoid death or differentiation and survive to give rise to CCAs, whereas with continuous CDE exposure the stem cells are forced to differentiate and give rise to HCCs and relatively few CCAs.
Introduction
The stem cell theory of cancer posits that cancers arise from tissue-determined stem cells present in various organs 1, 2. One of the processes inherent in aging is a loss in the number or potential of these tissue-renewing stem cells 3–5, believed to be mediated by a decreasing ability to repair ongoing DNA damage 6, 7, such as that which occurs when organs are exposed to carcinogens. In the present report, we compare cellular responses in the livers of rats exposed to hepatocarcinogenesis beginning at 3 weeks of age, beginni at 8 weeks of age, and in retired breeders (10 to 12 months), using a previously well studied chemical regimen, choline deficiency-ethionine (CDE) 8–10.
Following the hierarchical model of Pierce et al. 11, combined with analysis of the cellular events during chemical hepatocarcinogenesis, we previously concluded that, in adults, liver cancers can arise from liver stem cells (oval cells), transit-amplifying cells (ducts or immature hepatocytes), or mature hepatocytes, depending on the stage of maturation arrest 12–15. Although that model fit the experimental observations on the cellular origin of hepatocellular carcinomas (HCC) and cholangiocarcinomas (CCA), it did not include the step between pluripotent stem cells (teratocarcinoma) and the liver lineage cells. The missing link is a cancer known as hepatoblastoma (HB) 16,17. HB completes the cellular lineage of liver cancer that extends from pluripotent stem cells to liver-determined stem cells to ductular stem cells to mature liver cells (Text Fig 1A).
Figure 1.
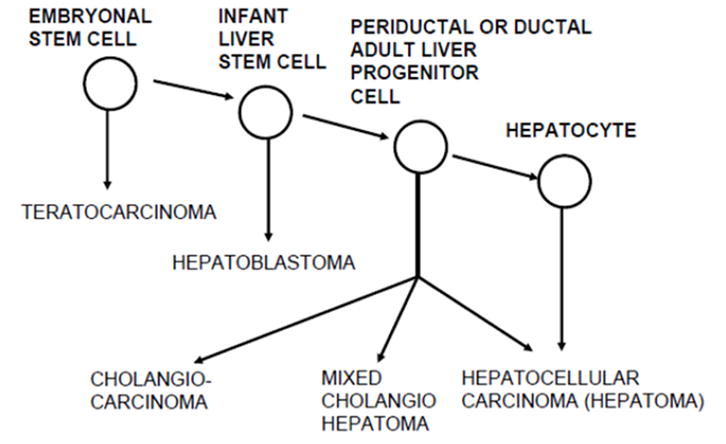
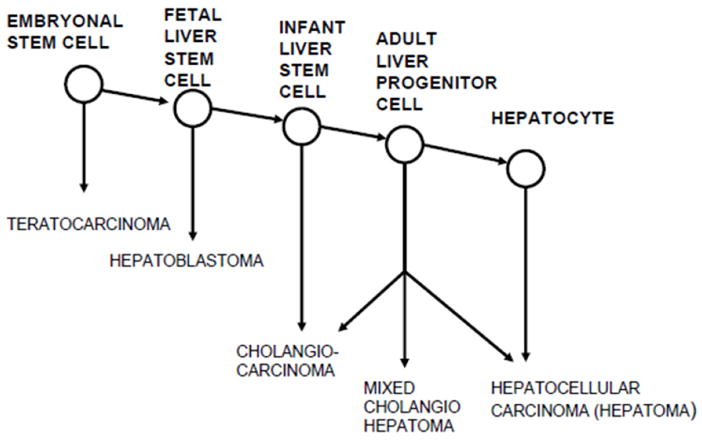
Figures 1A and 1B. Maturation arrest models of hepatocarcinogenesis. A. Experimental hepatocarcinogenesis in rats is explained by maturation arrest at the adult liver stem cell or hepatocyte level. The missing link to pluripotent stem cells is the postulated cell giving rise to hepatoblastoma. From reference 16. Figure 1B shows revised liver-cell lineage and maturation arrest model of hepatocarcinogenesis. We now postulate that in rats, between birth and 3 weeks of age, liver stem cells differentiate to a stage of bile duct determination, such that exposure to the cyclic CDE regimen results in induction of CCAs. Our model predicts that at a younger age, there still exists a more primitive liver determined stem cell that under appropriate conditions could give rise to hepatoblastomas.
We reasoned that if more liver stem cells are present, or if liver stem cells have greater potential in young rats as compared to old rats, then treatment of very young rats with a potent hepatocarcinogenic regimen should induce a more intense oval cell response and result in production of less well-differentiated HCCs, possibly such as HBs, than does treatment of older rats 18. We here report that cyclic CDE treatment of rats beginning at 3 weeks of age indeed caused a much higher level proliferation of oval cells than did treatment of rats beginning at 8 weeks of age and older. However, unexpectedly, exposure of the younger rats resulted in a high incidence of cholangiofibrosis and bile duct cancers, rather than HBs, suggesting that by 3 weeks of age the reactive liver-specific stem cells of the rat that give rise to cancer are already determined beyond the hepatoblast stage to ductal cell precursors.
