

1. Introduction
During the past three decades, nitric oxide (NO) has been shown to be an important signaling molecule in a wide variety of physiological processes, including blood pressure control, neurotransmission, immune response, and cell death [1–11]. Since these discoveries, research efforts have been directed towards development of exogenous NO donors that can deliver NO to biological targets to elicit desired responses [12–15]. Various NO donors have been developed in this regard, including organic nitrites and nitrates [16], nitrosothiols [17], diazeniumdiolates (NONOates) [18,19], and lastly transition metal-based NO donors such as sodium nitroprusside [20,21]. Organic NO donors such as glyceryl trinitrate and isosorbide dinitrate have been successfully used to treat hypertension and episodes of angina pectoris. Nitrosothiols have shown some promise in regulating immune response, while selected NONOates were shown to improve neurotransmission. Use of exogenous NO donors as potential anti-cancer agents has also been explored [22–25]. Indeed, NO has been shown to induce both apoptosis (programmed cell death) and cell destruction at elevated concentrations (mM range) [26–31]. Precise targeting of malignant sites versus healthy tissues however remains as a challenge in the use of systemic NO donors in anticancer therapy. Most NO donors in current use are non-specific in that they release NO spontaneously, although in some cases the rate of NO release can be modulated by ubiquitous stimuli such as temperature, pH or enzymes. Controlled (favorably triggered) release of NO at a selected site is the key for successful employment of an NO donor in the treatment of tumors and localized malignancy.
With the advent of photodynamic therapy (PDT) [32,33] as a common treatment for certain (especially skin) cancers [34–38], light-activated NO donors have gained much attention. The site-specificity provided by laser treatment allows for more precise targeting than systemic drugs alone. Early on, it was recognized that NO complexes of transition metals (metal nitrosyls) could release NO when exposed to light. For example, several iron-based nitrosyls including sodium nitroprusside (SNP, Na2[Fe(NO)(CN)5]) [39–44] and Roussin’s salts [20,41,45–48] were found to release NO when exposed to light. However, these complexes also release NO spontaneously (i.e. in the dark), and often changes in pH and temperature also induce loss of NO, rendering them non-specific for PDT. Additionally, side effects from labile ancillary cyanide ligands often limit the use of SNP [49–51]. Chelating ligands provide some relief from these problems. For example, the iron complex [(PaPy3)Fe(NO)](ClO4)2 was the first of many NO donors to be studied by Mascharak and co-workers [52–54]. This nitrosyl is derived from a tightly coordinating, pentadentate ligand that imparts high stability in donor solvents like MeCN or DMF, and it was shown to cleanly release NO when exposed to low-intensity visible light. Unfortunately, like many other iron nitrosyls, [(PaPy3)Fe(NO)](ClO4)2 exhibits unpredictable stability under biological conditions. In general, iron nitrosyls like Roussin’s salts and [(PaPy3)Fe(NO)](ClO4)2 undergo hydrolytic decomposition in aqueous solutions under physiological conditions (pH ~7, presence of oxygen) [55–58] and problems like NOx disproportionation [59–64] or ferric hydroxide (or oxide) precipitation limit the use of such iron-based NO donors. Several NO-releasing complexes of chromium [65–67] and manganese [68,69] have also been described, but are limited by similar effects. The only exception is the manganese nitrosyl [(PaPy3)Mn(NO)](BF4) [70–72]. This photoactive NO donor has been used to deliver NO to biological targets like myoglobin, cytochrome c oxidase, and papain [73,74]. Clearly, the number of metal nitrosyls that release NO exclusively when triggered by light and exhibit stability under physiological conditions is very limited. Researchers have therefore looked into more stable transition metal analogues, such as ruthenium nitrosyls to achieve these goals during the past several years. The results of such studies are included in this review.
Much like other complexes of ruthenium, the ruthenium nitrosyls are substitutionally inert at room temperature. However, some of these nitrosyls release NO when exposed to light. For example, nitrosyls of simple compositions such as K2[Ru(NO)(Cl)5] readily release NO when exposed to UV light. This property of ruthenium nitrosyls has been known for some time. One must note at this point that many coordination complexes of ruthenium (without NO) are also sensitive to light and undergo light-driven substitution reactions [75–78]. Typically, upon exposure to light, one coordinated ligand (L) is replaced by a solvent molecule, as indicated in Eq. (1).
| (1) |
Sauvage and co-workers have studied the photochemistry of such ligand replacement reactions quite extensively [75]. The reaction is usually driven by ultraviolet (UV) light in the region of 200–450 nm and the photosensitivity stems from accessibility to substitutionally active excited states with UV irradiation. The oxidation state of the metal center is crucial for such photoactivity. Only complexes with Ru(II) centers experience photosubstitution reactions. Also, photosubstitution reactions occur only with a select group of ligands, primarily neutral N donors. For example, ligands such as ammine (NH3) [76,79–82], nitriles (R–C≡N) [83,84] as well as pyridine (py) and related molecules (bpy, phen) [84–86] have been reported in the literature as photoactive ligands. The photochemistry of the ruthenium complexes of these ligands, studied by various groups, constitutes a significant part of the coordination chemistry of ruthenium. Indeed, many exciting developments in the area of fluorescence, luminescence, electron transfer, and molecular dynamics have their origin in such studies and have been reviewed elsewhere [87–90].
As mentioned above, the quest for photoactive and stable NO donors has raised interest in the photochemistry of ruthenium complexes with one or more NO ligands. The nature of metal-NO bond is however more involved compared to a simple dative bond. Free NO is an odd electron molecule and contains one unpaired electron in its π* orbital. When bound, NO can either donate to or accept significant amount of electron density from the metal center. As a result, NO binds metal centers in different formal states, such as NO+, NO•, or NO−. In 1974, Enemark and Feltham noted the difficulty of assigning oxidation states to both NO and the metal center [91]. A special notation of {M-NO}n was therefore devised to denote a metal-NO bond where n = the total number of electrons in the metal d plus the NO π* orbital. For example, in the case of ruthenium, a {Ru-NO}6 unit could represent one of two possible combinations of formal oxidation states of the metal center and NO, namely Ru(III)–NO• and Ru(II)–NO+.
In ruthenium chemistry, the {Ru-NO}6 configuration is generally accepted as NO+ bound to a Ru(II) center. This is largely based on the high NO stretching frequencies (νNO = 1820–1960 cm−1) noted with {Ru-NO}6 nitrosyls, versus that of either free NO (~1750 cm−1) [1,2] or bound NO• (1650–1750 cm−1) in {Ru-NO}7 species [92–97]. Additionally, {Ru-NO}6 nitrosyls exhibit spectroscopic properties similar to those of true Ru(II) species (low-spin, diamagnetic, sharp 1H-NMR spectra, and EPR silent). Mössbauer [53,98] and K-edge X-ray absorption [99] spectroscopic data on analogous {Fe-NO}6 nitrosyls with similar NO stretches have unequivocally established their formal {Fe(II)–NO+} description. Although similar data on {Ru-NO}6 species have not been reported, the {Ru(II)–NO+} formulation best describes most of the {Ru-NO}6 nitrosyls. However, photorelease of NO from {Ru-NO}6 nitrosyls often results in Ru(III) photoproducts since the photoreleased NO leaves as NO• (Eq. (2), vide infra).
| (2) |
To date, several ruthenium nitrosyls with {Ru-NO}7 configuration have also been isolated and characterized [92–96]. These NO complexes exhibit lower νNO values (1650–1750 cm−1) and a characteristic S = ½ EPR signal of the bound NO• radical near g ≈ 2. However, no structural data are available on any of these nitrosyls and their photochemistry appears to be more involved (vide infra). This review is more focused on NO complexes with the {Ru-NO}6 configuration. In the following sections, the properties, photoactivity, and biological uses of such photoactive ruthenium nitrosyls are described. Also, the parameters of electronic absorption spectra and quantum yield (φ) values of NO photorelease for these nitrosyls are listed in Table 1.
Table 1.
Quantum yield (φ) values and absorption parameters of {Ru-NO}6 nitrosyls. Solvent conditions and irradiation wavelengths are shown.
| Complex | Quantum Yield φ (λirr in nm) | Solvent (pH) | λmax in nm (ε in M−1cm−1) | Reference |
|---|---|---|---|---|
| (1) K2[Ru(NO)(Cl)5] | 0.06 (320) | H2O (7.0) | 335 (6600) 520 (50) |
[107] |
| (2) [Ru(NO)(Cl)3]n | 0.012 (320) | H2O (7.0) | [107] | |
| (5) trans-[Ru(NH3)4(NO)(py)](BF4)3 | 0.17±0.01 (310) 0.11±0.01 (330) 0.13±0.01 (330) 0.18±0.01 (330) 0.07±0.02 (370) |
H2O (3.0) (3.0) (4.4) (6.4) (4.5) |
324 (160) | [116,118] |
| (6) trans-[Ru(NH3)4(NO)(Im)](BF4)3 | 0.042±0.002 (313 0.063±0.003 (313) 0.079±0.003 (313) 0.033±0.004 (330) 0.017±0.003 (370) |
H2O (2.13) (3.45) (4.32) (2.13) (2.13) |
324 (660) | [118] |
| (7) trans-[Ru(NH3)4(NO)(4-acpy)](BF4)3 | 0.10±0.02 (313) 0.13±0.03 (313) 0.19±0.03 (313) 0.07±0.01 (334) 0.04±0.02 (370) |
H2O (2.13) (3.45) (4.32) (2.13) (2.13) |
330 (340) | [116,118] |
| (8) trans-[Ru(NH3)4(NO)(P(OEt)3)](PF6)3 | 0.30±0.05 (310) | H2O (2.0) | 316 (216) | [118] |
| trans-[Ru(NH3)4(NO)(nic)](BF4)3 | 0.07±0.01 (310) 0.08±0.01 (334) <0.001 (370) <0.001 (480) |
H2O (2.0) (2.0) (2.0) (2.0) |
320 (160) | [118] |
| trans-[Ru(NH3)4(NO)(isn)](BF4)3 | 0.04±0.01 (310) 0.05±0.01 (330) 0.05±0.01 (330) 0.07±0.01 (330) <0.01 (370) |
H2O (3.0) (3.0) (4.63) (4.95) (3.0) |
268 (1300) | [118] |
| trans-[Ru(NH3)4(NO)(pz)](BF4)3 | 0.13±0.03 (313) 0.17±0.03 (313) 0.23±0.03 (313) 0.10±0.01 (334) 0.07±0.01 (450) |
H2O (2.13) (3.45) (4.32) (2.13) (2.0) |
302 (660) | [116,118] |
| trans-[Ru(NH3)4(NO)(L-Hist)](BF4)3 | 0.045±0.003 (313) 0.067±0.004 (313) 0.086±0.004 (313) 0.039±0.005 (334) 0.022±0.002 (370) |
H2O (2.13) (3.45) (4.32) (2.13) (2.13) |
320 (410) | [116,118] |
| trans-[Ru(NH3)4(NO)(4-pic)](BF4)3 | 0.06±0.01 (310) 0.08±0.01 (330) 0.12±0.01 (330) 0.26±0.02 (330) 0.40±0.01 (330) 0.04±0.02 (370) <0.001 (410) <0.001 (480) |
H2O (2.0) (2.0) (3.45) (5.0) (6.0) (2.0) (2.0) (2.0) |
325 (220) | [116,118] |
| (9) trans-[(cyclam)Ru(NO)(Cl)](PF6)2 | 0.008±0.001 (334) 0.010±0.002 (313) 0.009±0.001 (334) <0.002 (370) 0.011±0.002 (313) 0.012±0.002 (334) <0.002 (370) 0.10±0.01 (334) 0.16±0.05 (355) |
H2O (1.0) (4.75) (4.75) (4.75) (4.9) (4.9) (4.9) (7.4) (7.4) |
263 (3011) | [126] |
| (14) [Ru(salen)(NO)(Cl)] | 0.13±0.01 (365) 0.09±0.01 (436) 0.07±0.02 (546) |
MeCN | 384 (6300) | [139a,139b] |
| (15) [Ru(salen)(NO)(ONO)] | 0.067±0.002 (365) 0.058±0.002 (436) |
MeCN | 370 (4600) | [139a] |
| (16) [Ru(salen)(NO)(H2O)]Cl | 0.005±0.001 (365) | MeCN | 354 (3800) | [139a] |
| (17) [Ru(tBu4salen)(NO)(Cl)] | 0.030±0.002 (365) 0.040±0.002 (436) |
MeCN | 370 (6900) | [139a] |
| (18) [Ru(tBu2salophen)(NO)(Cl)] | 0.077±0.008 (365) 0.033±0.002 (436) 0.014±0.001 (546) |
MeCN | 458 (10300) | [139a] |
| (19) [Ru(tBu4salophen)(NO)(Cl)] | 0.055±0.003 (365) 0.027±0.002 (436) 0.011±0.002 (546) |
MeCN | 468 (11100) | [139a] |
| (25) [Ru3O(CH3CO2)6(pic)2(NO)]PF6 | 0.038±xx (365) 0.019±xx (468) |
MeCN | [150] | |
| (26) [(PaPy3)Ru(NO)](BF4)2 | 0.11±0.02 (355) 0.06±0.01 (300) 0.05±0.01 (410) <0.01 (532) 0.042 (300) |
H2O (7.4) MeCN MeCN MeCN DMF |
410 (1550) | [164,179] |
| (27) [(PaPy2Q)Ru(NO)](BF4)2 | 0.20±0.03 (355) 0.14±0.02 (300) 0.17±0.02 (410) 0.075±0.012 (300) |
H2O (7.4) MeCN MeCN DMF |
420 (1030) | [164] |
| (28) [(bpb)Ru(NO)(Cl)] | 0.062±0.010 (300) | MeCN | 380 (5960) | [164] |
| (31) [(Me2bpb)Ru(NO)(Cl)] | 0.06±0.02 (300) | MeCN | 395 (5300) | this work |
| (32) [(Me2bpb)Ru(NO)(OH)] | 0.05±0.01 (300) <0.01 (500) |
MeCN DMF |
390 (5100) | [this work, 191] |
| (33) [(Me2bpb)Ru(NO)(py)]BF4 | 0.02±0.005 (300) | MeCN | 394 (7170) | this work |
| (34) [(Me2bpb)Ru(NO)(Im)]BF4 | 0.07±0.02 (300) | MeCN | 394 (7160) | this work |
| (35) [(Me2bpb)Ru(NO)(4-vypy)]BF4 | 0.18±0.02 (300) 0.11±0.01 (300) |
EtOH HEMAgel |
392 (7800) | [168] |
| (43) [(SBPy3)Ru(NO)](BF4)3 | 0.18±0.03 (300) <0.01 (532) |
MeCN |
315 (6060) | this work |
| (44) [Ru(terpy)(bpy)(NO)](PF6)3 | 0.14±0.02 (355) | H2O (2.0) | 332 (10470) 358 (8320) 480 (690) |
[180] |
| (45) [Ru(terpy)(NH2-NH2ph)(NO)](PF6)3 | 0.46±0.02 (355) | H2O (2.0) | 325 (12880) 355 (9550) |
[180] |
| (46) [Ru(terpy)(NH-NHq)(NO)](PF6)3 | 0.47±0.02 (355) 0.0065±0.001 (532) |
H2O (2.0) (2.0) |
324 (18620) 358 (14130) 510 (4470) |
[180] |
| (47) [Ru(NO2)(bpy)(terpy)]PF6 | 0.036±0.009 (355) | H2O (7.4) | 330 (9330) 420 (4900) 448 (5250) |
[183] |
| (50) cis-[Ru(NO2)(py)(bpy)2]PF6 | 0.007±0.001 (355) | H2O (7.4) | 332 (7240) 416 (3720) |
[183] |
| (51) cis-[Ru(NO2)(4-pic)(bpy)2]PF6 | 0.009±0.001 (355) | H2O (7.4) | 342 (5500) 418 (4170) |
[183] |
| (52) cis-[Ru(NO2)(pz)(bpy)2]PF6 | 0.037±0.009 (355) | H2O (7.4) | 408 (4790) | [183] |
| (53) [(Py3P)Ru(NO)]BF4 | 0.15±0.01 (300) 0.050±0.004 (532) |
MeCN | 530 (400 sh) | [179] |
| [(Py3P)Ru(NO)(Cl)] | 0.17±0.01 (300) 0.064±0.003 (532) |
MeCN | 520 (330 sh) | [179] |
| (56) [(Me2bpb)Ru(NO)(Resf)] | 0.052±0.008 (500) | DMF | 395 sh (5940) 500 (11920) |
[191] |
| (57) [(Me2bQb)Ru(NO)(Resf)] | 0.102±0.009 (500) | DMF | 510 (12300) | [191] |
| (58) [Ru(NH3)5(pz)Ru(bpy)2(NO)](PF6)5 | 0.08±0.01 (355) 0.025±0.002 (532) |
H2O (4.5) (4.5) |
408 (5010) 486 (7410) 530 (9550) |
[192,193]] |
| (59) [Ru(py)(NH3)4(pz)Ru(bpy)2(NO)] (PF6)5 | 0.05±0.01 (355) 0.038±0.002 (532) |
H2O (4.5) (4.5) |
400 (5010) 556 (18620) |
[193] |
| (60) [Ru(4-acpy)(NH3)4(pz)Ru(bpy)2(NO)](PF6)5 | 0.07±0.01 (355) 0.036±0.002 (532) |
H2O (4.5) (4.5) |
560 (17380) | [193] |
2. UV photoactivity
2.1 Photoactive {Ru-NO}6 nitrosyls with monodentate ligands
2.1.1 Ruthenium nitrosyl chlorides
The earliest accounts of photoactive ruthenium nitrosyls indicate the photosensitivity of the {Ru-NO}6 moiety [100]. Various ruthenium nitrosyls with chlorides as additional ligands have been studied. Two nitrosyls of this kind, namely K2[Ru(NO)(Cl)5] (1) and RuNOCl3 (2), have been thoroughly characterized and are commercially available. Although 1 is a monomeric {Ru-NO}6 nitrosyl [101,102], 2 is most possibly a chloro-bridged polymeric species {μ-(Cl)2-[RuNO(Cl)2]}n containing waters of hydration [103]. Related solvato species [Ru(NO)(Cl)3(solv)2] (solv = acetonitrile for 3a; DMSO for 3b) have also been reported [104].
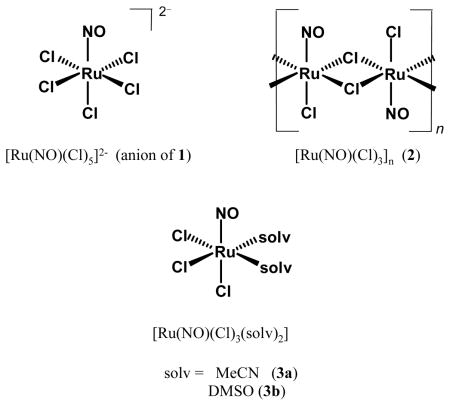
In 1971, Cox and Wallace observed that an acidic aqueous solution of potassium pentachloronitrosylruthenate(III) (1) turned brown over a period of hours when exposed to light, yet was thermally stable under dark conditions [100]. Controlled experiments confirmed that when an aqueous solution of 1 was exposed to UV light, the aqua complex [RuIII(H2O)(Cl)5]2− was formed (Fig. 1). Several years later, Vasilevskii and co-workers reported that the photoproduct of UV photolysis of 1 did not exhibit the characteristic νNO band in its IR spectrum thus confirming the loss of NO ligand [105].
Fig. 1.

