

Abstract
Alpha-synuclein is the major protein component of Lewy bodies, a cardinal pathological feature of the degenerating Parkinsonian brain. Alpha-synuclein has been reported to be able to intercalate into membranes via formation of an alpha-helical structure at its N-terminal end. Recent in vitro studies from various laboratories have demonstrated that alpha-synuclein can physically associate with mitochondria and interfere with mitochondrial function. α-Syn predominantly associates with the inner mitochondrial membrane, where it can apparently interact with complex I resulting in reduced mitochondrial complex I activity and increased free radical production. However, the effect of in vivo alpha-synuclein accumulation within dopaminergic neurons on mitochondrial function has not been thoroughly studied. Examination of transgenic animals which overexpress the familial mutant A53T form of the protein selectively within dopaminergic neurons reveals that A53T localizes to the mitochondrial membranes as monomers and oligomers particularly under conditions of proteasomal inhibitory stress, and that this localization coincides with a selective age-related mitochondrial complex I inhibition and decreased substrate-specific respiration along with increases in mitochondrial autophagy (mitophagy).
Keywords: Alpha-synuclein (α-syn), mitochondria, oligomers, Parkinson’s disease, proteasomal inhibition, mitochondrial complex I (CI) activity, mitophagy
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by preferential loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) [20]. PD is also accompanied by the hallmark formation of intracytoplasmic protein aggregates termed “Lewy bodies” which include alpha-synuclein (α-syn) as a major protein component [29]. Monomeric α-syn is a 14 kD presynaptic protein with as yet enigmatic cellular function. Due to the presence of six imperfect 11-amino acid repeat sequences within its N-terminus, this results in formation of an alpha helical structure which mediates the protein’s binding to phospholipid vesicles. α-syn can also aggregate into higher molecular weight oligomers. It has been proposed that oligomerization results in induction of a beta-pleated sheet structure within the protein that aids in its further aggregation [32]. α-Syn has been reported to undergo degradation by the proteasome and by alternative pathways such as autophagy [7]. The physiological significance of protein aggregates associated with dying neurons in PD is hotly debated. Although mutations in α-syn (A30P, E46K and A53T; triplication and duplication) have been implicated in rare forms of familial autosomal dominant Parkinsonism [24, 14], how these mutations may participate in neuronal cell loss is still unclear. Overexpression of mutant α-syn induces a significant increase in sensitivity of dopaminergic neurons to mitochondrial toxins such as MPP+ and 6-hydroxydopamine, resulting in increased protein carbonylation and lipid peroxidation both in vitro and in vivo [30, 22]. Conversely, studies with α-syn-knockout mice demonstrate a marked resistance to MPTP as well as other mitochondrial toxins including malonate and 3-nitropropionic acid [8, 13]. Recently, several laboratories have published in vitro evidence suggesting localization of α-syn to mitochondria coinciding with increased mitochondrial dysfunction [10, 6, 23, 26]. Based on these studies, α-syn appears to predominantly associate with the inner mitochondrial membrane where it may interact with complex I and reduce mitochondrial complex activity. This effect appears to be enhanced by the pathogenic A53T α-syn mutant [10]. Further supporting this direct functional link between α-syn and mitochondria, a recent study demonstrated that abrogation of mitochondria DNA in yeast prevented α-syn-induced ROS formation and apoptotic cell death [4]. However, the majority of these in vitro studies were performed in non-dopaminergic cell lines. Furthermore, while in vivo studies have demonstrated that overexpression of WT α-syn may lead to enhanced abnormalities in mitochondrial morphology [28], evidence coupling this to physical presence within dopaminergic neuronal mitochondria is lacking. Here, we report in transgenic animals overexpressing A53T specifically within dopaminergic neurons [25], that the protein localizes to the mitochondria in the form of both monomers and oligomers, the latter particularly under conditions of proteasome inhibition-mediated stress, and that this coincides with a selective age-related inhibition of mitochondrial complex I function as well as increased levels of mitochondrial turnover (mitophagy).