Materials and methods
Animals
Male Fischer 344 rats were obtained from Taconic Farms, Germantown, NY. Rats were housed in the Wadsworth Center’s animal facility and had free access to standard laboratory chow and water when not in the cyclic CDE regimen. All animal experiments were approved by and performed under the guidelines of the Wadsworth Center’s Institutional Animal Care and Use Committee (IACUC).
Materials
A choline-deficient diet was obtained from Dyets, Inc., Bethlehem, Pennsylvania (catalogue # 518753). D,L-ethionine was purchased from Sigma Chemical Company, St. Louis, MO. (catalogue # E5139).
Dietary manipulation
A cyclic feeding regimen of the choline-deficient, ethionine-supplemented diet (CDE) was followed. One cycle consisted of 2 weeks on the CD diet, followed by 1 week off. During the time when the rats were fed the CD diet, the drinking water was supplemented with D, L-ethionine at 1 g/L.
Three groups of rats were fed the cyclic CDE diet. Group 1 consisted of weanling rats that were 3 weeks old at the start of feeding; some of these rats were left untreated to serve as controls for both the 3 and 8 week old rats that were exposed to CDE feeding. Group 2 were 8 weeks old at the start of feeding and the group 3 were retired breeders (Age 10–12 months). Some retired breeders were also left untreated to serve as controls for this third group. The 3-week cycle was repeated 5 times. At the end of cycles 1, 3 and 5, 1 to 6 rats from each of the experimental groups were euthanized for pathological analysis (Table 1). All surviving rats were left for long-term observation of tumor development and were only sacrificed when found moribund or at the termination of the study. Depending upon the group, final groups of rats were 17.5 to 30 months old at study termination. See text below for details.
Table 1.
Summary of changes in liver and pancreas after cyclic CDE feeding for 3 age groups of male F344 rats.
| LIVER | PANCREAS | ||||||
|---|---|---|---|---|---|---|---|
| No. Rats | Oval cells | Duct Metaplas. | Mitosesa | Small Hepatocytesb | Loss of Glands | Oval Cells | |
| 3-week old rats | |||||||
| Cycle 1 | 4 | 1.8 | 0 | 1.5 | 0 | 0 | 1.2 |
| Cycle 3 | 6 | 4 | 2 | 1 | 0.3 | 2.5 | 2.8 |
| Cycle 5 | 5 | 4.2 | 3 | 2 | 0.7 | 3 | 3.5 |
| 8-week old rats | |||||||
| Cycle 1 | 3 | 0.5 | 0 | 2.3 | 1 | 0 | 0 |
| Cycle 3 | 6 | 2.2 | 0.5 | 1 | 1 | 2.0 | 2.0 |
| Cycle 5 | 4 | 2 | 1.8 | 0.8 | 0.8 | 1.5 | 1.3 |
| 12–18 mos old rats | |||||||
| Cycle 1 | 1 | 1 | 0 | 1 | 0 | NS | NS |
| Cycle 3 | 3 | 0.3 | 0 | 0.3 | 0 | 0.3 | 0.3 |
| Cycle 5 | 5 | 1 | 0.5 | 0.4 | 0.2 | 1 | 1 |
For grading criteria see Figure 1.
If any mitotic cell seen on liver section, graded as 1. If several mitotic cells seen, graded as 2
If a few clusters of small hepatocytes seen, graded as 1.
NS. Tissue not on slide.
(Individual grading for each rat listed in Supplemental Table 1)
Tissue collection and analysis
At selected times or when found ill as defined by IACUC criteria, rats were sacrificed by CO2 exposure/cervical dislocation. Samples of organs were fixed in formalin, processed and embedded in paraffin; 5 μM sections were then cut and stained with hematoxylin and eosin. Selected sections were immunostained for EpCAM, HNF6 and C-Met (See supplemental Fig 5 for methods). Images were captured on an Olympus BX 51 microscope equipped with fluorescence detection and Optronics PictureFrame Version 1.2 software. Over 600 individual microscopic slides were examined.
Results
Early oval cell response
The histologic grading of early changes for each experimental animal fed the CDE diet is presented in Supplemental Table 1, and the results are summarized in Text Table 1 and Text Figures 2A and 2B. The early changes associated with feeding of CDE are mainly seen in the liver and pancreas, and take the form of replacement of the normal cells with various numbers of oval cells. The extent of the oval cell response was graded as shown in Figure 1A, and is clearly related to the age at the time of initiation of the CDE diet. Thus, up to 80% of the liver was replaced by oval cells after 5 cycles of CDE feeding to 3 week old rats. A quarter or less of the liver was involved in rats fed CDE at 8 weeks of age, and very little response was seen in the retired breeders. A similar correlation with age was seen in the oval cell response in the pancreas (Fig. 2B). The oval cell response increased from CDE cycle 1 to 3 to 5 in the rats started at 3 weeks of age, but it decreased after 3 cycles in the rats started at 8 weeks and in the retired breeders (about 1 year of age). Early bile duct hyperplasia and metaplasia were also more frequent in the younger rats, but much more extensive bile duct changes were seen in the rats surviving until spontaneous death or euthanasia. Consistent lesions were not identified in other organs in the rats from the early-euthanized groups.
Figure 2A. EFFECT OF AGE ON DEGREE OF OVAL CELL RESPONSE OF LIVER TO CD-ETHIONINE.