Changes (indicated by arrows) in the electronic spectrum of [Ru(NO)(Cl)5]2− in an aqueous solution containing 1 M HClO4 when exposed to UV light over the course of 30 minutes [105]. Spectra in set a indicate absorbance in the far UV, while those in set b are enlarged view in near UV region; exposure times are as follows: 1 (0 min), 2 (7 min), 3 (17), 4 (27), 5 (10), 6 (20), 7 (30). Inset: X-ray structure of [Ru(NO)(Cl)5]2− (anion of 1) [101]. Selected bond distances and angle: Ru–N(O) = 1.738(2) Å, N–O = 1.131(3) Å, Ru–N–O = 176.7(5)°.
In 1983, Sinitsyn and co-workers investigated the nature of the product(s) following UV irradiation of 1 in aqueous solution by EPR spectroscopy [106]. The axial EPR signal (Fig. 2), characteristic of a paramagnetic species, confirmed the presence of low-spin d5 Ru(III) in the photoproduct. The resulting stoichiometry agreed with previous conclusions that irradiation resulted in homolysis of the {Ru-NO}6 moiety, generating Ru(III) and NO• (Eq. (2)). Subsequent EPR studies (vide infra) have confirmed that photorelease of NO from most {Ru-NO}6 nitrosyls (see polypyridine section for exceptions) affords Ru(III) photoproduct(s) regardless of the solvent (H2O, MeCN, MeOH, DMSO or DMF).
Fig. 2.

EPR spectrum of a photolyzed solution of K2[Ru(NO)(Cl)5] (1) in DMSO with UV light, indicating the formation of Ru(III) species (g-values as indicated, DPPH as standard marker at g = 2.0036) [106].
In 1996, the efficiency of NO release from 1 was quantified by Trentham and coworkers [107]. The {Ru-NO}6 nitrosyl was found to release NO in aqueous solution upon exposure to UV light (355 nm) with a quantum yield value of φ = 0.06. The related complex 2 released NO much less efficiently, exhibiting a quantum yield nearly five times lower (φ = 0.012). This difference in φ value indicates that an increase in the number of negatively charged ligands in 1 (containing five chlorides per Ru) versus 2 (only three chlorides per Ru) improves the quantum efficiency. The photochemistry of other chloro {Ru-NO}6 species such as [Ru(NO)(Cl)4(H2O)]− and [Ru(NO)(Cl)2(H2O)3]+ [108] has not been explored. Both 1 and 2 undergo clean NO photorelease in aqueous 1 M HCl solutions. However, typical physiological conditions invariably lead to spontaneous dissociation of the Cl− ligand(s) and/or redox reactions affording Ru(II) or Ru(IV) species [104,105]. Many Ru(II) species with water as ligands readily form new bonds at room temperature with biological molecules like DNA [109,110] leading to further complication. As a result, the biological utility of ruthenium nitrosyls of this type as NO donors is quite limited.
2.1.2 Ruthenium nitrosyl ammines
This class of photoactive ruthenium nitrosyls has been extensively studied by Franco and co-workers in recent years. As an extension of earlier work on the light-induced dissociation of ammonia (NH3) [76,79–82], Franco and co-workers also noted photorelease of NO from such complexes (when NO is present as a ligand) in acidic aqueous solutions [111]. The general structure of this class of NO donors, depicted below, includes the {Ru-NO}6 core ligated to four equatorial ammine ligands, and a sixth ligand X (trans to NO) which is varied. Although the simplest of these nitrosyls, namely [Ru(NH3)5(NO)](BF4)3 (4) was synthesized as far back as 1952 [112–114], the photochemistry of such species was not quantitatively investigated until some forty years later [111].
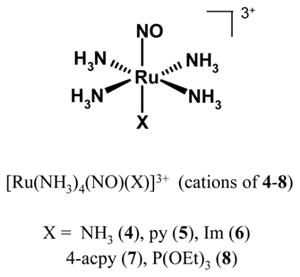
As part of their photochemical investigation on NO from complexes of type trans- [Ru(NH3)4(NO)(X)]3+, Franco and co-workers have synthesized a series complexes (examples include 4–8) to estimate the effects of the ligand X on NO photolability [115–117]. These complexes are soluble in H2O, and stable under acidic conditions (pH < 5). Exposure to UV light (300–370 nm) results in NO release (Eq. (2)) which can be monitored by using an NO-sensitive electrode (Fig. 3). In acidic solution, the photoproduct trans-[Ru(NH3)4(X)(H2O)]3+, exhibits its EPR signal at g ≈ 2.14 [118]. At pH ~ 6.0, it is converted into the hydroxo species trans-[Ru(NH3)4(X)(OH)]2+ [119,120]. Quantum yield (φ) measurements on this series of nitrosyls have afforded values ranging from φ = 0.04 (6, Im) to 0.23 (7, 4-acpy) [115,116]. Significant NO photolability (φ = 0.30) has been observed when X = phosphite ligand such as P(OEt)3 (8) [121,122]. The dπ(Ru) → π*(NO) metal-to-ligand charge transfer (MLCT) transition around 330–450 nm is believed to be responsible for the NO photolability of these nitrosyls. Results of theoretical studies support this view [123]. More discussion on their spectroscopic parameters could be found in published reviews [115,116]. It is important to note that results from studies on trans-[Ru(NH3)4(NO)(X)]3+ type of species have clearly shown that the peripheral ligands, particularly the ligand trans to NO, have a significant effect on NO release rates from ruthenium nitrosyls.
Fig. 3.

A typical NO amperogram obtained with an NO-sensitive electrode in aqueous solution, as mentioned for the series of complexes 4–8 by Franco and co-workers. X-ray structure of [Ru(NH3)4(NO)(P(OEt)3)]3+, the cation of 8. Selected bond distances and angle: Ru–N(O) = 1.774(8) Å, N–O = 1.130(1) Å, Ru–N–O = 175.1(8)° [122].
Results of extensive redox and kinetic studies have also shown that trans-[Ru(NH3)4(NO)(X)]3+ type of nitrosyls also release NO upon reduction and the redox potential of the NO+/NO couple and the lability of the NO ligand from the reduced species can be controlled by judicious choice of the trans ligand [116]. Since phosphite ligands raise the reduction potential (and makes it biologically accessible) and stabilize Ru(II) center, the authors have used trans-[Ru(NH3)4(NO)(P(OEt)3)](PF6)3 (8) to induce hypotensive effects in hippocampus slices of mice and live animals [124]. The toxicity of this type of ruthenium nitrosyl is less than SNP [49–51]. Such biological effects are however not related to NO photolability and will not be discussed further.
2.2 Photoactive nitrosyls derived from polydentate ligands
2.2.1 Cyclam (1,4,8,11-tetraazacyclotetradecane)
The use of polydentate ligands in coordination chemistry provides several advantages over monodentate ligands such as Cl− or NH3. Multidentate ligands allow greater control over stability, solubility and structural features of the resulting complexes. Complexes derived from tetra- or pentadentate ligand are often stable in biological media. Such stability is required to (a) sustain pharmacological activity, (b) reduce the toxicity of free metal ion(s), and (c) avoid non-specific binding of the partially-ligated metal ion(s) to other biological molecules. One early example of a ruthenium nitrosyl derived from a macrocyclic amine is the complex trans-[(cyclam)Ru(NO)(Cl)]X2 (9), (cyclam = 1,4,8,11-tetrazacylcotetradecane, and X = Cl−, ClO4−) reported by Clarke and co-workers [96].

This nitrosyl spontaneously releases NO following reduction in aqueous solution (Fig. 4). A metastable {Ru-NO}7 species is formed in situ (EPR signal with gx = 1.995, gy = 2.035 and gz = 1.883) [96] followed by slow NO release over the course of several hours (k = 6.40 × 10−4 s−1 at 25 °C). Although similar {Ru-NO}7 species have previously been observed in solution, thermal decomposition of Ru–NO• unit has prevented structural characterization of any {Ru-NO}7 species so far. Reductive NO release from 9 in cellular tests has shown some promise. Administration of 9 in hypertensive rats results in reduction of blood pressure, much like SNP, although the duration of such effect is much longer in case of 9 [125]. The photochemistry of 9 has been reported in a recent account by da Silva and co-workers [126]. This nitrosyl releases NO with φ values ranging from <0.002 to 0.16 depending on pH and the wavelength of irradiation.
Fig. 4.
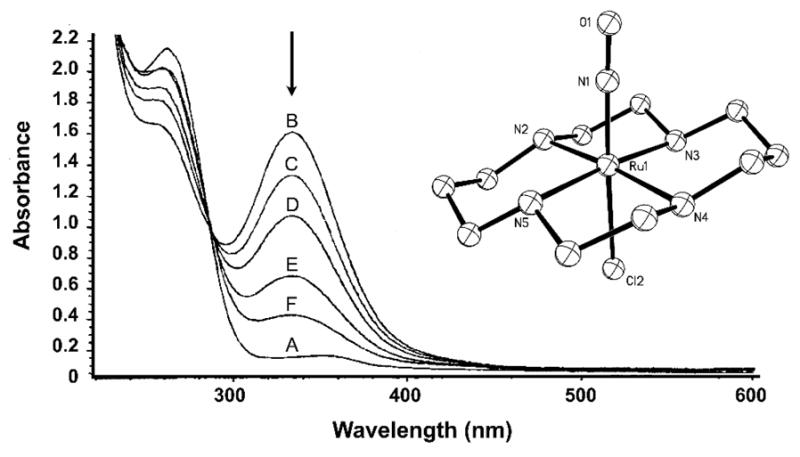
Electronic absorption spectra of 9 before reduction (A, λmax = 263 nm), after reduction (B), and subsequent loss of NO (C through F) driven by thermal processes. Inset: X-ray structure of trans-[(cyclam)Ru(NO)(Cl)]2+, the cation of 9. Selected {Ru-NO}6 bond distances and angle: Ru–N(O) = 1.747(4) Å, N–O = 1.128(5) Å, Ru–N–O = 178.0(5)° [96].
2.2.2 {Ru-NO}6 nitrosyls derived from porphyrins
Since the principal in vivo targets of NO are heme-containing enzymes such as guanylate cyclase, myoglobin, and cytochrome c oxidase, researchers have looked into the possibility of designing NO donors based on metal nitrosyls derived from natural macrocycles like porphyrins and corroles. Photosensitivity of the Fe-NO bond(s) in heme-based nitrosyls was detected at the very early phase of such research [127–129]. However, most iron complexes of this type are highly reactive toward oxygen and hardly suitable for photochemical delivery of NO to biological targets. Consequently, {Ru-NO}n nitrosyls of porphyrin ligands have received more attention as possible photoactive NO donors. Ford and co-workers have extensively studied such nitrosyls (along with the iron analogues) in establishing the photochemical features of the {Ru-NO}n (n = 6, 7) moiety. During the past few years, this group has synthesized a variety of ruthenium nitrosyls derived from porphyrin ligands of the type [Ru(P)(NO)(X)] (P = TPP, OEP; X = Cl−, ONO−) and determined the kinetic parameters of NO photorelease via the technique of time-resolved flash photolysis [130–135]. Richter-Addo and co-workers have synthesized two similar porphyrin complexes and have studied their redox behavior [136–138]. Through a series of carefully performed experiments, Ford and co-workers have shown that exposure of both [Ru(TPP)(NO)(Cl)] (10) and [Ru(OEP)(NO)(Cl)] (11) to UV light (either 300 W Xe lamp or Nd:YAG pulsed laser) leads to photolysis of the Ru–N(O) bond (Fig. 5) to afford a single transient species (presumably [RuIII(P)(Cl)]) which decays by a single NO-dependent pathway back to the starting materials [133,134]. Unlike the previous {Ru-NO}6 nitrosyls, the porphyrin-based nitrosyls of ruthenium (and iron) undergo a fast NO-recombination reaction (Eq. (3)). The fast rate of the NO capture (k > 107 M−1 s−1) renders these nitrosyls unsuitable for efficient NO delivery. The nitrito derivatives of the heme-nitrosyls, namely, [Ru(P)(NO)(ONO)] (P = TPP, OEP, FTTP), also release NO upon UV irradiation and undergo rapid NO recombination, although via a more complicated reaction scheme (reviewed elsewhere, see ref. 130 and 131). The chemical basis for the fast NO recombination (in case of porphyrin complexes) versus irreversible NO photorelease (in case of species described in other sections) is currently unclear.
Fig. 5.