pTH-A53T transgenics expressing high (HE) and low (LE) levels of mutant A53T synuclein mRNA in the midbrain were a gift from Dr. Dino Dimonte (Parkinson’s Institute, Sunnyvale, CA) [25]. For proteasomal inhibition studies, animals were injected with 0.5mg/kg epoxomicin (IP); six injections were made over the course of 2 weeks. The animals were sacrificed 6 weeks after the final injection [19]. Transgenic animals and non-transgenic littermate controls were anaesthetized and transcardially perfused with 4% paraformaldehyde. Nigrostriatal sections of 40 μM thickness from these transgenic mice were treated first with mouse anti human α-syn (1:500; Chemicon) followed by biotinylated antimouse IgG secondary antibody and Texas Red avidin D (Vector Laboratories). The sections were reincubated with mitochondrial markers either mouse Voltage Dependent Anion Selective Channel protein 1 (VDAC/Porin) (1; 1000, Calbiochem) or mouse anti-ATP synthase β chain antibody (1:1000, Chemicon) followed by biotinylated anti-mouse IgG secondary antibody and fluorescein avidin D (Vector Laboratories). The sections were visualized under fluorescence confocal microscopy. For EM analyses, dissected SN sections were post fixed in 2% PFA/0.2gluteraldehyde/0.2% picric acid in PBS for 2–4 hours RT. After dehydration with 50% to 100% alcohol, the sections were in embedded in gelatin capsules at 58°C for 24 h. Ultrathin sections (70 nm) were cut on an RMC MT7000 ultramicrotome, collected onto nickel grids, and the sections were imaged on FEI Tecnai 12 TEM at 80 kV accelerating voltage. To assess mitophagy levels in HE versus LE mice, sections on different grids were counted manually for the presence of mitochondria in autophagic vacuoles (AVs) which are considered a characteristic component of autophagy [31]. Under high magnification, the structures of all mitochondria, AVs and any mitochondria within those AVs were manually counted in each field.
For western blot analysis, mitochondria were prepared by as described previously [16]. Briefly, freshly dissected mouse nigrostriatal tissues from A53T and A53T+epoxomicine groups were washed and homogenized in ice-cold isolation buffer and centrifuged at 1000 X g for 5 min at 4°C, followed by centrifugation of the supernatant at 8500 X g for 10 min at 4°C. The mitochondrially enriched pellet was resuspended in isolation buffer and layered on top of 6% (w/v) Ficoll solution (F-4375; Sigma-Aldrich) and centrifuged at 75,000 X g for 30 min at 4°C to remove myelin. The pellet was solubilized in lysis buffer containing protease inhibitors and fractionated into soluble and insoluble fractions by centrifuging at 12,000 x g. Insoluble membrane fractions were directly analyzed on SDS PAGE followed by western blot using anti- α-syn antibody (Chemicon, USA). The purity of mitochondrial fractions were verified by western blotting with antibodies to mitochondrial marker ATPase (1:1000, millipore) and cytosolic marker actin (1:1500, Santa Cruz Biotechnology).
For respiration studies, dopaminergic and non-dopaminergic striatal synaptosomes were isolated using a previously published protocol from our laboratory [5]. Briefly, synaptosomes were prepared from dissected striatal tissue (n = 5 animals in each group) [3] and dopaminergic synaptosomes were then purified using a novel magnetic bead approach using antibody against the cell surface dopamine transporter (DAT) protein as described previously [5]. From a single striatum, we were able to isolate 20–25 μg of DA synaptosomal protein. These were used to measure both CI and CIV activities and substrate-specific respiration in HE versus LE α-syn transgenics. For respiration experiments, isolated DA vs non-DA striatal synaptosomes were incubated in serum free medium (pH 7.0) at 37°C in a Clark-type oxygen electrode (Hansatech). Respiration was calculated as the rate of oxygen consumption driven by pyruvate-malate (for CI) or TMPD/ascorbate (CIV) in the medium as a substrate. Carbonyl cyanide ptrifluoromethoxy phenylhydrazone (FCCP) was used as uncoupler to assess maximum respiration rates. Absolute values for uncoupled respiration rate were calculated as the difference between FCCP driven respiration and state IV respiration.