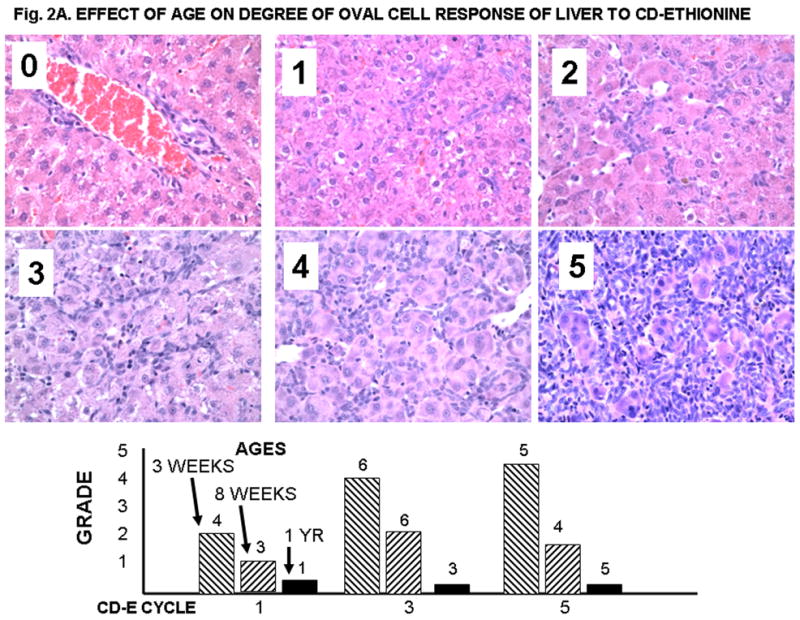
Histologic grading of oval cell reaction in liver (20X). 0. Normal 4 month old liver; 1, Grade 1; 2, Grade 2; 3, Grade 3; 4, Grade 4; 5, Grade 5. In A, small round cells are limited to the portal plate. Grades 1 and 2 show short extension of cords of small oval cells into the hepatic parenchyma. Grades 3 thorough 5 show increasing replacement of the normal hepatocytes with small oval cells. In livers with grade 5 lesions up to 80% of the liver may be replaced by oval cells. The bars show the average grade for the rats from each group; the numbers at the top of the bars indicate the number of animals examined.
Figure 2B. EFFECT OF AGE ON OVAL CELL RESPONSE OF PANCREAS TO 5 CYCLES OF CD-ETHIONINE.
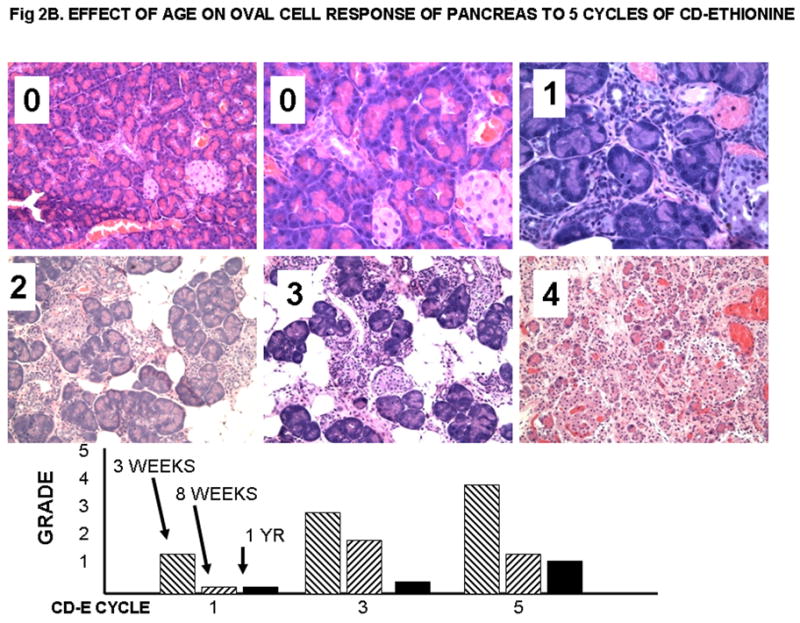
Oval cell response in rat pancreas. 0, normal, 20x and 40X; 1, 8 weeks-old, 5 cycles, 40X; 2, 3 weeks-old 5 cycles; 3, 3 weeks-old, 5 cycles, 20X; 4, 3 weeks-old, 5 cycles. Islets of Langerhans may remain after acinar structures are lost. The grading indicates that from 20 to 90 % of the pancreatic acinar cells are lost. The bars show the average grade for the rats from each group; the number of rats is the same as in Figure 2A.