Left: X-ray structure of [Ru(OEP)(NO)(Cl)] (11) [136]. Selected bond distances: Ru–N(O) = 1.803(7) Å, N–O = 1.093(9) Å, Ru–N–O = 176.6(6)°. Right: Reaction scheme proposed by Ford and coworkers describing the photochemical reactions of 10 and 11: light-induced NO dissociation followed by rapid Ru–NO back reaction [134].
| (3) |

2.2.3 Ruthenium nitrosyls derived from N2O2 ligands
The back-reactions observed with porphyrin-based {Ru-NO}6 nitrosyls led several investigators to synthesize {Ru-NO}6 complexes with non-heme multidentate ligands. In 2002, Ford and co-workers synthesized several {Ru-NO}6 nitrosysls derived from the tetradentate ligand N,N′-bis(salicylidene)ethylenediamine (H2salen) and related Schiff base ligands (14–17) and studied their photochemistry in water and other non-aqueous solvents [139,140]. Much like the porphyrin system, the dianion of this planar ligand provides four in-plane donors – two imine nitrogens and two phenolate oxygens to the ruthenium center. The photochemistry of the [Ru(salen)(NO)(Cl)] (14) has also been studied (and subsequently characterized by X-ray crystallography) by da Cunha and coworkers [141].

In donor solvents like acetonitrile, UV irradiation (λirr = 365 nm) of 14 results in release of NO with a quantum yield of φ = 0.13 (Eq. (2)) [139], with a slow recombination rate (kNO = 0.22 M−1 s−1). In other solvents like THF or toluene, much faster back-reaction (NO recombination) is observed (Eq. (3)) [139]. Low temperature EPR studies have confirmed that the release of NO from these {Ru-NO}6 nitrosyls affords a low-spin Ru(III) photoproduct with g = 2.27 and 2.06 [141], which readily reacts with NO to regenerate the starting compound (Fig. 6). Although 14 is soluble in water and exhibits moderate extent of NO photolability under visible light (546 nm), its utility as NO donor has not been established.
Fig. 6.
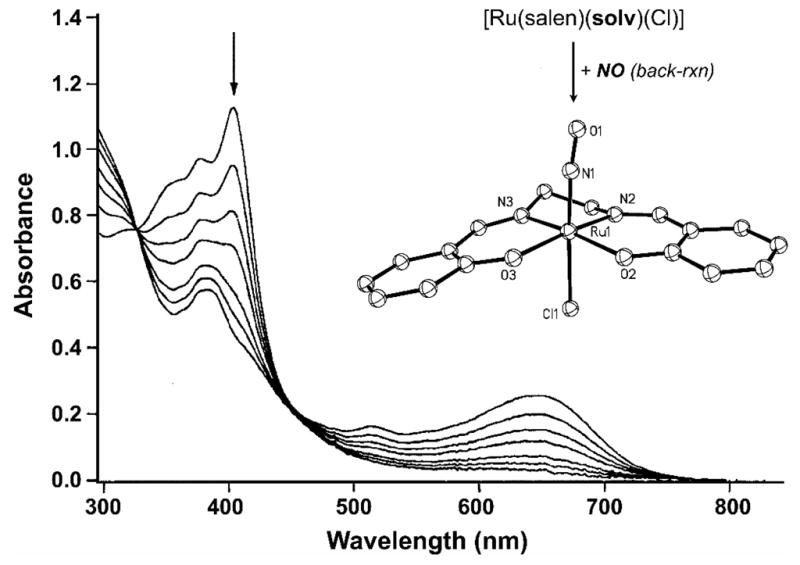
Changes in the electronic absorption spectrum observed upon rapid back-reaction of NO with a photolyzed solution of [Ru(tBu4salen)(NO)(Cl) (17) in THF [139b]. Inset: X-ray structure of complex 14. Selected bond distances and angle: Ru–N(O) = 1.728(6) Å, N–O = 1.149(7) Å, Ru–N–O = 173.7(5)° [141].
Additional {Ru-NO}6 nitrosyls with substituted salen ligands as well as different sixth ligands have also been synthesized and characterized by Ford and co-workers [139]. Interestingly, when the axial chloride ligand trans to NO is changed to O-bound nitrite (as in [Ru(salen)(NO)(ONO)] (15)) or a neutral ligand like water (as in [Ru(salen)(NO)(OH2)]+ (cation of 16)), an interesting trend in photochemical efficiency is observed. When irradiated with 365 nm light, the three nitrosyls exhibit a clear trend in quantum yields in the series Cl− > ONO− > H2O (φ = 0.13 > 0.067 > 0.005, respectively), which clearly corresponds to decreasing σ-donor strength of the trans ligand. The nitrosyls with substituted salen ligands like [Ru(tBu4salen)(NO)(Cl)] (17), [Ru(tBu2salophen)(NO)(Cl)] (18) and [Ru(tBu4salophen)(NO)(Cl)] (19) all exhibit decreased quantum efficiencies (φ = 0.030, 0.077, 0.055 respectively in acetonitrile) compared to the initial complex [Ru(salen)(NO)(Cl)] (14). Finally, the rates of NO recombination for the salen-based nitrosyls are slower (k = 10−2 to 10−4 M−1 s−1) compared to those of the porphyrin-based nitrosyls.

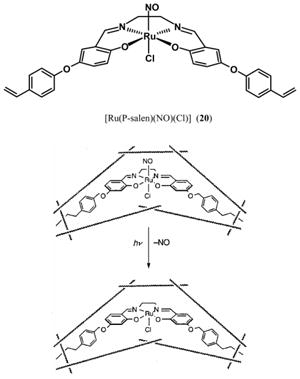
The persisting problem of NO recombination of the salen-based {Ru-NO}6 nitrosyls (and failure to serve as efficient NO donor) has recently been circumvented by Borovik and co-workers by incorporating the nitrosyl in a polymer matrix [142]. As shown below, an O-phenylvinyl appended derivative of 14 containing pendant vinyl groups, namely 20, was synthesized, and then covalently incorporated into a methacrylate-based polymer. The composite porous material exhibits photochemical behavior similar to that of [Ru(salen)(NO)(Cl)] (14) by (a) releasing NO upon exposure to UV light and (b) generating a Ru(III) photoproduct as observed by EPR spectroscopy. Borovik and co-workers have demonstrated that an aqueous suspension of the composite material can deliver NO to a biological target like myoglobin (Mb) when exposed to 370 nm UV light. In a typical experiment, the transfer of NO to Mb was complete after ~20 minutes of UV irradiation. The slow rate of NO transfer could arise from the highly cross-linked nature of the polymer. Nonetheless, this work demonstrates the utility of polymer-embedded nitrosyls as NO donors. Tfouni and co-workers have also isolated silicate sol-gel (SG) encapsulated [Ru(salen)(NO)(Cl)] (SG-14) as a brown-red vitreous material that displays a broad νNO at 1856 cm−1 and releases NO when irradiated with a Xe lamp [143]. The NO-spent material can be regenerated by reaction with NaNO2, 1 M HClO4 and Eu2+ as the reducing agent. Recently, the same group has also isolated SG-entrapped trans-[(cyclam)Ru(NO)(Cl)](PF6)2 (SG-9) and a trans-[Ru(NH3)4(NO)(X)]- modified silica gel conjugate [144]. Both materials release NO when illuminated with 334 nm light. SG-9 can be regenerated with NO while the latter nitrosyl-silica material requires NaNO2 and acid for its renewal.
In selected cases, irradiation of ruthenium nitrosyls leads to ligand isomerization and complex photochemical behavior. Miki and co-workers have studied the effects of light on ruthenium nitrosyls derived from 8-quinolinolato ligands [145–149]. These nitrosyls of the general formula [Ru(X)(2-R-qn)2(NO)] (where X = Cl−, OAc−; H2qn = 8-quinolinol derivatives with 2 position substituted with Me, Et or Cl) exhibit different extents of photoisomerization and NO photorelease under visible light. For example, the nitrosyl with 2-methyl substituted ligand (21) isomerizes to 22 both photochemically and thermally [149], while the nitrosyl with 2-chloro substituted ligand undergoes mostly thermal isomerization [147]. The thermal stability of the latter nitrosyls also depends on the isomer. For example, Miki and co-workers have recently reported the photochemical behavior of two geometrical isomers cis-1-[Ru(OAc)(2cqn)2(NO)] (23) and cis-2- [Ru(OAc)(2cqn)2(NO)] (24) derived from 2-chloro-8-quinolinol (H2cqn) [146–147]. These two structurally similar isomers exhibit very different photoreactivity under light. For example, 23 displays reversible NO photolability, even in strong donor solvents like DMSO. It undergoes ligand exchange with free 15NO upon irradiation with light (as noted by IR spectroscopy) and does not undergo isomerization [147]. Contrary to such behavior, 24 isomerizes to 23 when exposed to light. In addition, it undergoes ligand exchange with free 15NO. This example highlights difficulties that may be encountered in {Ru-NO}6 photochemistry when the complex is derived from asymmetric bidentate ligands. In general, the photochemical behavior of this class of nitrosyls changes dramatically with change in solvents, ligands (such as Cl− vs. acetate), and the substituents on the 8-quinolinolato frame. The use of a single polydentate ligand prevents this problem of photoisomerization and provides a basis for clean NO photorelease.

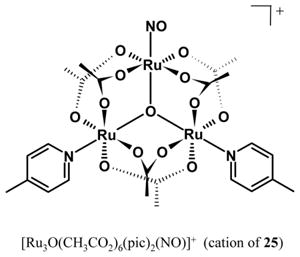
A triruthenium nitrosyl cluster with mixed N,O-coordination has recently been reported as a photochemical NO donor. Tomazela and co-workers have synthesized the complex [Ru3O(CH3CO2)6(pic)2(NO)]PF6 (25), that contains a single NO ligand [150]. The NO is released on exposure to UV light with a quantum yield of φ = 0.038 (λirr = 365 nm).
2.2.4 Ruthenium nitrosyls derived from ligands with carboxamide group(s)
Mascharak and co-workers have investigated the role of the carboxamide group in transition metal chemistry for a number of years [52–56,59,70–74,151,152]. The primary motivation behind this research was the non-heme iron enzyme nitrile hydratase [153–156]. Notably, the metal center is ligated by two carboxamido N donors (from peptide backbone) and the activity of the enzyme is regulated by NO. In 1990, Endo and coworkers discovered that the Fe–NO bond of the “dark” form of nitrile hydratase is photolyzed under light, a step that leads to enzyme activity [157–159]. In 1997 and 1998, two research groups separately reported the structures of the “light” isoform (NO photolyzed) [160] and the “dark” form of the enzyme (NO bound to active site iron) [161], proving that NO can be photolabile in non-heme systems with carboxamide group(s) as donor(s). In 2002, Mascharak and co-workers reported, for the first time, NO photolability in [(PaPy3)Fe(NO)](ClO4)2, a {Fe-NO}6 nitrosyl that mimics the photochemical characteristic of the iron site in nitrile hydratase [52]. As discussed above, the need for more stability in biological media prompted this group to synthesize the corresponding ruthenium nitrosyl namely, [(PaPy3)Ru(NO)](BF4)2 (26) [162]. As shown above, this nitrosyl is derived from a pentadentate polypyridine ligand with a single carboxamide group. It is important to note that the deprotonated carboxamido nitrogen (a strong σ-donating negatively charged donor) is trans to NO in 26. Unlike the reported water-soluble ammine complexes 4–8 (described above) that are stable only under acidic conditions (pH < 5), 26 is both soluble in water and indefinitely stable in aqueous solution in the pH range 5–9, a prerequisite for biological use. Many {Ru-NO}6 nitrosyls undergo hydrolysis of the nitrosyl moiety under neutral conditions to form coordinated nitrite (Eq. (4)). The stability of 26 even in presence of hydroxide clearly demonstrates that the strong donor ability of the carboxamido nitrogen provides protection to the {Ru-NO}6 core by reducing the electrophilicity of the NO moiety. The most desirable property of 26 is its NO photolability. This nitrosyl rapidly releases NO when exposed to UV light of low intensity (5–10 mW, 300–450 nm). Upon exposure to UV light, the yellow solution turned orange (Fig. 7) and NO is detected in the solution by NO-sensitive electrode. Unlike the porphyrin species [Ru(P)(NO)(X)] (10–11), there is no back reaction. All these features render 26 an ideal NO donor for biological use. Indeed, 26 has been successfully used to deliver NO to Mb [73,162] and cytochrome c oxidase [73]. In addition, this nitrosyl has been employed to activate soluble guanylate cyclase and relaxation of rat aorta muscle rings via light triggering [163]. The quantum yield value of this NO donor has recently improved by incorporation of a quinolyl-carboxamide moiety in place of the pyridyl-carboxamide group. The resulting nitrosyl, namely [(PaPy2Q)Ru(NO)](BF4)2 (27), releases NO more efficiently under low-intensity (5–10 mW) UV light of 410 nm wavelength (φ410 = 0.17 versus 0.05 for 26) [164].
Fig. 7.

Changes in the electronic absorption spectrum of [(PaPy3)Ru(NO)](BF4)2 in (26) in H2O (pH 7) upon exposure to UV light; no back-reaction of Ru–NO recombination was observed Inset: X-ray structure of the cation of 26. Selected bond distances and angle: Ru–N(O) = 1.779(2) Å, N–O = 1.142(3) Å, Ru–N–O = 170.9(2)° [162].
| (4) |

Mascharak and co-workers have also systematically investigated the effects of axial and in-plane ligands in ruthenium nitrosyls derived from ligands with carboxamide groups. In their recent work, this group has utilized the di-carboxamide ligand 1,2-bis(pyridine-2-carboxamido)benzene and its methylated analogue (H2bpb and H2Me2bpb respectively) to synthesize photoactive {Ru-NO}6 nitrosyls [165]. As shown below, these nitrosyls consist of the {Ru-NO}6 core coordinated by two carboxamido nitrogens and two pyridine nitrogens in addition to an exogenous ligand X (X = Cl−, py, Im, OH−) trans to NO. In general, these {Ru-NO}6 nitrosyls exhibit lower νNO values (≤ 1870 cm−1) than 26 discussed above (νNO = 1899 cm−1), owing to the greater σ-donation from two carboxamido nitrogens. Within this series, the exogenous ligand X has a clear role in controlling the rate of NO release. For example, the pyridine-bound complex 33 releases NO at an apparent rate of 0.004 s−1 (under 302 nm illumination), while the chloride-bound (31) or hydroxide-bound (32) species released NO at 0.012 s−1 – nearly three times faster (Fig. 8). Richter-Addo and co-workers have recently reported the μ-oxo bridged complex {μ-O-[(bpb)Ru(NO)]2} (30) in which two bpb-nitrosyl moieties are connected by an O2− bridge [166]. It will be interesting to compare the photochemistry of 30 versus the hydroxide-bound 29. Shepherd and co-workers have synthesized several di-carboxamide ruthenium nitrosyls derived from H2bpe or H2bpp (containing ethyl or propyl diamines, resepectively, substituted for phenylene diamine as in 28–30) [167]. Although the resulting nitrosyls like [(bpp)Ru(NO)(Cl)] exhibit structural parameters similar to 28 and 31, their NO releasing ability under UV light has not yet been explored.
Fig. 8.