Complex I activities were assayed in isolated dopaminergic and non-dopaminergic synaptosomal fractions from HE versus LE α-syn transgenics as rotenone-sensitive NADH dehydrogenase activity by measuring DCPIP (2, 6-dichlorophenolindophenol) reduction in synaptosomal extracts following addition of 200 mM NADH, 200 mM decylubiquinone, 2 mM KCN, and 0.002% DCPIP in the presence and absence of 2 mM rotenone [18]. Complex IV activity was assayed as cytochrome c oxidase activity by observing the rapid (1–2s) oxidation of freshly reduced 40 mM ferrocytochrome c in a 10 mM K-PO4 buffer pH 7.2 containing 100 mM KCl, 0.025% maltoside at 30°C, at 550 nm. Values for all assays were normalized/protein using BioRad reagent.
Data were expressed as means ± S.D. from at least three independent experiments. Significant differences between mean values were determined by Student’s t-test. Differences were considered to be statistically significant when P <0.05.
To assess whether expression of A53T within dopaminergic midbrain neurons in vivo results in its localization to mitochondrial membranes as has been previously reported in vitro [10, 6, 23, 26], we performed immunocytochemistry on A53T transgenic mice that specifically express the protein within tyrosine-hydroxylase-positive cells (Fig. 1). α-Syn was found to co-localize in both outer and inner mitochondrial membrane fractions within TH-positive midbrain neurons from HE synuclein animals (Fig. 1A&B). However, no significant co-localization of α-Syn was detected in either outer or inner mitochondrial membrane markers in LE synuclein mice or in control wild type animals (data not shown). Furthermore, α-Syn western analyses on isolated nigrostriatal mitochondria revealed the existence of both monomeric and oligomeric forms of synuclein within the mitochondrial fraction (Fig 1C). The levels of oligomeric forms of synuclein were found to increase in the presence of proteasomal inhibition-induced stress. The purity of mitochondrial preparations was subsequently confirmed via western blot analysis of mitochondrial and cytosolic markers (Fig 1B).
Fig. 1.

Expression of A53T α-syn in vivo results in its localization to the mitochondria in the form of monomers and oligomers. A, dual immunoytochemistry of TH-positive SN neuron using antibodies against synuclein (red) and outer mitochondrial membrane protein (VDAC, green). yellow = merged. B, Dual immunoytochemistry of TH-positive SN neuron using antibodies against synuclein (red) and inner mitochondrial membrane protein, complex V (ATPase, green). yellow= overlay. C. Anti-synuclein western blots of mitochondrial membrane fractions (75 μg each) isolated from the ST of 12 month old A53T (1) and A53T + epoxomicin (2) treated transgenics (M = molecular weight markers). *indicates monomer and red arrows indicate oligomers +/− proteasomal inhibition. Enrichment of the mitochondrial fractions was verified via western immunoblotting with the mitochondrial inner membrane protein ATPase and the cytoplasmic marker actin.
Using a novel synaptosomal isolation technique recently developed by our laboratory [5,18] we next assessed the impact of α-syn localization on mitochondrial function in striatal DA vs non-DA synaptosomes isolated from young (4 month) and old (12 month) HE versus LE A53T transgenic mice. In young mice, elevation of A53T synuclein had no significant impact on mitochondrial function (neither complex I vs CIV substrate-specific respiration nor mitochondrial complex I vs IV activities were impacted in either DA or non-DA synaptosomal populations; data not shown). However by 12 months of age, elevated expression of A53T was found to result in a significant decrease (nearly 40%) in mitochondrial complex I activity and subsequent complex I substrate-mediated respiration in DA synaptosomes from HE in comparison with LE α-syn- animals (Table 1); in contrast, no affect on CIV activity or substrate-mediated respiration was observed in DA populations from these two groups. In addition, no differences were noted in either CI or CIV functions in non-DA synaptosomal populations from animals expressing HE or LE α-syn.
Table 1.