Late cholangiofibrosis and bile duct cancer
The histologic grading of the lesions for each of the rats followed to death is listed in Supplemental Table 2, and the results are summarized in Text Table 2. As expected, major lesions were present in the liver in the form of bile duct hyperplasia, metaplasia, and fibrosis, also known as cholangiofibrosis and termed “tubuloform degeneration” in the older literature14. Intestinal metaplasia is a common feature of cholangiofibromas seen after oval cell proliferation in response to a chemical hepatocarcinogen 19. In the furan model of cholangiocarcinoma (CAA) 20, intestinal metaplasia preceding CAA is associated with expression of CDX1, a caudal-type homeobox intestine-specific transcription factor 21, as well as overexpression of the tyrosine kinase growth factor receptors, C-NEU and C-Met 22 and hepatocyte growth factor/scatter factor 23. Although not tested in this paper, it is likely that these factors play a critical role in the ductal differentiation of oval cells. The degree of cholangiofibrosis correlated with the age of initiation of the CDE feeding. Up to 30% of the liver was replaced by cholangiofibrosis in four of eight rats of the 3-weeks age group, whereas this occurred in only two of 15 of the 8-weeks age group, and in none of the retired breeder group. A striking finding is that seven of eight rats in the 3-weeks age group had bile duct cancers, whereas only one of 15 of the 8 weeks age group, and none in the retired breeder group demonstrated this cancer. Bile duct cancer (cholangiocarcinoma, CCA) was identified on the basis of infiltration of small bile ductules into the liver, such that mature hepatocytes became entrapped between the expanding bile ducts (Fig. 2C/F)24. By contrast, in cholangiofibrosis involving small ducts, the ducts were surrounded by fibrous tissue (Figure 2C/E). In some of the rats, large zones of the liver were occupied by CCA (see 3-weeks age group in Supplemental Table 2). Unexpectedly, no HCCs were seen.
Table 2.
Summary of lesions of long term survivors at euthanasia or death.
| No rats. | Age range at death | LUNG | LIVER | PANCREAS | KIDNEY | TESTES | |||
|---|---|---|---|---|---|---|---|---|---|
| (CIP) | Scaring | BDH&M | BD Cancer | Atrophy | (CIN) | Atrophy | Ca | ||
| Control rats for 3 and 8 weeks old groups. No CDE treatment | |||||||||
| 6 | 17.5–22.5 | 2* | 0 | 0 | 0 | 0.3 | 2.5* | 3* | 5/6 |
| 3 weeks old rats, completed 5 cycles of CDE | |||||||||
| 8 | 12.5–17.5 | 2.2* | 1 | 2.5 | 7/8 | 2.3 | 1.0* | 2* | 0 |
| 8 weeks old rat, completed 5 cycles of CDE | |||||||||
| 15 | 14–23.5 | 2.5* | 0 | 1.1 | 1/15 | 1.2 | 1.9* | 2.7* | 0 |
| Retired breeder controls, No CDE treatment | |||||||||
| 20 | 13–25 | 1.8* | 0 | 0 | 0 | 0.4 | 1.5* | 3* | 11/20a |
| 1 to 11/2 years old mice, completed 5 cycles of CDE | |||||||||
| 16 | 18–30 | 1.5* | 0.2 | 0.3 | 0 | 0.7 | 3.7* | 2.8* | 11/14b |
For grading of bile duct hypertrophy and metaplasia (BDH&M), lung, pancreas, kidney and testes lesions, see supplemental Figure 2.
Higher grades in older rats at time of death.
All rats over the age of 18.5 months had an interstitial cell cancer of the testes
There was no testes tissue on slides from two rats; two rats over the age of 20 months did not have interstitial cell carcinomas of the testes.
(Individual grading for each rat listed in Supplemental Table 2.).
Figure 2C.
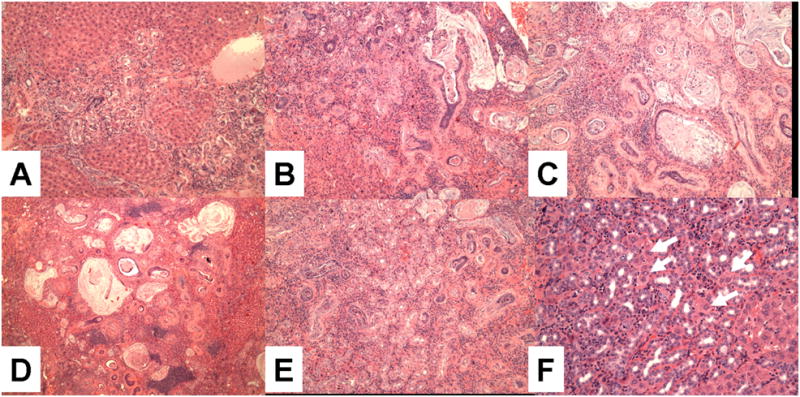
Grading of bile duct hyperplasia and metaplasia. A, Grade 1 (09–216, 10X); B, Grade 2 (09–696, 10X); C, Grade 3 (09–202, 20X); D, Grade 4 (09–208, 4X) E, Focus of proliferation of small ducts (09–207, 10X); F, CCA. Grading is based on extent of the proliferation. For example, in grade 4, larger zones of the liver are involved than was the case for grade 3. There is extensive gastrointestinal metaplasia, with production of mucus (B,C, D). The difference between E (small duct proliferation) and F (CCA) is the entrapment of hepatocytes between the ducts as shown in F (Arrows).
Other late lesions
Lesions of the lung (chronic interstitial pneumonitis), pancreas (atrophy and fibrosis), kidney (chronic interstitial nephritis) and testes (atrophy and interstitial cell carcinomas), were commonly seen (Text Table 2). In addition there were cancers of various tissue origin in single rats (Supplemental Table 2). Severe chronic interstitial pneumonia (Suppl. Fig. 1) and nephritis (Suppl. Fig. 2), as well as testicular atrophy (Suppl. Fig. 3) were seen in both the control and experimental rats, but were marginally more severe in the experimental rats. These lesions have been previously reported in normal aged Fischer 344 rats 25. In fact, a major cause of death and decision to euthanize is renal failure in the aged Fischer rat. Interstitial cell cancers of the testes (Suppl. Fig. 4A) are also commonly noted in aging Fischer 344 male rats 25. Indeed, those tumors were also found in high incidence in our control rats (15/26, Text Table 2), but were absent in the 3-week and 8-week groups that underwent five cycles of CDE. On the other hand, 11 of 14 rats in the retired breeder group fed CDE had these tumors. The reason for the low number of such tumors in the younger rats is not clear, but could be due to sampling error or the fact that rats receiving CDE die at an earlier age than the controls or retired breeders fed CDE and thus could be dying before the interstitial cell tumors develop.