Kinetic traces of NO photorelease from the complexes [(Me2bpb)Ru(NO)(Cl)] (31, ■) and [(Me2bpb)Ru(NO)(py)]+ (33, ▲) at similar concentrations (~0.1 mM) upon exposure to the same UV light source. The plots show the significant effect of the axial ligand (trans to NO) on the efficiency of NO photolability. Selected bond distances and angles for 31: Ru–N(O) = 1.742(3) Å, N–O = 1.154(4) Å, Ru–N–O = 173.9(3)° [165].

Very recently, Mascharak and co-workers have isolated nitrosyl-polymer hybrid materials (for site-specific NO delivery) that includes [(PaPy3)Ru(NO)](BF4)2 (26) and [(Me2bpb)Ru(NO)(py)]BF4 (33) [168]. These nitrosyls have been encapsulated into methacrylate-based polymers because these materials are both robust and inert enough for drug delivery [169]. The nitrosyl-polymer hybrids are stable, resistant to biological buffer components and exhibit excellent NO donor ability under UV light. Unfortunately, the nitrosyls slowly diffuse out of the porous materials and contaminate the biological media within hours. The leakage problem led this group to pursue the covalent attachment of the nitrosyls to the methacrylate-based polymer. In such attempt, the researchers have synthesized a nitrosyl [(Me2bpb)Ru(NO)(4-vypy)](BF4) (35) (4-vypy = 4 vinyl-pyridine) that contains a pendant vinyl group as a “tether” for covalent attachment to the polymer backbone. When nitrosyls such as 35 or 36 are co-polymerized with the polymer components, the resulting material retains its NO delivery capacity and exhibits no sign of leakage of any ruthenium-containing species [168]. The therapeutic worth of these materials has not been evaluated so far.

2.2.5 Ruthenium nitrosyls derived from thiolate ligands
Sellmann and co-workers have synthesized a number of sulfur-rich nitrosyl complexes and studied their photochemistry. Many of these complexes contain thiolate (examples include 37 and 38) and/or thioether donor group(s) which bind the {Ru-NO}6 core quite tightly [170,171]. The thiolato sulfur donors in the five-coordinate nitrosyl 37 are reactive and readily undergo subsequent reactions to afford complexes with extended S4N coordination such as [Ru(NO)(pyS4)](Tos) (Tos = tosylate, 39, 40) [172–174].

In 2004, Sellmann and co-workers reported the photoactivity of nitrosyls of the type [Ru(NO)(pytBuS4)]+ [174]. Upon exposure to UV light, [Ru(NO)(pytBuS4)]Br (40) releases NO and is converted into the corresponding Ru(III) species [Ru(Br)(pytBuS4)] (Fig. 9). Much like the nitrosyls with carboxamido nitrogen coordination discussed above, 40 exhibits a low νNO value of 1841 cm−1 (strong σ-donation from the thiolato sulfurs) and good stability in protic solvents such as methanol. Interestingly, 40 reacts with NaBH4 in methanol to afford the “nitroxyl” (HNO) complex [Ru(HNO)(pytBuS4)] [175].
Fig. 9.

Changes in the IR spectrum of [Ru(NO)(pytBuS4)]Br (40) in the νNO region in MeOH solution upon photolysis with UV light for ~2 hours [174]. The reaction generates the Ru(III) species [RuIII(Br)(pytBuS4)]. Selected bond distances and angle of {RuNO}6 moiety of 40: Ru–N(O) = 1.721(6) Å, N–O = 1.156(6) Å, Ru–N–O = 178.1(6)° [172].
2.3 Alternative photochemical pathways
2.3.1 Ru(II) photoproducts
While photolysis of many {Ru-NO}6 nitrosyls generate Ru(III) photoproducts following loss of NO (as described above, Eq. (2)), some ruthenium nitrosyls with polypyridine ligands exhibit alternate photoproducts. As early as 1977, Meyer and coworkers reported examples of such photosensitivity [93]. When a solution of cis- [(bpy)2Ru(NO)(Cl)](PF6)2 (bpy = bipyridine, 41) in acetonitrile (flushed with argon) is exposed to UV light, a mixture of Ru(III) and Ru(II) species namely, [(bpy)2Ru(MeCN)(Cl)]2+ and [(bpy)2Ru(MeCN)(Cl)]+ is obtained as the photoproduct. However, when the acetonitrile solution is saturated with oxygen, only the Ru(III) photoproduct is isolated. Interestingly, photolysis of 41 in aqueous solution does not promote NO release, but instead leads to very different photochemical product(s) (vide infra). The trans isomer 42 has also been synthesized and structurally characterized, but its photochemistry has not yet been studied [176,177].

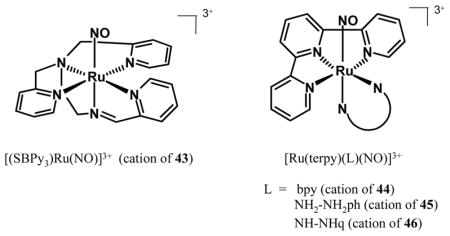
Recently, Mascharak and co-workers have synthesized a {Ru-NO}6 nitrosyl derived from the neutral, polypyridine Schiff base ligand SBPy3 (SBPy3 = N,N-bis(2-pyridylmethyl)amine-N-ethyl-2-pyridine-2-aldimine) that resembles the ligand PaPy3H except for the carboxamide group [178]. The photoactivity of [(SBPy3)Ru(NO)](BF4)3 (43) is somewhat similar to that of 41. When a solution of 43 in acetonitrile is exposed to UV light, NO is released (detected by NO electrode) and the solvent-bound Ru(II) species [(SBPy3)Ru(MeCN)]2+ is isolated as the only photoproduct even in presence of oxygen [178]. Spectroscopic (1H-NMR, EPR, UV-vis) measurements during photolysis confirm that expulsion of NO from 43 leads to the diamagnetic Ru(II) photoproduct [(SBPy3)Ru(MeCN)]2+ almost instantaneously. In absence of any time-resolved studies, the overall photochemical transformation appears to proceed via spontaneous reduction of a transient Ru(III) species (Eq. (5)). Indeed, analogous auto-reduction reaction is well-documented in iron chemistry of SBPy3 [179] and is also likely in case of ruthenium [178]. Since neutral polypyridyl ligands provide high stability to Ru(II) centers, it is quite possible that photorelease of NO in case of 43 (and species like 41) affords Ru(II) photoproducts in selected solvents. The exact nature of the reducing equivalent in Eq. (5) is unknown although NO itself could act as the source [93]. Despite rapid release of NO under low-intensity UV light, the utility of 43 as an exogenous photoactive NO donor is limited because of its instability in aqueous solution above pH 5. Dissolution of 43 in aqueous solution of pH 7 results in conversion of the NO group to NO2− (nitrosyl to nitrite, Eq. (6)). Recently, da Silva and co-workers have reported {Ru-NO}6 nitrosyls derived from terpyridine (terpy) and ancillary ligands like bpy (44), 1,2-phenylenediamine (NH2-NH2ph, 45) and its quinone derivative (NH-NHq, 46) [180]. These complexes release NO under UV light, but also suffer from facile NO → NO2− conversion at physiological pH.
| (5) |
In aqueous solution, the NO → NO2− conversion can be accelerated under illumination in case of {Ru-NO}6 complexes derived from neutral pyridine-based ligands. Earlier photochemical studies on cis-[(bpy)2Ru(NO)(Cl)]2+ (41) revealed this pathway. For example, Shimizu and co-workers found that photolysis of aqueous solutions of 41 did not result in NO generation [181]. Instead, excitation by UV light promoted conversion of the NO moiety into bound nitrite (Eq. (6)). The researchers suggested that the bound NO must be electron-deficient in the excited state, due to excitation of the dπ(Ru) → π*(bpy) transition. As electron density moves from ruthenium to bpy in the excited state, NO becomes more electron-deficient thereby allowing attack by water. The quantum yield of the reaction was unchanged (φ = 0.04) in the UV range from 254–334, which is the accepted range for the dπ(Ru) → π*(bpy) transition (in addition to π → π* transition(s) of the bpy ligand). It is important to note that this transition is not present in the ammine-based nitrosyls described above, which contain primarily σ-donating NH3 ligands. As a result, this reactivity has not been observed such {Ru-NO}6 nitrosyls under UV light. In a recent report, da Silva and co-workers have reported that laser flash-photolysis (355 nm) of 41 in aqueous solution at pH 5.7 affords cis-[(bpy)2Ru(OH)(Cl)]+ as the predominant photoproduct following photorelease of NO (detected by NO-sensitive electrode) [182]. However, above pH 6, the nitro species cis-[(bpy)2Ru(NO2)(Cl)]+ is present in significant amount at equilibrium.
| (6) |
2.3.2 NO generation from Ru(II)-NO2
During photolysis, some ruthenium complexes could release NO as a result of disproportionation of other nitrogen oxides, albeit with much reduced photochemical efficiency. Ligands such as nitrite (NO2−) and nitrate (NO3−) and loosely-bound nitrous oxide (N2O) have been implicated in some photochemical processes generating NO. For example, da Silva and co-workers have shown that nitrite ion (NO2−) bound to Ru(II) center could act as a photochemical NO source. Their results suggest that such bound nitrite undergoes photochemical disproportionation to produce NO in aqueous solution, the stoichiometry of which is described by Eq. (7). Indeed, the nitrosyls 44–46 exist as complexes [Ru(terpy)(L)(NO2)]+ (L = bpy, NH2-NH2ph, NH-NHq, 47–49) at physiological pH 7 [180]. Additionally, a series of polypyridine complexes of the type cis-[Ru(NO2)(L)(bpy)2]+ (where L = py (50), 4-picoline (51) or pz (52)) were studied in this regard [183]. All these nitro complexes release NO in aqueous solution at pH 7.4 upon exposure to UV irradiation, as determined by NO-sensitive electrode. The ancillary ligand L modulates the efficiency of NO release, as evidenced by quantum yields which range from φ = 0.007 (L = py) to 0.037 (L = pyrazine). The related polypyridine complex [Ru(NO2)(bpy)(terpy)]2+ also releases NO with a quantum yield of 0.036. Although these quantum yields of NO production are lower than the {Ru-NO}6 complexes described in previous sections, these nitro complexes are stable in aqueous solution at physiological pH (~7.4).
| (7) |

2.3.3 Photochemical intermediates in Ru–NO photodissociation
Studies on transient intermediates during photolysis of {Ru-NO}6 nitrosyls have revealed different modes of binding (and dissociation) of coordinated NO at the ruthenium centers [184]. The pioneering work of Coppens and co-workers in this area has demonstrated this aspect of NO photochemistry in iron and ruthenium nitrosyls. In their early work, this group irradiated crystals of SNP with 488 nm light (100 mW/cm2 Ar laser) at 50 K and determined the crystal structures of irradiated sample (also at 50 K) following equilibration of the metastable states at 165 K. The structures of the light-induced metastable state were then compared with the ground state structure. In the ground state structure, the NO ligand is coordinated via nitrogen in standard η1-N fashion (νNO = 1950 cm−1). In metastable state MS1, the NO moiety is also linear, but coordinated in an η1-O (or isonitrosyl) fashion (accompanied by decrease in νNO to 1835 cm−1). Lastly, in MS2, the NO moiety is in a perpendicular “side-on” geometry, i.e. bonded to the metal center in a η2-NO fashion (νNO = 1663 cm−1). Clearly, these results indicate that the mode of binding of NO to the metal center in a nitrosyl undergoes changes (in orientations) and such changes can be monitored by crystallography as well as by following changes in νNO values [185].

Similar changes in νNO values have also been observed in low-temperature Raman and IR experiments with {Ru-NO}6 nitrosyls by Woike and co-workers [186]. For example, the archetypal nitrosyl K2[Ru(NO)(Cl)5] (1) exhibits its ground state νNO at 1893 cm−1 at 20 K. Photoexcitation (Ar laser or Xe lamp with filters) at 410–500 nm (108 K) changes this value to 1768 cm−1. A nearly identical shift (from 1886 cm−1 in ground state to 1772 cm−1 in MS1) is observed in case of K2[Ru(NO)(NO2)4(OH)] upon excitation with 410–500 nm light [186]. In this case, additional laser excitation of MS1 leads to the MS2 isomer, further decreasing the νNO value to 1623 cm−1. These results along with crystallographic studies [185,187] confirm the formation of the η1-O and η2-NO states in these ruthenium nitrosyls. More recent work by Richter-Addo and co-workers with porphyrin-based {Ru-NO}6 nitrosyls (11 and its derivatives) has shown the same trend in νNO shift upon laser excitation [188]. Franco and co-workers have also reported similar changes in νNO values (1916→1823→1798 cm−1) upon excitation of [Ru(NO)(NH3)5]3+ (4) with 480 nm light in the solid state [189]. In all these studies with solid samples of nitrosyls, laser excitation at low temperature generates the different NO metastable state linkage isomers which eventually revert to the ground state {M-NO}6 (M = Fe, Ru) nitrosyls when the samples are warmed to ambient temperature. No photorelease of NO has been reported in these experiments.
In a recent report, Einarsdóttir and co-workers have detected similar transient species in their time-resolved flash photolysis studies on 26 in aqueous solution at 298 K [73]. In this study, a short-lived intermediate (formed within ≤ 100 ns of laser flash, 355 nm) with λmax ≈ 550 nm has been attributed to formation of either η1-O isonitrosyl (MS1) or η2-NO (MS2) linkage isomer. NO is rapidly released from this metastable state (τ ≈ 2 μs) to afford the Ru(III) photoproduct. Although photorelease of NO from the metastable state isomers of ruthenium (or any other metal) nitrosyls has not been studied systematically by any group so far, it is apparent that photorelease of NO from {Ru-NO}6 nitrosyls most possibly proceeds through one or more of these metastable states. The existence (and role) of such species in biological media however remains as an open question at this time.
2.4 Overview of the photochemical pathways of {Ru-NO}6 nitrosyls
Studies on several sets of {Ru-NO}6 nitrosyls have now revealed that these species release NO mainly via two photochemical pathways. As shown in Scheme 1a, some {Ru-NO}6 nitrosyls release NO upon illumination and afford Ru(III) photoproducts. This pathway is promoted by strong, usually negatively charged, σ-donors such as amines, phenolates, carboxamides and thiolates. These donors stabilize the Ru(III) photoproduct, and as such promote this photochemical pathway in most solvent medium including water at physiological pH. Several nitrosyls of this category require moderate to low intensity light (mostly in the 300–450 nm range) to release significant amounts of NO. The notable exceptions are the nitrosyls derived from porphyrin-based ligands (Scheme 1b). Fast NO recombination rates severely limit the NO donor capacity of such {Ru-NO}6 nitrosyls. High intensity light sources are required to release NO from these species. The second photochemical pathway affords Ru(II) photoproducts following photorelease of NO. {Ru-NO}6 nitrosyls derived from polypyridine ligands and neutral Schiff bases fall in this category. Photochemical loss of NO from these nitrosyls is accompanied by auto-reduction of the ruthenium center in selected solvents (Scheme 2a). In aqueous solutions, exposure to light can promote NO → NO2− conversion (Scheme 2b). Although some of the resultant nitro complexes photorelease NO, the starting {Ru-NO}6 nitrosyls are not considered efficient NO donors due to their complex photochemistry in aqueous media.
Scheme 1.

Scheme 2.