CI vs CIV activities and substrate-specific respiration in ST DA vs non DA synaptosomes isolated from 12 month old high vs low (HE vs LE) A53T α-syn expressing transgenics. Uncoupled complex I-mediated oxygen consumption was measured in isolated synaptosomes using glutamate/malate as substrate in the presence of ADP and carbonyl cyanide p-trifluoromethoxyphexylhydrazone. Rotenone-sensitive respiration was calculated as the difference between oxygen consumption rate pre- and post-rotenone addition (2 μM). KCN-sensitive complex IV-mediated respiration was similarly measured using TMPD/ascorbate as substrate. n = 3 for each condition.
| CI-mediated oxygen consumption (rotenone inhibitable, nM O2/min) | CIV-mediated oxygen consumption (KCN inhibitable, nM O2/min) | |
|---|---|---|
| LE DA | 7.8 +/− 0.4 | 33.4 |
| HE DA | 3.3 +/− 0.8a | 37.1 |
| LE NDA | 17.9+/− 0.6 | 34.5 |
| HE NDA | 17.7+−0.4 | 35.1 |
| CI activity (μM NADH/μg/min | CIV activity (μM Fe-Cyt/μg/min | |
| LE DA | 17.15+/− 2.81 | 10.16+/− 1.68 |
| HE DA | 9.92+/− 0.29a | 9.0 +/−0.72 |
| LE NDA | 20.16+/− 2.33 | 8.24 +/− 0.34 |
| HE NDA | 19.38+/− 1.7 | 8.6 +/− 1.2 |
p <0.05 compared with LE controls.
Earlier studies in these transgenic lines demonstrated the presence of abnormal nerve terminals, age-related impairments in motor coordination, and age-related reductions in DA and its metabolites although no frank nigral dopaminergic neuronal loss was observed even with age [25]. In addition to these alterations, our results suggest that HE coincides with a significant age-related decrease in mitochondrial CI activity and substrate-mediated respiration. In order to assess how this impacted on mitochondrial morphology, we assessed dopaminergic SN neuron in HE versus LE via electron microscopy. Ultrastructural studies of dopaminergic neuronal mitochondria from HE versus LE A53T syn mice demonstrated a significant increase in lysosome-mediated mitochondrial autophagy (mitochondria sequestered into double-membraned autophagosomes) within the HE mice (Fig 2).
Fig. 2.
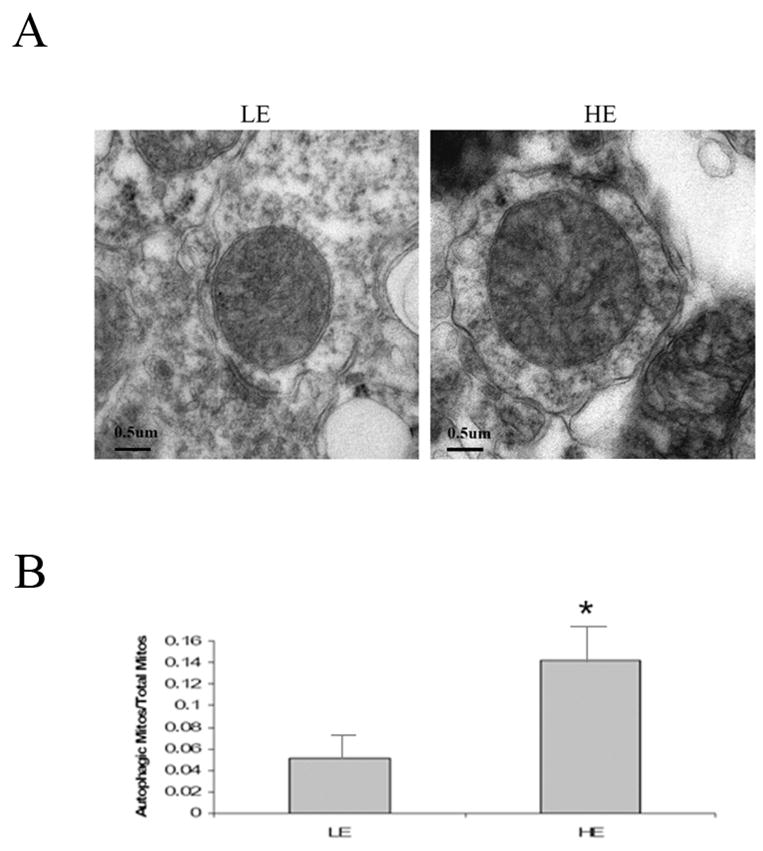
Quantitation of autophagic mitochondria in midbrain dopaminergic neurons of A53T high versus low-expressing α-syn-expressing transgenic mice (HE vs LE) at 12 months of age. A, Representative electron micrographs prepared from SN sections from pTH-A53T synuclein HE vs LE-expressing transgenics at 12 months of age. B, autophagic mitochondria versus total mitochondria were counted in at least three separate EM fields of midbrain dopaminergic neurons. n = 3; *p, 0.05 vs Low expressing transgenic (LE).