Various other cancers
Other cancers found in single rats (Supplemental Table 2, Supplemental Figure 2 B–F) included leukemia, lung adenocarcinomas, renal cell carcinoma, mucinous carcinomatosis of the peritoneum, transitional cell carcinoma of the bladder, and an osteogenic sarcoma. These tumors are also found characteristically in aged Fischer 344 rats 25, and did not show any clear association with CDE feeding. For example, we found two leukemias in the control groups and one in the experimental. The incidence of leukemia here was lower than what has been reported in the literature, but we found that a number of both control and experimental rats showed extramedullary hematopoiesis in the liver; that condition could have been reported as leukemia in other studies.
Immunostaining
Localization of EpCAM, HNF6 and C-Met were done to determine expression in oval cells, cholangiofibrosis (CF) and CAA (Supplemental Table 3 and Supplemental Fig 5). EpCAM was present in normal ducts, oval cells, epithelial cells in CF and CAA, but not in normal or reactive hepatocytes, thus showing consistency of expression in biliary cell types. HNF6 was present in oval cells and CF, but unexpectedly not in CAA. C-Met was expressed weakly in normal hepatocytes, but was highly expressed in focal hepatocytes (notably in mitotic hepatocytes) in CDE fed rats, as well as in oval cells, CF and CAA.
Discussion
The cellular response of rats to an hepatocarcinogenic regimen is age-dependent. When fed five cycles of CDE diet beginning at age 3 weeks, seven of eight rats developed CCA, preceded by florid oval cell proliferation. In rats fed the CDE diet beginning at 8 weeks of age, the oval cell response was much lower and a CCA developed in only one of 15 rats. When rats were fed the diet beginning at 1 year of age there was minimal oval cell proliferation and no bile duct carcinomas were seen. This result supports the concept that cancers arise from tissue-determined stem cells, and that the number or potential, or both characteristics, of tissue stem cells to respond to carcinogens declines with aging.
Unexpectedly, our cyclic CDE-fed rats did not develop either HBs, as predicted by our working hypothesis, or HCCs. In the literature, oval cell proliferation has generally been considered to be a precursor for development of HCC (for an extensive review, see ref. 16). Early studies clearly showed that a combined deficiency of lipotropic factors enhanced the chemical induction of tumors of the liver 8, 9, 26. When Lombardi et al. combined CD with azaserine 27 (not known to be an hepatocarcinogen) or with either of the liver carcinogens ethionine 10 and acetylaminofluorine (AAF) 28 and fed to Sprague-Dawley rats at about 8 weeks of age, they reported rapid development of HCC, with an incidence of up to 80% after 6 months, and only a 38% incidence of CCAs 28. When initiation by diethylnitrosamine (DEN) was followed by CDE, Takahashi et al. obtained similar results 29. Mikol et al. 30 found a 90–100% incidence of HCC in Fischer 344 rats initiated with DEN and fed a diet devoid of methionine and choline. We have no clear explanation for why our present finding of a high incidence of CCA in rats fed cyclic CDE beginning at 3 weeks of age but no HCC in either the rats started on CDE at 3 weeks or 8 weeks, is different from other results previously reported by others. Clearly, most investigators have reported HCC in the various hepatocarcinogenic regimens that induce oval cell proliferation. However, to the best of our knowledge, no other study used cyclic CDE exposure. Perhaps the week off during each cycle allows putative liver stem cells to evade death or differentiation and thus be able to give rise to CCAs; in contrast, with continuous CDE exposure, the stem cells would be forced to differentiate, such that they give rise to relatively few CCAs and more HCCs. An alternative explanation is that the default differentiation pathway of oval cells is to form ducts. If this is true, then when hepatocarcinogenic regimens induce large numbers of oval cells, CCAs would be expected rather than HCCs.
We chose a cyclic CDE dietary schedule for two principal reasons. The main reason was to reduce morbidity and mortality during a chronic feeding schedule, based on previous studies using a cyclic feeding of AAF 31, 32. Rats continuously fed CDE for more than 2 weeks in our previous studies died with massive oval cell proliferation 33. A secondary reason was as an attempt to augment oval cell proliferation and tumor development, by repetitive exposure to a CD diet 10,28. Choline deficiency induces a state of fat deposition, apoptosis, and compensatory regeneration; in this abnormal situation, hepatocytes are forced to divide repeatedly, such that the deficiency has been termed a nutritional partial hepatectomy 34. The aberrant hepatocyte proliferation within the liver is believed to be the fundamental alteration that is ultimately responsible for the development of HCC from a methyl-deficient diet 35.