3. Strategies for photosensitization of the Ru–NO bond to lower-energy (visible and near-IR) light
One key requirement for phototherapeutics in PDT is the possibility of drug delivery upon exposure to visible or near IR light, mainly to avoid tissue damage and allow skin penetration in case of skin malignancies. In case of photoactive ruthenium-based NO donors, it is therefore desirable that the {Ru-NO}6 nitrosyls undergo facile NO photorelease upon illumination with light of longer (500–850 nm) wavelengths. However, most photoactive {Ru-NO}6 nitrosyls, discussed so far, release NO when irradiated with light in the UV region since the photosensitivity of these nitrosyls is largely governed by the intrinsic location of the dπ(Ru) → π*NO) transition in their electronic absorption spectra (λmax = 300–450 nm, Table 1). Such UV photoactivation is of course incompatible with many medical applications. During the past few years, researchers have therefore shifted their attention to designed ruthenium nitrosyls that could deliver NO under visible (and/or near IR) light.
To date, two strategies for photosensitizing the Ru–NO bond to visible light have been pursued. They include (a) alteration (or “tuning”) of the donor set and/or the ligand frame to modulate the energy of the dπ(Ru) → π* (NO) transition, thereby shifting the λmax of the photoband into the visible region and (b) the use of strongly colored dyes as pendant (not bonded to metal center) or as coordinating (directly bonded to the metal center) ligands and utilization of the harvested light (by the added chromophore) to facilitate NO photorelease.
3.1 Strategy A: Increase in the number of anionic donors and/or charge
As far back as 1975, researchers have documented the effect of increasing donor strength/charge on the spectroscopic properties of {Ru-NO}6 nitrosyls. One instructive example is the simple set of chloro complexes [Ru(NO)(Cl)x(H2O)y]m (where x = 1–5; y = 0–4; x + y = 5; and m = 2− to 2+) [108]. The nitrosyl with a low number of chloride ligands, such as [Ru(NO)(Cl)2(H2O)3]+, exhibits a clear absorption band at 440 nm (Fig. 10). The next nitrosyl in the series, namely [Ru(NO)(Cl)3(H2O)2] exhibits its band at 460 nm. A systematic trend is observed in the red shift of the λmax values from 420 nm to 520 nm as the number chloride ligands increases from one to five (Fig. 10). Although poor stability in aqueous solution and readily exchanging ligands limit the use of this type of {Ru-NO}6 nitrosyls as NO donors, it is evident that more electron-donating ligands cause red shift of the absorption band of simple {Ru-NO}6 nitrosyls.
Fig. 10.

Electronic absorption spectra of ruthenium nitrosyls in the series [Ru(NO)(Cl)x(H2O)y]m. The λmax values for x = 1 through 5 (ε-values listed in Table 1) increase systematically from ~420 nm to 520 nm as the number of chloride ligands (x) increases from 1 to 5.
Modulation of λmax via addition of more charged donors has been observed in more complicated ligand systems. For example, in a recent study, Mascharak and co-workers have isolated [(Py3P)Ru(NO)]BF4 (53), a photoactive {Ru-NO}6 nitrosyl derived from the pentadentate ligand Py3PH2 with two carboxamide groups. Inspection of the electronic absorption spectra of [(SBPy3)Ru(NO)]3+ (43), [(PaPy3)Ru(NO)]2+ (26), and [(Py3P)Ru(NO)]+ (53), a series of polypyridine N5 complexes in which the number of carboxamido nitrogen donor center(s) systematically increases from zero (43), to one (26), to two (53), reveals increase in absorption in the visible range with increasing number of carboxamido nitrogen(s) [178]. For example, 43 exhibits its major absorption with λmax at 315 nm while 26 displays its band at 410 nm. Interestingly, 53 exhibits additional absorption with a feature at 530 nm (ε = 400 M−1 cm−1) and NO photorelease is observed (unlike 43) when it is exposed to visible light (500–550 nm, φ532 = 0.05).

Addition of electron-rich substituents to the ligand frame around the metal center can also shift the position of the photoband of {Ru-NO}6 nitrosyls. For example, Sellmann and co-workers have recently reported photorelease of NO via use of visible light from nitrosyls derived from thiolate ligands, similar to 37–40. In their approach, several bulky triphenylsilyl (−SiPh3) groups were appended to the ligand frame, as shown for [Ru(NO)(pysiS4)]Br (54) [190]. The structure of this nitrosyl is similar to that of [Ru(NO)(pybuS4]+ (40). When the red solution of 54 in methanol is exposed to visible light (≥ 455 nm), NO is released and the corresponding Ru(III) bromo complex [Ru(Br)(pysiS4)] is obtained as the photoproduct. The strongly electron-donating silyl groups most likely increase the electron density at the metal center and shift the dπ(Ru) → π*(NO) transition to lower energy in this case. Additionally, the bulky SiPh3 groups prevent formation of a thiolate-bridged dimer in solution (as is the case with 40), thereby facilitating NO release. Insolubility of 54 in aqueous solution however prevents use of this nitrosyl as a NO donor to biological targets.
Mascharak and co-workers have utilized extended conjugation in the ligand frame to move the photobands of {Ru-NO}6 nitrosyls to visible region. In general, ruthenium nitrosyls with nitrogen-based ligands exhibit strong absorption bands in the UV range (300–400 nm, Table 1). For example, the nitrosyl [(Me2bpb)Ru(NO)(Cl)] (31) has a strong absorption band with λmax at 395 nm (ε ≈ 5 000 M−1 cm−1) and it releases NO under low-intensity UV light (5–10 mW). However, the lack of absorbance at wavelengths greater than 400 nm with nitrosyls of this type restricts their use in PDT under visible light. In order to overcome this limitation, Mascharak and co-workers have synthesized the designed ligand Me2bQb2− in which the pyridine rings of Me2bpb2− ligand are replaced by the larger quinoline groups [165]. In the resulting {Ru-NO}6 nitrosyl, namely [(Me2bQb)Ru(NO)(Cl)] (55), the λmax value is shifted to 455 nm (Fig. 11) and exposure of this nitrosyl to visible light (≥ 455 nm) indeed results in rapid release of NO. In contrast, the less-conjugated complexes such as 28 and 31 undergo minimal loss of NO (~1%) under identical conditions. It is therefore reasonable to expect that ruthenium nitrosyls could be sensitized to visible light via judicious modifications of the ligand frames.
Fig. 11.

Electronic absorption spectra showing the effect of increasing ligand frame conjugation on λmax position upon going from [(bpb)Ru(NO)(Cl)] (28, pyridine based-ligand) to [(Me2bQb)Ru(NO)(Cl)] (55, quinoline-based ligand). The structure of 55 is shown. Selected {RuNO}6 bond distances and angle for 55: Ru–N(O) = 1.7389(19) Å, N–O = 1.146(3) Å, Ru–N–O = 177.77(19)° [165].

3.2 Strategy B: Attachment of pendant or coordinated chromophores as sensitizers
Research in past three decades has indicated synthetic difficulties encountered in shifting the dπ(Ru) → π*(NO) transitions of {Ru-NO}6 nitrosyls well in the visible (or near IR) region. Although a fewl nitrosyls that exhibit absorption bands around 500 nm have been isolated, the extinction coefficients of these transitions are usually low (ε ≤ 500 M−1 cm−1). As a consequence, their capacities for NO delivery are somewhat restricted particularly under light of moderate intensity. In recent years, the strategy of efficient harvesting of visible light via “antennae” to facilitate NO photorelease has been pursued by several groups to circumvent this problem. Ford and co-workers first employed this strategy in sensitizing Cr(III) nitrite complexes (species containing Cr–NO2 bonds) derived from cyclam-type ligands which release NO upon exposure to UV light [65a]. In such attempts, this group appended “far-UV” chromophores (like pyrene, anthracene) to the periphery of the ligand frame as light harvesting units. The alkyl linkers keep the pendant chromophores close (although not directly bonded) to the metal center. Although the intrinsic quantum yield (φ) value is unaffected by the high absorbance (ε in M−1 cm−1) of the pendant chromophore, the “effective” utility of the complexes (ε × φ) is improved significantly due to the conjugation. As an extension of this work, Ford and co-workers have used a similar alkyl linker to tether fluorescein (a near-UV chromophore) to the Roussin’s salt Na2[Fe2S2(NO)2] again allowing peripheral photoactivation of the metal center. [46a, 46b].
Mascharak and co-workers have recently reported a set of {Ru-NO}6 nitrosyls in which a dye molecule has been employed both as a ligand and for harvesting energy from visible light (i.e. as a sensitizer) and facilitating NO photorelease. In this research, the UV-active nitrosyl [(Me2bpb)Ru(NO)(Cl)] (31) was derivatized with the bright red dye Resorufin (Resf) to afford [(Me2bpb)Ru(NO)(Resf)] (56) [191]. This dye-conjugate {Ru-NO}6 nitrosyl exhibits a very strong absorption band at 500 nm (ε = 12 000 M−1 cm−1, Fig. 12) and rapidly releases NO (detected by NO-sensitive electrode) when exposed to visible light (≥ 455 nm) with a quantum yield (φ) value of 0.05 at 500 nm. The authors suggest that direct coordination of the dye molecule is responsible for raising the quantum yield of 56 in the visible range via energy transfer along the {(Resf)O–Ru–NO} axis. In order to support this hypothesis further, this group has also isolated the corresponding {Ru-NO}6 nitrosyl-Resf conjugate [(Me2bQb)Ru(NO)(Resf)] (57) in which the dπ(Ru) → π*(NO) transition almost merges with the strong dye absorption band at 510 nm (Fig. 12). In line with the hypothesis, 57 exhibits a higher quantum yield value in the visible range (φ500 = 0.10). Both 56 and 57 are stable in aqueous solutions (no observed NO → NO2− conversion in the pH range 3–9) and rapidly photorelease NO under visible light, thus fulfilling two crucial requirements for effective NO donors for PDT.
Fig. 12.

Absorption profiles in the UV/Vis region of the dye-modified complexes [(Me2bpb)Ru(NO)(Resf)] (56, solid line), [(Me2bQb)Ru(NO)(Resf] (57, dash-dot line), and the analogous hydroxide-bound complex [(Me2bpb)Ru(NO)(OH)] (32, dashed line). The structure of 56 is shown on the right. Selected Ru–NO structural parameters for dye-bound complex 56: Ru–N(O) = 1.7347(16) Å, N–O = 1.159(2) Å, Ru–N–O = 178.13(15)° [191].

Da Silva and co-workers have pursued a related strategy for visible light sensitization of ruthenium nitrosyls. In their work, a second ruthenium complex has been utilized as the sensitizer [192,193]. The dinuclear {Ru-NO}6 nitrosyl [Ru(NH3)5(pz)Ru(bpy)2(NO)](PF6)5 (58), with a pyrazine (pz) bridge between the two metal centers, exhibits a strong absorption band in the visible region (λmax = 530 nm, ε= 10 000 M−1 cm−1) due to a metal-to-ligand charge transfer (MLCT) transition of dπRu(II) → π*(pz) origin [192]. When 58 is exposed to 532 nm light (Nd:YAG laser), the MLCT transition in the Ru-pz moiety followed by an intramolecular electron transfer to the Ru-NO unit causes photorelease of NO (φ532 = 0.025) under acidic conditions. Although da Silva and co-workers have reported a few dinuclear nitrosyls of this type (58–60), no information on their structures or biological utility has been reported so far [193].