α-Syn-containing intraneuronal inclusions are found in both PD itself and in animal models of the disease employing either mitochondrial or proteasomal inhibitors [11, 19]. Previous studies had demonstrated that increased expression or mutation of α-Syn has an impact on mitochondrial function [1, 2, 27] but the specific mechanism involved has remained relatively unexplored. In addition to enhanced DA SN mitochondrial pathology in α-syn expressing mice following MPTP treatment [28], exposure to pesticides such as paraquat and maneb have also been reported to lead to the presence of disorganized and degenerative mitochondria in A53T human α-syn expressing but not WT α-syn-expressing transgenic mice, suggesting the involvement of particularly mutant α-syn in mitochondrial dysfunction [21].
Recent in vitro studies have demonstrated that alterations in intracellular pH conditions (acidic conditions) can result in translocation of α-Syn to the mitochondria [6]. Accumulated α-Syn was found to be associated with selective mitochondrial complex I inhibition, decreased mitochondrial membrane potential, increased cytochrome C release and increased production of reactive oxygen species indicating that mitochondrial accumulated α-syn may interact with complex I and interfere with its function [10, 23, 26]. This effect has been reported to be further enhanced following expression of the pathogenic mutant A53T versus the wild type (WT) form of the protein [10]. However, most of these in vitro studies were performed in non-dopaminergic cell lines (except Devi et al., 2008, where they used human PD autopsy samples) and the effects of A53T mutant versus WT forms on mitochondrial function were not generally explored. A recent report suggested that a portion of α-syn may be present in the membrane of mitochondria within normal dopaminergic neurons of the SNpc [15]. Another study using western blot analysis of mitochondrial fractions isolated from different regions of rat brain demonstrated that α-syn could be detected in mitochondria from many brain regions, including olfactory bulb, striatum, hippocampus, cerebral cortex, and brain stem [16]. Although, these in vivo studies have demonstrated the presence of α-syn within dopaminergic neuronal mitochondria, the consequences of protein accumulation on mitochondrial function has yet to be assessed in an in vivo model particularly in the context of aging.
In the present study, we demonstrated that localization of A53T α-syn to the mitochondria of SN DA neurons in vivo was found to coincide with a selective age-related decline in dopaminergic CI activity and subsequent substrate-mediated respiration. The observed selective complex I inhibition and mitochondrial respiration in these older mice may be due to relatively lower threshold inhibition for synaptic mitochondrial CI compared to other OXPHOS enzymes [9]. Our results are in contrast to a recent in vivo study using transgenic mice overexpressing A53T α-syn under the general neuronal PrP promoter where the authors reported that CI inhibition in these mice was neither age-related nor dependent on the levels of α-syn oligomers [17]. The discrepancy between these findings and our current results may be due pan-neuronal expression and the use of whole brain mitochondria to measure CI activity in the former. Transgenics utilized for our analyses overexpress A53T α-syn specifically within dopaminergic nigral neurons and moreover CI activity was assessed in isolated nerve terminals (synaptosomes) specifically from this neuronal population.
We also observed an increase in lysosome-mediated mitochondrial autophagy (mitophagy) in A53T overexpressing midbrain dopaminergic neurons, likely a compensatory attempt to remove defective mitochondria; the mechanism of mitochondrial turnover is predominantly autophagic sequestration and delivery to lysosomes for hydrolytic degradation. Timely elimination of aged and dysfunctional mitochondria is essential to protect cells from elevated levels of ROS and release of proapoptotic proteins [12]. This may in part explain lack of actual midbrain dopaminergic cell loss in these lines. Ongoing studies are assessing the impact of agents that alter the rate of mitophagy (lithium, rapamycin) on mitochondrial function in these lines are currently underway.