As discussed above, our results are in contrast to those found previously. In a study entailing a CD diet, only 51% of male F344 rats fed that CD diet for 13–24 months had developed HCC by the end of the 24-month period 36. Three months duration on a continuous CD diet has been considered to be the minimum period required to induce HCC 34. It is thus possible that we did not expose the rats to total CDE for a sufficiently long enough period. In addition, ethionine was administered in the drinking water in the present study; consequently, dosing may not be comparable to a regimen in which ethionine is incorporated into the diet, given that rats drink less water when it contains ethionine.
Many others have used various carcinogenic regimens to study the origin of oval-cell proliferation in rats, assuming that such proliferation is a precursor to development of HCC, but without actually following the treated rats to determine whether any cancers subsequently develop 37–40. However, after rapid proliferation, most oval cells, including those involved in bile-duct proliferation, are either lost by apoptosis, or differentiate into mature liver cells 41–43 or duct-like cells 44,45. Thus, oval cells are part of a normal response to liver injury, producing progeny to replace hepatocytes and ducts. It is not known where the critical step occurs in this process that results in induction of cancers. However, shared marker phenotypes between oval cells and hepatocellular carcinomas identified by monoclonal antibodies to cells in the liver lineage support the concept that oval cells could give rise to both HCC and CCA 14, 46,47.Our maker studies utilizing EpCAM, HNF6 and C-Met (Supplemental Fig. 5) are not definitive but are consistent with an oval cell to duct cell lineage in development of CAA. Bile ducts, oval cells, CF and CAA are all EpCAM positive, whereas hepatocytes are not. In human liver cancer EpCAM expression defines hepatocellular carcinomas with stem cell features 48. C-Met is known to be positive in mucin producing cholangiocarcinomas 49. The unexpected finding is that oval cells and bile duct cells in CF are positive for HNF6, but CAAs are negative. HNF6 is a transcription factor proposed to drive cholangiocyte differentiation 50. Thus, there appears to be a loss of this ductal phenotype with malignant transformation.
Although a direct comparison of our results to the human liver is not possible, it is likely that there is a decrease of functional liver stem cells in humans with aging. The major confounding factor is that there is no generally accepted marker for putative stem cells in the adult human liver. In fact, the adult human liver stem cell is functionally defined as “facultative”. That is, such cells are not identifiable in normal liver, but there are cells in the liver that respond to injury 51. OV 6 has been proposed as a marker for “transitional hepatocytes” that may serve this function 52. It has also been proposed that stem cells are located in the canals of Hering 53. There is a decrease in the number of biliary cells expressing the putative liver stem cell marker CD133 from 96.3% at 3 years of age to 59% 54.
Supplementary Material
Acknowledgments
Supported by NIH grant R01-112481 (Dr. Sell)
References
- 1.Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6–22. [PubMed] [Google Scholar]
- 2.Sell S. On the stem cell origin of cancer. Am J Pathol. 2010;176:2584–2594. doi: 10.2353/ajpath.2010.091064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Troen BR. The biology of aging. Mt Sinai J Med. 2003;70:3–22. [PubMed] [Google Scholar]
- 4.Kim M, Moon H-B, Spangrude GJ. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann N Y Acad Sci. 2003;996:195–208. doi: 10.1111/j.1749-6632.2003.tb03247.x. [DOI] [PubMed] [Google Scholar]
- 5.Dykstra B, de Haan G. Hematopoietic stem cell aging and self-renewal. Cell Tissue Res. 2008;331:91–101. doi: 10.1007/s00441-007-0529-9. [DOI] [PubMed] [Google Scholar]
- 6.Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–690. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 7.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 8.Rogers AE. Variable effects of a lipotrope-deficient, high-fat diet on chemical carcinogenesis in rats. Cancer Res. 1975;35:2469–2474. [PubMed] [Google Scholar]
- 9.Rogers AE, Newberne PM. Dietary effects on chemical carcinogenesis in animal models for colon and liver tumors. Cancer Res. 1975;35:3427–3431. [PubMed] [Google Scholar]
- 10.Shinozuka H, Lombardi B, Sell S, Iammarino RM. Enhancement of DL-ethionine-induced liver carcinogenesis in rats fed a choline-devoid diet. J Natl Cancer Inst. 1978;61:813–817. [PubMed] [Google Scholar]
- 11.Pierce G. Cancer: A Problem of Developmental Biology. Englewood Cliffs, NJ: Prentice Hall, Inc; 1978. pp. 1–242. [Google Scholar]