4. Biological utility of ruthenium NO donors
To date, no ruthenium nitrosyl has been introduced clinically as an exogenous NO donor. Research in this area has focused more on their photochemistry and their utility as research tools in studying reactions of NO with proteins and DNA. A few studies of such kind with {Ru-NO}6 nitrosyls are discussed here to establish the potential of such species as convenient agents for nitrosylation of biological targets.
4.1 NO transfer to proteins
The capacity of ruthenium nitrosyls to photodeliver NO to proteins was explored by Trentham and co-workers as early as 1990 [107]. This group employed the commercially available salts Ru(NO)Cl3•xH2O (often designated as RuNOCl3) and K2[Ru(NO)(Cl)5] to study the process of delivery of NO to hemoglobin (Hb). Reduction of Hb with sodium ascorbate under N2 atmosphere generates deoxyHb, which reacts rapidly with NO to form nitrosylHb (Eq. (8)). In presence of RuNOCl3, deoxyHb is completely converted to nitrosylHb within several minutes of UV irradiation. Conversion of oxyHb to metHb by the action of photolyzed NO within a similar timeframe is also observed. More in-depth flash photolysis studies reveal that NO binds to Hb in less than 4 milliseconds, with a second-order rate constant of 8 × 106 M−1 s−1 (dependent on both [Hb] and [NO]).
| (8) |
Einarsdóttir and co-workers have recently explored the use of [(PaPy3)Ru(NO)](BF4)2 (26) to photodeliver NO to heme proteins such as myoglobin (Mb) and cytochrome c oxidase (CcO), both of which react with NO in fashion analogous to Hb (Eq. (8)) [73]. Time-resolved flash photolytic studies have shown that the fast kinetics of NO binding to Mb (τ = 2.7 ms; k = 3 × 107 M−1 s−1) is comparable to that of Hb. NO reacts more rapidly with CcO (τ = 940 μs; k = 1 × 108 M−1 s−1). This study has demonstrated that kinetic resolution of such fast biological reactions is possible with photoactive ruthenium nitrosyls.
There are several advantages that one enjoys with this type of NO donors over NO gas. First, all the complications and precautions related to handling toxic NO gas via manometry as well as the lengthy procedures for the purification of NO gas are all absent in experiments with ruthenium nitrosyls as NO donors. The compounds are all stable, pure, and can be handled easily in the absence of light. Second, one can add the NO donor to the target protein in the desired medium and then trigger the photorelease of NO (and start the reaction) at one’s convenience. As a result, these nitrosyls are excellent research tools for exploring the mechanism(s) of fast nitrosylation reactions of proteins. As mentioned before, results from both Borovik and Mascharak’s laboratory have shown that polymer-embedded {Ru-NO}6 nitrosyls can also deliver NO to Mb [142,162,167].
Mascharak and co-workers have also utilized 26 to activate soluble guanylate cyclase (sGC) [163] and papain [74] although the kinetics of NO binding have not been explored yet. There is much interest in exploring the reactions of NO with heme proteins like soluble guanylate cyclase (sGC), nitric oxide reductase (NOR), and nitric oxide synthase (NOS). Studies on such nitrosylation by ruthenium nitrosyls (often referred to as “caged-NO” complexes of ruthenium by several biophysical groups) have been recently reviewed [194].
4.2. Delivery of NO to cells/tissues
Light-induced NO delivery to tissues from {Ru-NO}6 nitrosyls have also been reported. Unlike the systemic NO donors like NONOates, nitrates, and SNP (SNP can also be activated by light [39,40,42]), release of NO can be better controlled in case of photoactive ruthenium nitrosyls. As early as 1993, the ruthenium analogue of SNP namely, [Ru(NO)(Cl)5]2− (referred to in some literature as ruthenium nitrosyl pentachloride, or RNP) and RuNOCl3 were used by several groups as a controlled source of NO in biological experiments. These commercially available nitrosyls have been extensively used during the period when the emerging roles of NO in biology were being realized. Stimulation of synaptic activity in rat hippocampus slices, relaxation of rabbit aortic rings, and induction of interneuronal activity in mollusks have been noted with RNP and RuNOCl3 under UV light (Xe flash lamp, λmax 320 nm) [195,196]. Although such physiological responses definitely arise from photoreleased NO, non-specific side reactions of RNP (much like the ones noted with SNP) have also been observed in complex biological systems. Ruthenium nitrosyls with chloride as ligands such as RNP and [(cyclam)Ru(NO)(Cl)]2+ experience substitution of Cl− for H2O under physiological conditions. The water-bound ruthenium species exhibit high affinity to DNA, especially guanine residues, which can lead to unwanted side effects. As discussed earlier, the instability of the {Ru-NO}6 unit in aqueous solution under physiological conditions leads to other species (such as nitro complexes) in many cases. As a result, “clean” examples of tissue experiments exhibiting the effect(s) of NO photoreleased from a ruthenium nitrosyl require extensive control studies. One such study has recently been reported by Fukuto and co-workers [163]. This group has demonstrated relaxation of rat aorta muscle rings with 26 under UV light. Similar studies with 27 and 57, both with high quantum yield values in water at physiological pH (φ = 0.20 and 0.35 respectively) are in progress.
In tissue studies, loss of NO from {Ru-NO}6 nitrosyls via cellular reduction often introduces complication particularly when the effect of light is under investigation. For example, Franco and co-workers have demonstrated that the hypotensive effects of trans-[Ru(NH3)4(NO)(P(OEt)3)](PF6)3 (8) in live mice arise from NO in complete absence of light [197,198]. Similarly, bolus injections of trans-[(cyclam)Ru(NO)(Cl)](PF6)2 (9) rapidly reduce the blood pressure of hypertensive rats in absence of light and the duration of the effect is longer than that of SNP [125]. The reducing environment inside the cell however could augment the overall effect of light in some cases. Indeed, in recent studies, da Silva and co-workers have shown that reducing conditions coupled with irradiation of the solution show improved biological activity of the photoreleased NO [180]. The complex [Ru(terpy)(NH-NHq)(NO)](PF6)3 (46, NH-NHq = quinonediimine) is readily reduced and undergoes NO loss. It also releases NO upon exposure to UV light (φ = 0.47) according to Eq. (2). To demonstrate the effect of NO release, the authors utilized thoracic rings that were pre-contracted with norephidrine. When the tissues were treated with the nitrosyl in the dark, complete relaxation of the muscle was achieved within ~60 minutes due to NO released from 46 via intracellular reductive mechanism(s) as reported for SNP. In contrast, the same dosage under light irradiation achieved complete muscle relaxation in just 4 minutes. Clearly, this study underscores the need for careful studies in case of tissue experiments.
5. Conclusions
The quest for a ruthenium nitrosyl that could photodeliver NO to biological targets on demand has produced interesting and encouraging results so far. It is now evident that clever design of ruthenium nitrosyls, preferably {Ru-NO}6 species derived from polydentate ligands (to avoid further speciation of the drug) with strong absorption bands in the 500–800 nm region, could perform such a job. Research efforts along these lines have provided clues regarding how to increase the stability of Ru-NO bonds in water at physiological pH and how to red shift the photobands of {Ru-NO}6 nitrosyls via alterations of the coordination spheres. The overall stability of metal-ligand bonds in ruthenium complexes is clearly an advantage in terms of integrity of the drug molecules in biological systems although extensive toxicity studies are required before any of these compounds could actually be employed to combat any malady. Recent research has also indicated that incorporation of the nitrosyls in biocompatible polymer matrices could be used to deliver NO to specific sites under the control of light exposure. Such materials have potential in PDT of localized malignancies such as skin cancer. As is the case with any drug discovery project, attempts to isolate the right drug have so far afforded much insight to the basic coordination chemistry and photochemistry of ruthenium and NO, and we anticipate this topic to have a direct impact on medicinal chemistry in the near future.
Acknowledgments
Studies in this laboratory on designed metal nitrosyls as exogenous NO donors have been supported by the US National Science Foundation (CHE-9818492 and CHE-0553405) and the US National Institute of Health (GM 61636).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ignarro LJ. Nitric Oxide: Biology and Pathobiology. Academic Press; San Diego: 2000. [Google Scholar]
- 2.Feelisch M, Stamler JS. Nitric Oxide Research. John Wiley and Sons; Chichester, UK: 1996. [Google Scholar]
- 3.Fukuto JM, Wink DA. Metal Ions Biol Syst. 1999;36:547. [PubMed] [Google Scholar]
- 4.Lincoln J, Burnstock G. Nitric Oxide in Health and Disease. Cambridge University Press; New York: 1997. [Google Scholar]
- 5.Fang FC. Nitric Oxide and Infection. Kluwer Academic/Plenum Publishers; New York: 1999. [Google Scholar]
- 6.Weissman BA, Allan N, Shapiro S. Biochemical, Pharmacological, and Clinical Aspects of Nitric Oxide. Plenum Press; New York: 1995. [Google Scholar]
- 7.Chiueh CC, Hong JS, Leong SK. Nitric Oxide: Novel Actions, Deleterious Effects, and Clinical Potential. Vol. 962. New York: Academy of Sciences; 2002. [Google Scholar]
- 8.Kalsner S. Nitric Oxide and Free Radicals in Peripheral Neurotransmission. Birkhäuser; Boston: 2000. [Google Scholar]
- 9.Goligorsky MS, Gross SS. Nitric Oxide and the Kidney. Chapman & Hall; New York: 1997. [Google Scholar]
- 10.Lancaster J., Jr . Nitric Oxide: Principles and Actions. Academic Press; San Diego: 1996. [Google Scholar]
- 11.Sitaramayya A. Introduction to Cellular Signal Transduction. Birkhäuser; New York: 1999. [Google Scholar]
- 12.Wang PG, Cai TB, Taniguchi N. Nitric Oxide Donors for Pharmaceutical and Biological Applications. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 13.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, Janczuk AJ. Chem Rev. 2002;102:1091. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 14.Cai TB, Wang PG. Expert Opin Therap Patents. 2004;14:84. [Google Scholar]
- 15.(a) Napoli C, Ignaro LJ. Ann Rev Pharmacol Toxicol. 2003;43:97. doi: 10.1146/annurev.pharmtox.43.100901.140226. [DOI] [PubMed] [Google Scholar]; (b) Ignarro LJ, Napoli C, Loscalzo J. Circ Res. 2002;90:21. doi: 10.1161/hh0102.102330. [DOI] [PubMed] [Google Scholar]
- 16.Thatcher GRJ. Curr Topics Med Chem. 2005;5:597. doi: 10.2174/1568026054679281. [DOI] [PubMed] [Google Scholar]
- 17.(a) Al’Sadoni HH, Ferro A. Rev Med Chem. 2005;5:247. [Google Scholar]; (b) Williams DLH. Acc Chem Res. 1999;32:869. [Google Scholar]
- 18.(a) Keefer LK. Curr Topics Med Chem. 2005;5:625. doi: 10.2174/1568026054679380. [DOI] [PubMed] [Google Scholar]; (b) Keefer LK. Ann Rev Pharmacol Toxicol. 2003;43:585. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]; (c) Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 19.Smith DJ, Chakravorthy D, Pulfer S, Simmons ML, Hrabie JA, Citro ML, Saavedra JE, Davis KM, Hutsell TC, Mooradian DL, Hanson SR, Keefer LK. J Med Chem. 1996;39:1148. doi: 10.1021/jm950652b. [DOI] [PubMed] [Google Scholar]
- 20.(a) Butler AR, Megson IL. Chem Rev. 2002;102:1155. doi: 10.1021/cr000076d. [DOI] [PubMed] [Google Scholar]; (b) Friederich JA, Butterworth JF. Anesth Analg. 1995;81:152. doi: 10.1097/00000539-199507000-00031. [DOI] [PubMed] [Google Scholar]
- 21.(a) Klink M, Sulowska Z. Lett Drug Design Disc. 2007;4:55. [Google Scholar]; (b) Degoute CS. Drugs. 2007;67:1053. doi: 10.2165/00003495-200767070-00007. [DOI] [PubMed] [Google Scholar]; (c) Thatcher GRJ. Curr Topics Med Chem. 2005;5:597. doi: 10.2174/1568026054679281. [DOI] [PubMed] [Google Scholar]
- 22.(a) Mocellin S, Bronte V, Nitti D. Med Res Rev. 2007;27:317. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]; (b) Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. Carcinogenesis. 1998;19:711. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- 23.(a) Ying L, Hofseth LJ. Cancer Res. 2007;67:1407. doi: 10.1158/0008-5472.CAN-06-2149. [DOI] [PubMed] [Google Scholar]; (b) Hirst D, Robson T. J Pharm Pharmacol. 2007;59:3. doi: 10.1211/jpp.59.1.0002. [DOI] [PubMed] [Google Scholar]
- 24.Hofseth LJ, Hussain SP, Wogan GN, Harris CC. Free Radical Biol Med. 2003;34:955. doi: 10.1016/s0891-5849(02)01363-1. [DOI] [PubMed] [Google Scholar]
- 25.(a) Xu W, Liu LZ, Loizidou M, Ahmed M, Charles IG. Cell Res. 2002;12:311. doi: 10.1038/sj.cr.7290133. [DOI] [PubMed] [Google Scholar]; (b) Abramson SB, Amin AR, Clancy RM, Attur M. Best Pract Res Clin Reumatol. 2001;15:831. doi: 10.1053/berh.2001.0196. [DOI] [PubMed] [Google Scholar]
- 26.Simeone A-M, Collela S, Krahe R, Johnson MM, Mora E, Tari AM. Carcinogenesis. 2006;27:568. doi: 10.1093/carcin/bgi233. [DOI] [PubMed] [Google Scholar]
- 27.Xie KP, Huang S. Free Radical Biol Med. 2003;34:969. doi: 10.1016/s0891-5849(02)01364-3. [DOI] [PubMed] [Google Scholar]
