

Abstract
The catalytic enantioselective SN2′ displacement of (Z)-allylic trichloroacetimidates catalyzed by the palladium(II) complex [COP-OAc]2 is a broadly useful method for the asymmetric synthesis of chiral branched allylic esters. A variety of experiments aimed at elucidating the nature of the catalytic mechanism and its rate- and enantiodetermining steps are reported. Key findings include: (a) the demonstration that a variety of bridged-dipalladium complexes are present and constitute resting states of the COP catalyst, however, monomeric palladium (II) complexes are likely involved in the catalytic cycle; (b) labeling experiments establishing that the reaction proceeds in an overall antarafacial fashion; (c) secondary deuterium kinetic isotope effects that suggest substantial rehybridization at both C1 and C3 in the rate-limiting step; and (d) DFT computational studies (B3-LYP/def2-TZVP) that provide evidence for bidentate substrate-bound intermediates and an anti-oxypalladation/syn-deoxypalladation pathway. These results are consistent with a novel mechanism in which chelation of the imidate nitrogen to form a cationic palladium (II) intermediate activates the alkene for attack by external carboxylate in the enantiodetermining step. Computational modeling of the transition-state structure for the acyloxy palladation step provides a model for enantioinduction.
Introduction
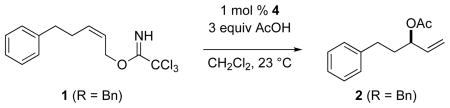
Enantioenriched allylic alcohols and their derivatives are widely employed intermediates in the enantioselective synthesis of a variety of chiral organic molecules.1,2 Recently, we reported the use of the enantiopure palladium(II) complex [(Rp,S)-COP-OAc]2 (4)3 and its enantiomer to catalyze the transformation of prochiral (Z)-allylic trichloroacetimidates 1 to enantioenriched branched allylic esters 2 of high enantiomeric purity (86–99% ee) (eq 1).4 Using the same palladium(II) catalyst, enantioenriched branched allylic aryl ethers 3 can be synthesized in an analogous manner in high enantiomeric purity (90–98%) and yield (61–97%, eq 2).5
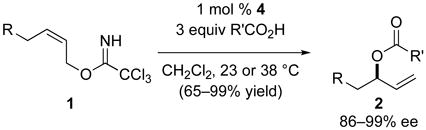 |
(1) |
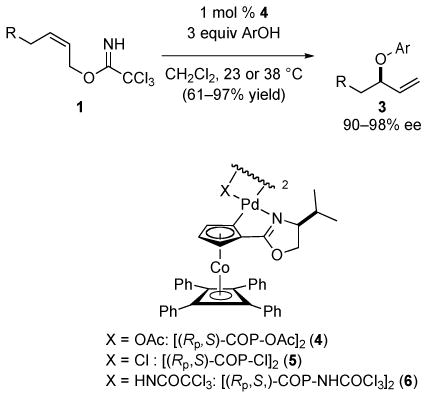 |
(2) |
The COP-catalyzed SN2′-displacement of prochiral (Z)-allylic trichloroacetimidates to form allylic esters and allylic aryl ethers is characterized by exceptionally high branched-to-linear ratios. The linear product is typically not seen by 1H NMR analysis of crude reaction products, with branched-to-linear ratios greater than 800:1 being established in several cases.4,5 Carboxylic acids with low pKa values (< 3.5) can generate allylic cations by protonolysis of the imidate leaving group; as a result of this background reaction, lower branched-to-linear selectivities are observed in the synthesis of chiral allylic esters with these stronger acids.4a
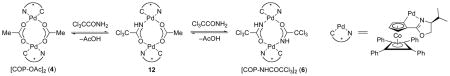
The [COP-OAc]2-catalyzed enantioselective allylic displacement reactions of carboxylic acids and phenols are limited to (Z)-allylic trichloroacetimidate precursors, because the corresponding E stereoisomers (7) preferentially undergo [COP-OAc]2-catalyzed [3,3]-sigmatropic rearrangement (eq 3). This enantioselective rearrangement to form chiral allylic amides, which is optimally catalyzed by [COP-Cl]2 (5),6 has been established to take place by a two-step, cyclization-induced rearrangement mechanism.7
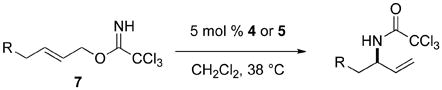 |
(3) |
In some cases, the nature of the bridging ligand of the COP complex can influence the partitioning between the SN2′ and [3,3]-sigmatropic rearrangement pathways. In the synthesis of allylic phenyl ethers, but not allylic esters, changing the COP catalyst to the di-μ-amidate dipalladium complex 6, allows prochiral (E)-allylic trichloroacetimidates 7 to be employed (eq 4).8 Success in this case derives from [COP-NHCOCCl3]2 (6) being a poor catalyst for allylic trichloroacetimidate rearrangements.
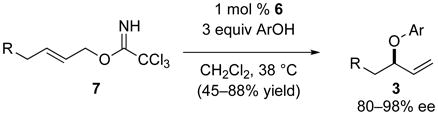 |
(4) |
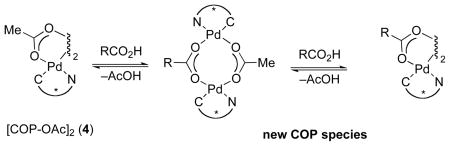
In our original report,4b a catalytic cycle for the [COP-OAc]2-catalyzed allylic ester synthesis that involves chelation of the allylic imidate substrate to the palladium(II) catalyst was proposed (Scheme 1). In this mechanism, reversible coordination of the imidate nitrogen to palladium gives complex 8. Displacement of acetate by the tethered C–C π-bond generates cationic palladium(II) complex 9, which is activated for attack by an external carboxylic acid to produce complex 10.9 Deoxypalladation of this palladacyclic intermediate delivers alkene complex 11, and after ligand exchange the allylic ester product 2.