- 12.Sell S. Alpha-fetoprotein, stem cells and cancer. The Abbott Award lecture. Tumor Biology. 2008;29:161–180. doi: 10.1159/000143402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sell S, Leffert H. An evaluation of the cellular lineages in the pathogenesis of experimental hepatocellular carcinoma. Hepatology. 1982;2:77–86. doi: 10.1002/hep.1840020113. [DOI] [PubMed] [Google Scholar]
- 14.Sell S, Dunsford HA. Evidence for the stem cell origin of hepatocellular carcinoma and cholangiocarcinoma. Am J Pathol. 1989;134:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 15.Sell S, Leffert HL. Liver cancer stem cells. J Clin Oncol. 2008;26:2800–2805. doi: 10.1200/JCO.2007.15.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sell S. Stem cells in hepatocarcinogenesis - the liver is the exception that proves the rule. Cell Science Reviews. 2006;3:302–341. [Google Scholar]
- 17.Turusov VS. Murine hepatoblastoma. Vopr Onkol. 2004;50:266–270. [PubMed] [Google Scholar]
- 18.Hixson DC, Brown J, McBride AC, Affigne S. Differentiation status of rat ductal cells and ethionine-induced hepatic carcinomas defined with surface-reactive monoclonal antibodies. Exp Mol Pathol. 2000;68:152–169. doi: 10.1006/exmp.2000.2302. [DOI] [PubMed] [Google Scholar]
- 19.Tatematsu M, Kaku T, Medline A, Farber E. Intestinal metaplasia as a common option of oval cells in relation to cholangiofibrosis in liver of rats exposed to 2-acetylaminofluorene. Lab Invest. 1985;52:354–362. [PubMed] [Google Scholar]
- 20.Hickling KC, HItchcock JM, Chipman JK, Hammond TG, Evans JG. Induction and progression of cholangiofibrosis in rat liver injured by oral administration of Furan. Toxicol Pathol. 2010;38:213–229. doi: 10.1177/0192623309357945. [DOI] [PubMed] [Google Scholar]
- 21.Ren P, Silberg DG, Sirica AE. Expression of an intestine-specific transcription factor (CDX1) in intestinal metaplasia and in subsequently developed intestinal type of cholangiocarcinoma in rat liver. Am J Pathol. 2000;156:621–627. doi: 10.1016/S0002-9440(10)64766-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radaeva S, Gerrira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: a link between biliary intestinal metaplasia and mucin-producing cholangiocarcinoma. Hepatol. 1999;29:1453–1462. doi: 10.1002/hep.510290524. [DOI] [PubMed] [Google Scholar]
- 23.Lai G-H, Radaeva S, Nakamura T, Sirica AE. Unique epithelial cell production of hepatocyte growth facto/scatter factor by putative precancerous intestinal metaplasias and associated “intestinal-type” biliary cancer chemically induced in rat liver. Hepatol. 2000;31:1257–1265. doi: 10.1053/jhep.2000.8108. [DOI] [PubMed] [Google Scholar]
- 24.MacSween R, Anthony P, Scheuer P, Burt AD, Partmann B. Pathology of the Liver. Edinburgh: Churchill Livingstone; 1994. p. 670. [Google Scholar]
- 25.Coleman GL, Barthold W, Osbaldiston GW, Foster SJ, Jonas AM. Pathological changes during aging in barrier-reared Fischer 344 male rats. J Gerontol. 1977;32:258–278. doi: 10.1093/geronj/32.3.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogers A, Newberne PM. Diet and aflatoxin B1 toxicity in rats. Toxicol Appl Pharmacol. 1971;20:113–121. doi: 10.1016/0041-008x(71)90095-0. [DOI] [PubMed] [Google Scholar]
- 27.Shinozuka H, Katyal SL, Lombardi B. Azaserine carcinogenesis: organ susceptibility change in rats fed a diet devoid of choline. Int J Cancer. 1978;22:36–39. doi: 10.1002/ijc.2910220109. [DOI] [PubMed] [Google Scholar]
- 28.Lombardi B, Shinozuka H. Enhancement of 2-acetylaminofluorene liver carcinogenesis in rats fed a choline-devoid diet. Int J Cancer. 1979;23:565–570. doi: 10.1002/ijc.2910230419. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi S, Lombardi B, Shinozuka H. Progression of carcinogen-induced foci of gamma-glutamyltranspeptidase-positive hepatocytes to hepatomas in rats fed a choline-deficient diet. Int J Cancer. 1982;29:445–450. doi: 10.1002/ijc.2910290414. [DOI] [PubMed] [Google Scholar]
- 30.Mikol YB, Hoover KL, Creasia D, Poirier LA. Hepatocarcinogenesis in rats fed methyl-deficient, amino acid-defined diets. Carcinogenesis. 1983;4:1619–1629. doi: 10.1093/carcin/4.12.1619. [DOI] [PubMed] [Google Scholar]
- 31.Teebor GW, Becker FF. Regression and persistence of hyperplastic hepatic nodules induced by N-2-Fluorenylacetamide and their relationship to hepatocarcinogenesis. Cancer Res. 1971;31:1–3. [PubMed] [Google Scholar]
- 32.Sell S. Distribution of alphafetoprotein and albumin containing cells in the livers of Fischer rats fed four cycles of N-2-fluorenylacetamid. Cancer Res. 1978;38:3107–3113. [PubMed] [Google Scholar]
- 33.Sell S, Leffert HL, Shinozuka H, Lombardi B, Gochman N. Rapid development of large numbers of alpha-fetoprotein-containing “oval” cells in the liver of rats fed N-2-fluorenylacetamide in a choline-devoid diet. Gann. 1981;72:479–487. [PubMed] [Google Scholar]
- 34.Lombardi B, Chandar N, Locker J. Nutritional model of hepatocarcinogenesis. Rats fed choline-devoid diet. Dig Dis Sci. 1991;36:979–984. doi: 10.1007/BF01297151. [DOI] [PubMed] [Google Scholar]