- 28.Brüne B. Cell Death Diff. 2003;10:864. doi: 10.1038/sj.cdd.4401261. [DOI] [PubMed] [Google Scholar]
- 29.Boyd CS, Cadenas E. Biol Chem. 2002;383:411. doi: 10.1515/BC.2002.045. [DOI] [PubMed] [Google Scholar]
- 30.Li CQ, Wogan GN. Cancer Lett. 2005;226:1. doi: 10.1016/j.canlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 31.Brüne B, von Knethan A, Sandau KB. Eur J Pharm. 1998;351:261. doi: 10.1016/s0014-2999(98)00274-x. [DOI] [PubMed] [Google Scholar]
- 32.(a) Castano AP, Mroz P, Hamblin MR. Nat Rev Cancer. 2006;6:535. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ogura S, Tabata K, Fukushima K, Kamachi T, Okura I. J Porph Phthal. 2006;10:1116. [Google Scholar]; (c) Detty MR, Gibson SL, Wagner SJ. J Med Chem. 2004;47:3897. doi: 10.1021/jm040074b. [DOI] [PubMed] [Google Scholar]
- 33.(a) Ackroyd R, Ketty C, Brown N, Reed M. Photochem Photobiol. 2001;74:656. doi: 10.1562/0031-8655(2001)074<0656:thopap>2.0.co;2. [DOI] [PubMed] [Google Scholar]; (b) Pandey RK. J Porph Phthal. 2000;4:368. [Google Scholar]
- 34.Fan BG, Andren-Sandberg A. Pancreas. 2007;34:385. doi: 10.1097/mpa.0b013e3180439c50. [DOI] [PubMed] [Google Scholar]
- 35.Moore CM, Hoh IM, Bown SG, Emberton M. BJU Intl. 2005;96:754. doi: 10.1111/j.1464-410X.2005.05709.x. [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Zuazaga J, Cooper KD, Baron ED. Rev Anticancer Ther. 2005;5:791. doi: 10.1586/14737140.5.5.791. [DOI] [PubMed] [Google Scholar]
- 37.Marmur ES, Schmults CD, Goldberg DJ. Derm Surg. 2004;30:264. doi: 10.1111/j.1524-4725.2004.30083.x. [DOI] [PubMed] [Google Scholar]
- 38.Lopez RFV, Lange N, Guy R, Bentley MVLB. Adv Drug Del Rev. 2004;56:77. doi: 10.1016/j.addr.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 39.(a) Kudo S, Bourassa JL, Boggs SE, Sato Y, Ford PC. Anal Biochem. 1997;247:193. doi: 10.1006/abio.1997.2097. [DOI] [PubMed] [Google Scholar]; (b) Singh RJ, Hogg N, Neese F, Joseph J, Kalyanaraman B. Photochem Photobiol. 1995;61:325. doi: 10.1111/j.1751-1097.1995.tb08616.x. [DOI] [PubMed] [Google Scholar]
- 40.(a) Szacilowski K, Wacyk W, Stochel G, Stasicka Z, Sostero S, Traverso O. Coord Chem Rev. 2000;208:277. [Google Scholar]; (b) Stochel G, Wanat A, Kulis E, Stasicka Z. Coord Chem Rev. 1998;171:203. [Google Scholar]
- 41.Clarke MJ, Gaul JB. Struct Bonding. 1993;81:147. [Google Scholar]
- 42.(a) Mitra RP, Jain DVS, Banerjee AK, Chari KVR. J Inorg Nucl Chem. 1972;34:3919. [Google Scholar]; (b) Mitra RP, Jain DVS, Banerjee AK, Chari KVR. J Inorg Nucl Chem. 1963;25:1263. [Google Scholar]
- 43.(a) Hurst RD, Chowdhury R, Clark JB. J Neurochem. 1996;67:1200. doi: 10.1046/j.1471-4159.1996.67031200.x. [DOI] [PubMed] [Google Scholar]; (b) Flitney FW, Kennovin G. J Physiol. 1986;380:37P. [Google Scholar]
- 44.Wolfe SK, Swinehart JH. Inorg Chem. 1975;14:1049. [Google Scholar]
- 45.Bourassa JL, Ford PC. Coord Chem Rev. 200–202. 2000. p. 887. [Google Scholar]
- 46.(a) Wecksler SR, Mikhailovsky A, Korystov D, Buller F, Kannan R, Tan LS, Ford PC. Inorg Chem. 2007;46:395. doi: 10.1021/ic0607336. [DOI] [PubMed] [Google Scholar]; (b) Wecksler SR, Hutchinson J, Ford PC. Inorg Chem. 2006;45:1192. doi: 10.1021/ic051723s. [DOI] [PubMed] [Google Scholar]; (c) Conrado CL, Bourassa JL, Egler C, Wecksler S, Ford PC. Inorg Chem. 2003;42:2288. doi: 10.1021/ic020309e. [DOI] [PubMed] [Google Scholar]
- 47.Ford PC, Bourassa J, Miranda K, Lee B, Lorkovic I, Boggs S, Kudo S, Laverman L. Coord Chem Rev. 1998;171:185. [Google Scholar]
- 48.(a) Flitney FW, Megson IL, Thomson JLM, Kennovin GD, Butler AR. Br J Pharmacol. 1996;117:1549. doi: 10.1111/j.1476-5381.1996.tb15320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flitney FW, Megson IL, Flitney DE, Butler AR. Br J Pharmacol. 1992;107:842. doi: 10.1111/j.1476-5381.1992.tb14534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.(a) Venugopal A, Topf JM. Am J Kidney Disease. 2006;168:A60. [Google Scholar]; (b) Alaniz C, Watts B. Ann Pharmacother. 2005;39:388. doi: 10.1345/aph.1E226. [DOI] [PubMed] [Google Scholar]
- 50.Mayer B, Brunner F, Schmidt K. Biochem Pharm. 1993;45:367. doi: 10.1016/0006-2952(93)90072-5. [DOI] [PubMed] [Google Scholar]
- 51.(a) Robin ED, McCauley R. Chest. 1992;102:1842. doi: 10.1378/chest.102.6.1842. [DOI] [PubMed] [Google Scholar]; (b) Vesy CJ, Krapez JR, Cole PV. J Pharm Pharmacol. 1980;32:256. doi: 10.1111/j.2042-7158.1980.tb12908.x. [DOI] [PubMed] [Google Scholar]; (c) Spiegel HE, Kucera V. Clin Chem. 1977;23:2329. [PubMed] [Google Scholar]; (d) Jack RD. Br J Anaesth. 1974;46:952. doi: 10.1093/bja/46.12.952. [DOI] [PubMed] [Google Scholar]
- 52.Patra AK, Afshar RK, Olmstead MM, Mascharak PK. Angew Chem Intl Ed. 2002;41:2512. doi: 10.1002/1521-3773(20020715)41:14<2512::AID-ANIE2512>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 53.Patra AK, Rowland JM, Marlin DS, Bill E, Olmstead MM, Mascharak PK. Inorg Chem. 2003;42:6812. doi: 10.1021/ic0301627. [DOI] [PubMed] [Google Scholar]
- 54.Afshar RK, Patra AK, Olmstead MM, Mascharak PK. Inorg Chem. 2004;43:5736. doi: 10.1021/ic040057c. [DOI] [PubMed] [Google Scholar]
- 55.(a) Afshar RK, Eroy-Reveles AA, Olmstead MM, Mascharak PK. Inorg Chem. 2006;45:10347. doi: 10.1021/ic0614115. [DOI] [PubMed] [Google Scholar]; (b) Patra AK, Afshar RK, Rowland JM, Olmstead MM, Mascharak PK. Angew Chem Intl Ed. 2003;42:4517. doi: 10.1002/anie.200352070. [DOI] [PubMed] [Google Scholar]
- 56.Harrop TC, Olmstead MM, Mascharak PK. Inorg Chem. 2005;44:6918. doi: 10.1021/ic050659b. [DOI] [PubMed] [Google Scholar]
- 57.(a) Conrado C, Bourassa JL, Egler C, Wecksler S, Ford PC. Inorg Chem. 2003;42:2288. doi: 10.1021/ic020309e. [DOI] [PubMed] [Google Scholar]; (b) Bourassa J, Lee B, Bernard S, Schoonover J, Ford PC. Inorg Chem. 1999;38:2947. doi: 10.1021/ic981282v. [DOI] [PubMed] [Google Scholar]; (c) Bourassa J, DeGraff W, Kudo S, Wink DA, Mitchell JB, Ford PC. J Am Chem Soc. 1997;119:2853. [Google Scholar]
- 58.Caulton KG. Coord Chem Rev. 1975;14:317. [Google Scholar]
- 59.Patra AK, Rose MJ, Olmstead MM, Mascharak PK. J Am Chem Soc. 2004;126:4780. doi: 10.1021/ja040015n. [DOI] [PubMed] [Google Scholar]
- 60.(a) Lim MD, Lorkovic IM, Ford PC. Inorg Chem. 2002;41:1026. doi: 10.1021/ic0108585. [DOI] [PubMed] [Google Scholar]; (b) Lorkovic IM, Ford PC. Inorg Chem. 1999;38:1467. [Google Scholar]
- 61.Ford PC, Lorkovic IM. Chem Rev. 2002;102:993. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- 62.Lin R, Farmer PJ. J Am Chem Soc. 2001;123:1143. doi: 10.1021/ja993643r. [DOI] [PubMed] [Google Scholar]
- 63.Franz KJ, Lippard SJ. J Am Chem Soc. 1998;120:9034. [Google Scholar]
- 64.Bottomley F. Acc Chem Res. 1978;11:158. [Google Scholar]
- 65.(a) DeRosa F, Bu X, Ford PC. Inorg Chem. 2005;44:4157. doi: 10.1021/ic048311o. [DOI] [PubMed] [Google Scholar]; (b) De Leo MA, Ford PC. J Am Chem Soc. 1999;121:1980. [Google Scholar]
- 66.De Leo MA, Ford PC. Coord Chem Rev. 2000;208:47. [Google Scholar]
- 67.Fischer M, Warneck F. J Phys Chem. 1996;100:18750. [Google Scholar]
- 68.Franz KJ, Lippard SJ. Inorg Chem. 2000;39:3722. doi: 10.1021/ic000360n. [DOI] [PubMed] [Google Scholar]
- 69.Suslick KS, Watson RA. Inorg Chem. 1991;30:912. [Google Scholar]
- 70.Eroy-Reveles AA, Leung Y, Mascharak PK. J Am Chem Soc. 2006;128:7166. doi: 10.1021/ja061852n. [DOI] [PubMed] [Google Scholar]
- 71.Ghosh K, Eroy-Reveles AA, Olmstead MM, Mascharak PK. Inorg Chem. 2005;44:8469. doi: 10.1021/ic0503535. [DOI] [PubMed] [Google Scholar]
- 72.Ghosh K, Eroy-Reveles AA, Avila B, Holman TR, Olmstead MM, Mascharak PK. Inorg Chem. 2004;43:2988. doi: 10.1021/ic030331n. [DOI] [PubMed] [Google Scholar]
- 73.Szundi I, Rose MJ, Sen I, Eroy-Reveles AA, Mascharak PK, Einarsdóttir Ó. Photochem Photobiol. 2006;82:1377. doi: 10.1562/2006-07-25-rc-984. [DOI] [PubMed] [Google Scholar]
- 74.Afshar RK, Patra AK, Mascharak PK. J Inorg Biochem. 2005;99:1458. doi: 10.1016/j.jinorgbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 75.Baranoff E, Collin J-P, Flamigni L, Sauvage J-P. Chem Soc Rev. 2004;33:147. doi: 10.1039/b308983e. [DOI] [PubMed] [Google Scholar]
- 76.Tfouni E. Coord Chem Rev. 2000;196:281. [Google Scholar]
- 77.(a) Durham B, Wilson S, Hodgson D, Meyer TJ. J Am Chem Soc. 1980;102:600. [Google Scholar]; (b) Durham B, Walsh JL, Carter CL, Meyer TJ. Inorg Chem. 1980;19:860. [Google Scholar]
- 78.Sizova OV, Ivanova NV, Ershov AY, Shashko AD. Russ J Gen Chem. 2003;73:1846. [Google Scholar]
- 79.Frugeri PM, Vasconcellos LCG, Mazzetto SE, Franco DW. New J Chem. 1997;21:349. [Google Scholar]
- 80.Mazzetto SE, Tfouni E, Franco DW. Inorg Chem. 1996;35:3509. [Google Scholar]
- 81.Tfouni E, Ford PC. Inorg Chem. 1980;19:72. [Google Scholar]
- 82.Malouf G, Ford PC. J Am Chem Soc. 1977;99:7213. [Google Scholar]
- 83.(a) Bonnet S, Collin JP, Sauvage JP, Schofield E. Inorg Chem. 2004;43:8346. doi: 10.1021/ic0491736. [DOI] [PubMed] [Google Scholar]; (b) Baranoff E, Collin JP, Furusho J, Furusho Y, Laemmel AC, Sauvage JP. Inorg Chem. 2002;41:1215. doi: 10.1021/ic011014o. [DOI] [PubMed] [Google Scholar]
- 84.Martinez MS, de Oliveira EC, Tfouni E. J Photochem Photobio. 1999;122:103. [Google Scholar]
- 85.Collin J-P, Laemmel AC, Sauvage J-P. New J Chem. 2001;25:22. [Google Scholar]
- 86.Laemmel A-C, Collin J-P, Sauvage J-P. Eur J Inorg Chem. 1999:383. [Google Scholar]
- 87.Schoonover JR, Bignozzi CA, Meyer TJ. Coord Chem Rev. 1997;165:239. [Google Scholar]
- 88.(a) Collin JP, Heitz V, Sauvage JP. Topics Curr Chem. 2005;262:29. [Google Scholar]; (b) Sauvage JP, Collin JP, Chambron JC, Guillerez S, Coudret C. Chem Rev. 1994;94:993. [Google Scholar]
- 89.Medlycott EA, Hanan GS. Chem Soc Rev. 2005;34:133. doi: 10.1039/b316486c. [DOI] [PubMed] [Google Scholar]
- 90.Roundhill DM. Photochemistry and Photophysics of Metal Complexes. Plenum Press; New York: 1994. [Google Scholar]
- 91.Enemark JH, Feltham RD. Coord Chem Rev. 1974;13:339. [Google Scholar]
- 92.McGarvey BR, Ferro AA, Tfouni E, Bezerra CWB, Bagatin I, Franco DW. Inorg Chem. 2000;39:3577. doi: 10.1021/ic9914701. [DOI] [PubMed] [Google Scholar]
- 93.(a) Pipes DW, Meyer TJ. Inorg Chem. 1984;23:2466. [Google Scholar]; (b) Callahan RW, Meyer TJ. Inorg Chem. 1977;16:574. [Google Scholar]
- 94.(a) Roncaroli F, Videla M, Slep LD, Olabe JA. Coord Chem Rev. 2007;252:1903. [Google Scholar]; (b) Videla M, Jacinto JS, Baggio R, Garland MT, Singh P, Kaim W, Slep LD, Olabe JA. Inorg Chem. 2006;45:8608. doi: 10.1021/ic061062e. [DOI] [PubMed] [Google Scholar]; (c) Roncaroli F, Ruggiero ME, Franco DW, Estiú GL, Olabe JA. Inorg Chem. 2002;41:5760. doi: 10.1021/ic025653q. [DOI] [PubMed] [Google Scholar]
- 95.Pandey KK, Ahuja SR, Poonia NS, Bharti S. Chem Comm. 1982:1268. [Google Scholar]
- 96.Lang DR, Davis JA, Lopes LGF, Ferro AA, Vasconcellos LCG, Franco DW, Tfouni E, Wieraszko A, Clarke MJ. Inorg Chem. 2000;39:2294. doi: 10.1021/ic9912979. [DOI] [PubMed] [Google Scholar]
- 97.Richter-Addo GB, Lagzdins P. Metal Nitrosyls. Oxford University Press; 1992. [Google Scholar]
- 98.(a) Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermüller T, Neese F, Wieghardt K. J Am Chem Soc. 2004;126:5138. doi: 10.1021/ja030645+. [DOI] [PubMed] [Google Scholar]; (b) Hauser C, Glaser T, Bill E, Weyhermuller T, Wieghardt K. J Am Chem Soc. 2000;122:4352. [Google Scholar]
- 99.Dey A, Chow M, Taniguchi K, Lugo-Mas P, Davin S, Maeda M, Kovacs JA, Odaka M, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2006;128:533. doi: 10.1021/ja0549695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cox AB, Wallace RM. Inorg Nucl Chem Letts. 1971;7:1191. [Google Scholar]
- 101.Khodashva TS, Bokil GB. Zh Strukt Khim. 1960;1:151. [Google Scholar]
- 102.Veal JT, Hodgson DJ. Inorg Chem. 1972;11:1420. doi: 10.1021/ic50118a603. [DOI] [PubMed] [Google Scholar]
- 103.A chloro-bridged dimer of the composition (PPh3Me)2[Ru(NO)Cl4]2•2CH2Cl2 has been reported. See Fenske D, Demant U, Dehnicke K. Zh Natursh B: Chem Sci. 1985;40:1672.
- 104.Paula QA, Batista AA, Castellano EE, Ellena J. J Inorg Biochem. 2002;90:144. doi: 10.1016/s0162-0134(02)00409-9. [DOI] [PubMed] [Google Scholar]
- 105.Nikol’skii AB, Popov AM, Vasilevskii IV. Koordinatsionnaya Khimiya. 1976;2:671. [Google Scholar]
- 106.Komozin PN, Kazakova VM, Miroshnichenko IV, Sinitsyn NM. Zh Neorganich Khim. 1983;28:3186. [Google Scholar]
- 107.Bettache N, Carter T, Corrie JET, Ogden D, Trentham DR. Methods Enzymol. 1996;268:266. doi: 10.1016/s0076-6879(96)68029-x. [DOI] [PubMed] [Google Scholar]
- 108.Mercer EE, Campbell WM, Wallace RM. Inorg Chem. 1964;3:1018. [Google Scholar]
- 109.(a) Clarke MJ. Coord Chem Rev. 2002;232:69. [Google Scholar]; (b) Clarke MJ. Adv Chem Ser. 1997;253:349. [Google Scholar]; (c) Clarke MJ, Stubbs M. Met Ions Biol Syst. 1996;32:727. [PubMed] [Google Scholar]
- 110.(a) Brabec V, Novakova O. Drug Res Update. 2006;9:111. doi: 10.1016/j.drup.2006.05.002. [DOI] [PubMed] [Google Scholar]; (b) Brabec V. Prog Nucl Acid Res Mol Biol. 2002;71:1. doi: 10.1016/s0079-6603(02)71040-4. [DOI] [PubMed] [Google Scholar]
- 111.Gomes MG, Davanzo CU, Silva SC, Lopes LGF, Santos PS, Franco DW. J Chem Soc, Dalton Trans. 1998:601. [Google Scholar]
- 112.Gleu K, Büddecker I. Z Anorg Allgem Chem. 1952;268:208. [Google Scholar]
- 113.Armor JN, Scheidegger HA, Taube H. J Am Chem Soc. 1968;90:5928. [Google Scholar]
- 114.Bottomley F. J Chem Soc Dalton Trans. 1974:1600. [Google Scholar]