In conclusion, our data suggests that α-syn physically localizes to dopaminergic mitochondria in vivo and impacts on mitochondrial function. The aberrant interaction of mutant α-syn with mitochondria may ultimately influence the age-related midbrain dopaminergic mitochondrial dysfunction and subsequent neuropathology.
Acknowledgments
We would like to thank Daniel Crippen for technical assistance with EM studies and Dr. Bharat Srinivas for early discussions on this project. This work was financially supported by NIH R01 AG12141 to JKA and American Parkinson's Disease Association to SJC.
Footnotes
Conflict of interest
The authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 3.Budd DC, Nicholls DG. Protein kinase C-mediated suppression of the presynaptic adenosine A1 receptor by a facilitatory metabotropic glutamate receptor. J Neurochem. 1995 Aug;65(2):615–21. doi: 10.1046/j.1471-4159.1995.65020615.x. [DOI] [PubMed] [Google Scholar]
- 4.Buttner S, Bitto A, Ring J, Augsten M, Zabrocki P, Eisenberg T, Jungwirth H, Hutter S, Carmona-Gutierrez D, Kroemer G, Winderickx J, Madeo F. Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J Biol Chem. 2008;283(12):7554–60. doi: 10.1074/jbc.M708477200. [DOI] [PubMed] [Google Scholar]
- 5.Chinta SJ, Kumar MJ, Hsu M, Rajagopalan S, Kaur D, Rane A, Nicholls DG, Choi J, Andersen JK. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci. 2007 Dec 19;27(51):13997–4006. doi: 10.1523/JNEUROSCI.3885-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008;314(10):2076–89. doi: 10.1016/j.yexcr.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 8.Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davey GP, Peuchen S, Clark JB. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem. 1998 May 22;273(21):12753–7. doi: 10.1074/jbc.273.21.12753. [DOI] [PubMed] [Google Scholar]
- 10.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283(14):9089–100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Sudhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007 Jun 15;462(2):245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klivenyi P, Siwek D, Gardian G, Yang L, Starkov A, Cleren C, Ferrante RJ, Kowall NW, Abeliovich A, Beal MF. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol Dis. 2006;21:541–548. doi: 10.1016/j.nbd.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 15.Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport. 2007;18:1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- 16.Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, Yang H, Uéda K, Chan P, Yu S. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett. 2009 May 1;454(3):187–92. doi: 10.1016/j.neulet.2009.02.056. [DOI] [PubMed] [Google Scholar]
- 17.Loeb V, Yakunin E, Saada A, Sharon R. The transgenic overexpression of alpha-synuclein and not its related pathology associates with complex I inhibition. J Biol Chem. 2010 Mar 5;285(10):7334–43. doi: 10.1074/jbc.M109.061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallajosyula JK, Kaur D, Chinta SJ, Rajagopalan S, Rane A, Nicholls DG, Di Monte DA, Macarthur H, Andersen JK. MAO-B elevation in mouse brain astrocytes results in Parkinson's pathology. PLoS ONE. 2008;20;3(2):e1616. doi: 10.1371/journal.pone.0001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- 20.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 21.Norris EH, Uryu K, Leight S, Giasson BI, Trojanowski JQ, Lee VM. Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol. 2007;170:658–666. doi: 10.2353/ajpath.2007.060359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orth M, Tabrizi SJ, Tomlinson C, Messmer K, Korlipara LV, Schapira AH, Cooper JM. G209A mutant alpha synuclein expression specifically enhances dopamine induced oxidative damage. Neurochem Int. 2004;45:669–676. doi: 10.1016/j.neuint.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 23.Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008;65(7–8):1272–84. doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 25.Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol. 2002 May;175(1):35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- 26.Shavali S, Brown-Borg HM, Ebadi M, Porter J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett. 2008;439(2):125–8. doi: 10.1016/j.neulet.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alphasynuclein- induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- 28.Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- 29.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 30.Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM, Schapira AH. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet. 2000;9:2683–2689. doi: 10.1093/hmg/9.18.2683. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 32.Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]