- 35.Lombardi B, Smith ML. Tumorigenesis, protooncogene activation and other gene abnormalities in methyl deficiency. J Nutr Biochemistry. 1994;5:2–9. [Google Scholar]
- 36.Ghoshal AK, Farber E. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis. 1984;5:1367–1370. doi: 10.1093/carcin/5.10.1367. [DOI] [PubMed] [Google Scholar]
- 37.Lenzi R, Liu MH, Tarsetti F, Slott PA, Alpini G, Zhai WR, et al. Histogenesis of bile duct-like cells proliferating during ethionine hepatocarcinogenesis. Evidence for a biliary epithelial nature of oval cells. Lab Invest. 1992;66:390–402. [PubMed] [Google Scholar]
- 38.Sarraf C, Lalani E-N, Golding M, Anikumar TV, Poulsom R, Alison MR. Cell behavior in the acetylaminofluorene-treated regenerating rat liver. Light and electron microscopic observations. Am J Pathol. 1994;145:1114–1126. [PMC free article] [PubMed] [Google Scholar]
- 39.Bisgaard HC, Nagy P, Santoni-Rugiu E, Thorgeirsson SS. Proliferation, apoptosis, and induction of hepatic transcription factors are characteristics of the early response of biliary epithelial (oval) cells to chemical carcinogens. Hepatology. 1996;23:62–70. doi: 10.1002/hep.510230110. [DOI] [PubMed] [Google Scholar]
- 40.Tee LB, Kirilak Y, Huang WH, Smith PG, Morgan RH, Yeoh GC. Dual phenotypic expression of hepatocytes and bile ductular markers in developing and preneoplastic rat liver. Carcinogenesis. 1996;17:251–259. doi: 10.1093/carcin/17.2.251. [DOI] [PubMed] [Google Scholar]
- 41.Alison M, Golding M, Lalani EN, Nagy P, Thorgeirsson S, Sarraf C. Wholesale hepatocytic differentiation in the rat from ductular oval cells, the progeny of biliary stem cells. J Hepatol. 1997;26:343–352. doi: 10.1016/s0168-8278(97)80051-7. [DOI] [PubMed] [Google Scholar]
- 42.Evarts RP, Nagy P, Marsden E, Thorgeirsson SS. A precursor-product relationship exists between oval cells and hepatocytes in rat liver. Carcinogenesis. 1987;8:1737–1740. doi: 10.1093/carcin/8.11.1737. [DOI] [PubMed] [Google Scholar]
- 43.Novikoff PM, Ikeda T, Hixson DC, Yam A. Characterization of and interactions between bile ductule cells and hepatocytes in early stages of rat hepatocarcinogenesis studies by ethionine. Am J Pathol. 1991;139:1352–1368. [PMC free article] [PubMed] [Google Scholar]
- 44.Tee L, Kirilak Y, Huang W, Morgan R, Yeoh GC. Differentiation of oval cells into duct-like cells in preneoplastic liver of rats placed on a choline-deficient diet supplemented with ethionine. Carcinogenesis. 1994;15:2747–2756. doi: 10.1093/carcin/15.12.2747. [DOI] [PubMed] [Google Scholar]
- 45.Yin L, Lynch D, Ilic Z, Sell S. Proliferation and differentiation of ductular progenitor cells and littoral cells during the regeneration of the rat liver to CCl4/2-AAF injury. Histol. Histopathol. 2002;17:65–81. doi: 10.14670/HH-17.65. [DOI] [PubMed] [Google Scholar]
- 46.Dunsford HA, Karnasuta C, Hunt JM, Sell S. Different lineages of chemically induced hepatocellular carcinoma in rats defined by monoclonal antibodies. Cancer Res. 1989;49:4894–4900. [PubMed] [Google Scholar]
- 47.Hixson DC, Chapman L, McBride A, Faris R, Yang L. Antigenic phenotypes common to rat oval cells, primary hepatocellular carcinomas and developing bile ducts. Carcinogenesis. 1997;18:1169–1175. doi: 10.1093/carcin/18.6.1169. [DOI] [PubMed] [Google Scholar]
- 48.Yamashita T, Forgues M, Want W, Kin JW, Ye Q, Jia H, Budhu A, Zanetti KA, CHen Y, Qin L-X, TAng Z-Y, Wang XW. EpCAM and α-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:a451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 49.Ferreira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: A link between biliary intestinal metaplasia and mucin-producing cholangiocarcinoma. Hepatol. 1999;29:1453–1462. doi: 10.1002/hep.510290524. [DOI] [PubMed] [Google Scholar]
- 50.Zaret KS, Grompe M. Generation and regeneration of cells of the lver and pancreas. Science. 2008;322:1490–1494. doi: 10.1126/science.1161431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teutsch HF. The nodular microacchitecture of human liver. Hepatol. 2005;42:317–325. doi: 10.1002/hep.20764. [DOI] [PubMed] [Google Scholar]
- 52.Crosby HA, Hubschler SG, JOpoin RE, Gelly DA, Srrain AJ. Immunolocalization of OV-6, a putative progenitor cell marker in human fetal and diseased pediatric liver. Hepatol. 1998;28:980–985. doi: 10.1002/hep.510280412. [DOI] [PubMed] [Google Scholar]
- 53.Zhang L, Theise N, Chua M, Reid LM. The stem cell niche of human livers:symmetry between development and regeneration. Hepatol. 2008;48:1598–1607. doi: 10.1002/hep.22516. [DOI] [PubMed] [Google Scholar]
- 54.Dezso K, Paku S, Papp V, Turanyi E, Nagy P. Architectural and immunohistochemical chearcterization of biliary ductules in normal human liver. Stem Cells & Develop. 2009;18:1417–1422. doi: 10.1089/scd.2009.0110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.