- 115.Toledo JC, Ledo BDSL, Franco DW. Coord Chem Rev. 2005;249:419. [Google Scholar]
- 116.Tfouni E, Krieger M, McGarvey BR, Franco DW. Coord Chem Rev. 2003;236:57. [Google Scholar]
- 117.Borges SSS, Davazno CU, Castellano EE, Schpector JZ, Silva SC, Franco DW. Inorg Chem. 1998;37:2670. doi: 10.1021/ic951563s. [DOI] [PubMed] [Google Scholar]
- 118.Carlos RM, Ferro AA, Silva HAS, Gomes MG, de Borges SSS, Ford PC, Tfouni E, Franco DW. Inorg Chim Acta. 2004;357:1381. [Google Scholar]
- 119.Silva WC, Castellano EE, Franco DW. Polyhedron. 2004;23:1063. [Google Scholar]
- 120.Bezerra CWB, da Silva SC, Gambardella MTP, Santos RHA, Plicas LMA, Tfouni E, Franco DW. Inorg Chem. 1999;38:5660. [Google Scholar]
- 121.Carlos RM, Cardoso DR, Castellano EE, Osti RZ, Camargo AJ, Macedo LG, Franco DW. J Am Chem Soc. 2004;126:2546. doi: 10.1021/ja0373263. [DOI] [PubMed] [Google Scholar]
- 122.Lopes LGF, Castellano EE, Ferreira AG, Davanzo CU, Clarke MJ, Franco DW. Inorg Chim Acta. 2005;358:2883. [Google Scholar]
- 123.(a) Sizova OV, Lyubimova OO. Ruus J Gen Chem. 2004;74:996. [Google Scholar]; (b) Sizova NV, Ivanova NV, Sizov VV, Nikolskii AB. Russ J Gen Chem. 2004;74:481. [Google Scholar]; (c) Sizova OV, Ivanova NV, Lyubimova OO, Nikolskii AB. Russ J Gen Chem. 2004;74:155. [Google Scholar]; (d) Gorelski SI, da Silva SC, Lever ABP, Franco DW. Inorg Chim Acta. 2000;300–302:698. [Google Scholar]
- 124.Torsoni AS, Barros BF, Toledo JC, Jr, Haun M, Krieger MH, Tfouni E, Franco DW. Nitric Oxide. 2002;6:247. doi: 10.1006/niox.2001.0409. [DOI] [PubMed] [Google Scholar]
- 125.Marcondes FG, Ferro AA, Souza-Torsoni A, Sumitani M, Clarke MJ, Franco DW, Tfouni E, Krieger MH. Life Sci. 2002;70:2735. doi: 10.1016/s0024-3205(02)01528-x. [DOI] [PubMed] [Google Scholar]
- 126.de Souza Oliveira F, Ferreira KQ, Bonaventura D, Bendhack LM, Tedesco AC, de Machado SP, Tfouni E, da Silva RS. J Inorg Biochem. 2007;101:313. doi: 10.1016/j.jinorgbio.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 127.Antonini A, Brunori M. Hemoglobin and Myoglobin in their Reaction with Ligands. North-Holland; Amsterdam: 1971. [Google Scholar]
- 128.Hoshino M, Laverman L, Ford PC. Coord Chem Rev. 1999;187:75. [Google Scholar]
- 129.(a) Vos MH, Lipowski G, Lambry JC, Martin JL, Liebl U. Biochemistry. 2001;40:7806. doi: 10.1021/bi010060x. [DOI] [PubMed] [Google Scholar]; (b) Eich RF, Li T, Lemon DD, Doherty DH, Curry SR, Aitken JF, Mathews AJ, Johnson KA, Smith RD, Phillips GN, Olson JS. Biochemistry. 1996;35:6976. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]; (c) Rose EJ, Hoffman BM. J Am Chem Soc. 1983;105:2866. [Google Scholar]; (d) Hoffman BM, Gibson QH. Proc Natl Acad Sci. 1978;75:21. doi: 10.1073/pnas.75.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ford PC, Laverman LE. Coord Chem Rev. 2005;249:391. [Google Scholar]
- 131.Lim MD, Lorkovic IM, Ford PC. J Inorg Biochem. 2005;99:151. doi: 10.1016/j.jinorgbio.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 132.Ford PC. Intl J Photoenergy. 2001;3:161. [Google Scholar]
- 133.Lorkovic IM, Ford PC. Inorg Chem. 1999;38:1467. [Google Scholar]
- 134.Lorkovic IM, Miranda KM, Lee B, Bernhard S, Schoonover JR, Ford PC. J Am Chem Soc. 1998;120:11674. [Google Scholar]
- 135.Miranda KM, Bu X, Lorkovic I, Ford PC. Inorg Chem. 1997;36:4838. doi: 10.1021/ic970065b. [DOI] [PubMed] [Google Scholar]
- 136.Xu N, Lee J, Powell DR, Richter-Addo GB. Inorg Chim Acta. 2005;358:2855. [Google Scholar]
- 137.Lee JY, Richter-Addo GB. J Inorg Biochem. 2004;98:1247. doi: 10.1016/j.jinorgbio.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 138.Kadish KM, Adamian VA, Camelbecke EV, Tan Z, Tagliateste P, Bianco P, Boschi T, Yi GB, Khan MA, Richter-Addo GB. Inorg Chem. 1996;35:1343. doi: 10.1021/ic950799t. [DOI] [PubMed] [Google Scholar]
- 139.(a) Works CF, Jocher CJ, Bart GD, Bu X, Ford PC. Inorg Chem. 2002;41:3728. doi: 10.1021/ic020248k. [DOI] [PubMed] [Google Scholar]; (b) Works CF, Ford PC. J Am Chem Soc. 2000;122:7592. [Google Scholar]
- 140.The {Ru-NO}6 nitrosyl derived from salen was first reported by Bosnich and coworkers. See Odenkirk W, Rheingold AL, Bosnich AL. J Am Chem Soc. 1992;114:6392.
- 141.Bordini J, Hughes DL, da Motto Neto JD, da Cunha CJ. Inorg Chem. 2002;41:5410. doi: 10.1021/ic011273d. [DOI] [PubMed] [Google Scholar]
- 142.Mitchell-Koch JT, Reed TM, Borovik AS. Angew Chem Intl Ed. 2004;43:2806. doi: 10.1002/anie.200352881. [DOI] [PubMed] [Google Scholar]
- 143.Bordini J, Ford PC, Tfouni E. Chem Commun. 2005:4169. doi: 10.1039/b507407j. [DOI] [PubMed] [Google Scholar]
- 144.(a) Ferreira KQ, Schneider JF, Nascente PAP, Rodrigues-Filho UP, Tfouni E. J Colloid Interface Sci. 2006;300:543. doi: 10.1016/j.jcis.2006.03.081. [DOI] [PubMed] [Google Scholar]; (b) Doro FG, Rodrigues-Filho UP, Tfouni E. J Colloid Interface Sci. 2007;307:405. doi: 10.1016/j.jcis.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 145.Harada F, Onozuka T, Tomizawa H, Tanaka M, Miki E. Inorg Chim Acta. 2006;359:665. [Google Scholar]
- 146.Wang HF, Onozuka T, Tomizawa H, Tanaka M, Miki E. Inorg Chim Acta. 2004;357:1303. [Google Scholar]
- 147.Wang H, Tomizawa H, Miki E. Inorg Chim Acta. 2004;357:4291. [Google Scholar]
- 148.Suganuma T, Tanada A, Tomizawa H, Tanaka M, Miki E. Inorg Chim Acta. 2001;320:22. [Google Scholar]
- 149.Wang H, Hagihara T, Ikezawa H, Tomizawa H, Miki E. Inorg Chim Acta. 2000;299:80. [Google Scholar]
- 150.Toma HE, Alexiou ADP, Formiga ALB, Nakamura M, Dovidauskas S, Eberlin MN, Tomazela DM. Inorg Chim Acta. 2005;358:2891. [Google Scholar]
- 151.Marlin DS, Mascharak PK. Chem Soc Rev. 2000;29:69. [Google Scholar]
- 152.Marlin DS, Olmstead MM, Mascharak PK. Inorg Chem. 2001;40:7003. doi: 10.1021/ic010523n. [DOI] [PubMed] [Google Scholar]
- 153.(a) Endo I, Nojiri M, Tsujimura M, Nakasako M, Nagashima S, Yohda M, Odaka M. J Inorg Biochem. 2001;83:247. doi: 10.1016/s0162-0134(00)00171-9. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi M, Shimizu S. Curr Opin Chem Biol. 2000;4:95. doi: 10.1016/s1367-5931(99)00058-7. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi M, Shimizu S. Eur J Biochem. 1999;261:1. doi: 10.1046/j.1432-1327.1999.00186.x. [DOI] [PubMed] [Google Scholar]; (b) Endo I, Odaka M, Yohda M. Trends Biotechnol. 1999;17:244. doi: 10.1016/s0167-7799(99)01303-7. [DOI] [PubMed] [Google Scholar]; (c) Yamada H, Kobayashi M. Biosci Biotech Biochem. 1996;60:1391. doi: 10.1271/bbb.60.1391. [DOI] [PubMed] [Google Scholar]
- 154.(a) Harrop TC, Mascharak PK. Acc Chem Res. 2004;37:253. doi: 10.1021/ar0301532. [DOI] [PubMed] [Google Scholar]; (b) Mascharak PK. Coord Chem Rev. 2002;225:201. doi: 10.1016/j.ccr.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kovacs JA. Chem Rev. 2004;104:825. doi: 10.1021/cr020619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.(a) Tyler LA, Noveron JC, Olmstead MM, Mascharak PK. Inorg Chem. 2003;42:5751. doi: 10.1021/ic030088s. [DOI] [PubMed] [Google Scholar]; (b) Noveron JC, Olmstead MM, Mascharak PK. J Am Chem Soc. 2001;123:3247. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- 157.Nagamune T, Kurata H, Hirata M, Honda J, Hirata A, Endo I. Photochem Photobiol. 1990;51:87. [Google Scholar]
- 158.Odaka M, Fujii K, Hoshino M, Noguchi T, Tsujimura M, Nagashima S, Yohda M, Nagamune T, Inoue Y, Endo I. J Am Chem Soc. 1997;119:3785. [Google Scholar]
- 159.Noguchi T, Hoshino M, Tsujimura M, Odaka M, Inoue Y, Endo I. Biochemistry. 1996;35:16777. doi: 10.1021/bi961562r. [DOI] [PubMed] [Google Scholar]
- 160.Huang W, Jia J, Cummings J, Nelson M, Schneider G, Lindqvist Y. Structure. 1997;5:691. doi: 10.1016/s0969-2126(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 161.Nagashima S, Nakasako M, Dohmae N, Tsujimora M, Takio K, Masafumi O, Yohda M, Kamiya N, Endo I. Nat Struct Biol. 1998;5:347. doi: 10.1038/nsb0598-347. [DOI] [PubMed] [Google Scholar]
- 162.Patra AK, Mascharak PK. Inorg Chem. 2003;42:7363. doi: 10.1021/ic030110h. [DOI] [PubMed] [Google Scholar]
- 163.Madhani M, Patra AK, Miller TW, Eroy-Reveles AA, Hobbs AJ, Fukuto JM, Mascharak PK. J Med Chem. 2006;49:7325. doi: 10.1021/jm0604629. [DOI] [PubMed] [Google Scholar]
- 164.Rose MJ, Olmstead MM, Mascharak PK. Polyhedron. 2007;26:4713. [Google Scholar]
- 165.Patra AK, Rose MJ, Murphy KM, Olmstead MM, Mascharak PK. Inorg Chem. 2004;43:4487. doi: 10.1021/ic040030t. [DOI] [PubMed] [Google Scholar]
- 166.Zahran ZN, Powell DR, Richter-Addo GB. Inorg Chim Acta. 2006;359:3084. [Google Scholar]
- 167.(a) Fortney CF, Shepherd RE. Inorg Chem Commun. 2004;7:1065. [Google Scholar]; (b) Fortney CF, Geib SJ, Lin F, Shepherd RE. Inorg Chim Acta. 2005;358:2921. [Google Scholar]
- 168.Halpenny GM, Olmstead MM, Mascharak PK. Inorg Chem. 2007;46:6601. doi: 10.1021/ic700694b. [DOI] [PubMed] [Google Scholar]
- 169.(a) Coradin T, Boissiere M, Livage J. Curr Med Chem. 2006;13:99. doi: 10.2174/092986706789803044. [DOI] [PubMed] [Google Scholar]; (b) Peppas NA, Bures P, Leobandung W, Ichikawa H. Eur J Pharm Biopharm. 2000;50:27. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 170.Sellmann D, Geck M, Moll M. Z Naturforsch. 1992;47b:74. [Google Scholar]
- 171.Sellmann D, Rohm C, Moll M, Knoch F. Z Naturforsch. 1995;50b:1229. [Google Scholar]
- 172.Sellmann D, Häuβinger D, Gottschalk-Gaudig T, Heinemann FW, Naturforsch Z. 2000;55b:723. [Google Scholar]
- 173.Sellmann D, Engl K, Heinemann FW. Eur J Inorg Chem. 2000:423. [Google Scholar]
- 174.Sellmann D, Prakash R, Heinemann FW, Moll M, Klimowicz M. Angew Chem Intl Ed. 2004;43:1877. doi: 10.1002/anie.200453717. [DOI] [PubMed] [Google Scholar]
- 175.Sellmann D, Gottschalk-Gaudig T, Häuβinger D, Heinemann FW, Hess BA. Chem Eur J. 2001;7:2099. doi: 10.1002/1521-3765(20010518)7:10<2099::aid-chem2099>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 176.Nagao H, Nishimura H, Funato H, Ichikawa Y, Howell FS, Mukaida M, Kakihana H. Inorg Chem. 1989;28:3955. [Google Scholar]
- 177.Nagao H, Ooyama D, Howell FS, Mukaida M, Mizumachi K. Anal Sci. 1998;14:645. [Google Scholar]
- 178.Rose MJ, Patra AK, Alcid EA, Olmstead MM, Mascharak PK. Inorg Chem. 2007;46:2328. doi: 10.1021/ic0620945. [DOI] [PubMed] [Google Scholar]
- 179.Patra AK, Olmstead MM, Mascharak PK. Inorg Chem. 2002;41:5403. doi: 10.1021/ic020373w. [DOI] [PubMed] [Google Scholar]
- 180.de Lima RG, Sauaia MG, Bonaventura D, Tedesco AC, Bendhack LM, da Silva RS. Inorg Chim Acta. 2006;359:2543. [Google Scholar]
- 181.Sugimori A, Uchida H, Akiyama T, Mukaida M, Shimizu K. K Chem Letts. 1982:1135. [Google Scholar]
- 182.Tognolio V, da Silva RS, Tedesco AC. Inorg Chim Acta. 2001;316:7. [Google Scholar]
- 183.de Lima RG, Sauaia MG, Bonaventura D, Tedesco AC, Lopez RFV, Bendhack LM, da Silva RS. Inorg Chim Acta. 2005;358:2643. [Google Scholar]
- 184.(a) Coppens P, Novozhilova I, Kovalevsky A. Chem Rev. 2002;102:861. doi: 10.1021/cr000031c. [DOI] [PubMed] [Google Scholar]; (b) Coppens P, Fomitchev DV, Carducci MD, Culp K. J Chem Soc Dalton Trans. 1998:865. [Google Scholar]
- 185.Bitterwolf TE. Coord Chem Rev. 2006;250:1196. [Google Scholar]
- 186.(a) Woike T, Haüssuhl S. Solid State Commun. 1993;86:333. [Google Scholar]; (b) Woike T, Zöllner H, Krasser W. Solid State Commun. 1990;73:149. [Google Scholar]
- 187.Fomitchev DV, Coppens P. Inorg Chem. 1996;35:7021. doi: 10.1021/ic9607144. [DOI] [PubMed] [Google Scholar]
- 188.Fomitchev DV, Coppens P, Li T, Bagley KA, Chen L, Richter-Addo GB. Chem Commun. 1999:2013. [Google Scholar]
- 189.Da Silva SC, Franco DW. Spectrochim Acta (A) 1999;55:1515. [Google Scholar]
- 190.Prakash R, Czaja AU, Heinemann FW, Sellmann D. J Am Chem Soc. 2005;127:13758. doi: 10.1021/ja053758x. [DOI] [PubMed] [Google Scholar]
- 191.Rose MJ, Olmstead MM, Mascharak PK. J Am Chem Soc. 2007;129:5342. doi: 10.1021/ja070247x. [DOI] [PubMed] [Google Scholar]
- 192.Sauaia MG, de Lima RG, Tedesco AC, da Silva RS. J Am Chem Soc. 2003;125:14718. doi: 10.1021/ja0376801. [DOI] [PubMed] [Google Scholar]
- 193.Sauaia MG, de Lima RG, Tedesco AC, da Silva RS. Inorg Chem. 2005;44:9946. doi: 10.1021/ic051346j. [DOI] [PubMed] [Google Scholar]
- 194.(a) Pavlos CM, Xu H, Toscano JP. Curr Topics Med Chem. 2005;5:635. doi: 10.2174/1568026054679326. [DOI] [PubMed] [Google Scholar]; (b) Pavlos CM, Xu H, Toscano JP. Dynamic Studies Biol. 2005;205:178. [Google Scholar]
- 195.(a) Carter TD, Bettache N, Ogden D, Trentham DR. J Physiol. 1993;467:165P. [Google Scholar]; (b) Williams JH, Bettache N, Trentham DR, Bliss TVP. J Physiol. 1993;467:166P. [Google Scholar]
- 196.(a) Murphy KPSJ, Williams JH, Bettache N, Bliss TVP. Neuropharm. 1994;33:1375. doi: 10.1016/0028-3908(94)90039-6. [DOI] [PubMed] [Google Scholar]; (b) Murphy KPSJ, Williams JH, Bettache N, Trentham DR, Bliss TVP. Brain Res Assoc Abstr. 1994;11:92. [Google Scholar]
- 197.Toledo JC, Jr, de Franca Lopes LG, Alves AA, da Silva LP, Franco DW. J Inorg Biochem. 2002;89:267. doi: 10.1016/s0162-0134(01)00414-7. [DOI] [PubMed] [Google Scholar]
- 198.(a) de Barros BF, Toledo JC, Jr, Franco DW, Tfouni E, Krieger MH. Nitric Oxide. 2002;7:50. doi: 10.1016/s1089-8603(02)00007-1. [DOI] [PubMed] [Google Scholar]; (b) Torsoni AS, de Barros BF, Toledo JC, Jr, Haun M, Krieger MH, Tfouni E, Franco DW. Nitric Oxide: Biol Chem. 2002;6:247. doi: 10.1006/niox.2001.0409. [DOI] [PubMed] [Google Scholar]