Scheme 1.

Originally Proposed Mechanism for [COP-OAc]2-Catalyzed Synthesis of Allylic Esters
The initial mechanistic proposal depicted in Scheme 1 was founded on three observations. First, allylic N-arylimidates are unsuitable leaving groups even though they are effective substrates for COP-catalyzed [3,3]-sigmatropic rearrangements.4a This difference in reactivity is attributed to reduced binding to the palladium center by the imidate nitrogen in the less-basic and more-sterically hindered arylimidate precursors. Second, substrates such as allylic esters having leaving groups that lack the strongly coordinating nitrogen atom are unreactive.4a Third, the exceptionally high branched-to-linear ratios strongly suggest that the reaction does not proceed via η3-allylpalladium intermediates.10
Our studies to further understand the nature of these mechanistically novel palladium(II)-catalyzed SN2′ reactions are the subject of this account. The scope and limitations of the [COP-OAc]2-catalyzed enantioselective synthesis of allylic esters are addressed in the preceding article.4a
Results
Catalytic Species in Solution
NMR observation of the reaction of imidate 1 (R = Et) with varying amounts of added acetic acid in the presence of 1 equiv of [COP-OAc]2 (4) revealed the presence of two new palladium(II) complexes: one the well-characterized di-μ-trichloroacetamidate dipalladium complex 6,11 and the second a structurally uncharacterized mixed dimer 12 having acetate and trichloroacetamidate as the two bridging ligands. To explore the composition of COP complexes, which changes during the course of the [COP-OAc]2-catalyzed reaction of (Z)-allylic trichloroacetimidates and carboxylic acids as trichloroacetamide is produced, COP catalysts 4 and/or 6 were mixed with acetic acid or trichloroacetamide in CD2Cl2 and the resulting solution was monitored by 1H NMR until equilibrium was reached (Table 1). Starting solutions of [COP-OAc]2 (4) and trichloroacetamide, or of [COP-NHCOCCl3]2 (6) and acetic acid, converged to the same equilibrium mixture (Table 1, entries 1–2). In addition, mixtures of [COP-OAc]2 and [COP-NHCOCCl3]2 also converged to similar equilibrium mixtures (Table 1, entries 3–4). Equilibrium was reached within 4 h, which is faster than the time frame of the SN2′ reaction (typically 17 h). The equilibrium mixture of catalyst species agrees with a simple statistical distribution, indicating no significant thermodynamic preference for forming either of the three palladacyclic complexes.
Table 1.
Equilibria of [COP-OAc]2 (4) and [COP-NHCOCCl3]2 (6)
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | initial concentration (mM)a | equilibrium concentration (mM)b | ||||||
| [COP-OAc]2 (4) | [COP-NHCOCCl3]2 (6) | HOAc | H2NCOCl3 | [COP-OAc]2 (4) | [COP]2-OAc/NHCOCCl3 (12) | [COP-NHCOCl3]2 (6) | Kc | |
| 1 | 9.28 | – | – | 18.8 | 2.16 | 2.81 | 4.32 | – |
| 2 | – | 9.28 | 18.5 | – | 2.06 | 2.68 | 4.52 | – |
| 3 | 4.65 | 4.65 | – | – | 2.07 | 4.55 | 2.69 | 3.7 |
| 4 | 6.20 | 3.10 | – | – | 3.64 | 4.31 | 1.35 | 3.8 |
Experiments conducted in CD2Cl2.
Determined by 1H NMR spectroscopy.
K = [12]2/([4][6]).
The rapid equilibration of [COP-OAc]2 (4) with [COP-NHCOCCl3]2 (6) in the presence of acetic acid provides a rationale for the inability of complex 6 to be an effective catalyst for enantioselective displacement of (E)-allylic trichloroacetimidates with acetic acid. As a result of this catalyst equilibration, the [3,3]-rearrangement of the allylic trichloroacetimidate substrate remains a competitive reaction even when the di-μ-amidate dipalladium complex 6 is employed.
The equilibration of [COP-OAc]2 with various carboxylic acids is summarized in Table 2. Both sterically demanding acids (Table 2, entries 1–2) and benzoic acids (Table 2, entries 3–4) readily formed new palladacyclic species. Addition of allylic imidate 1 (R = Et) to these mixtures resulted in the formation of the branched allylic ester products 2 (R = Et) with high enantiomeric excess, suggesting that any of the dimers could be the active catalyst, or could be easily converted to the active catalyst, in the presence of the allylic imidate substrate. No obvious correlation between the equilibrium of the various di-μ-carboxylate dipalladium complexes with either the acidity of the carboxylic acid or the rate of the [COP-OAc]2-catalyzed reaction of the acid with allylic imidate 1 (R = Et) is apparent.4a
Table 2.
New Palladium Species Formed by Exposure of [COP-OAc]2 (4) to Carboxylic Acidsa
 | ||||
|---|---|---|---|---|
| entry | R | 4:new COP speciesb | SN2′ reactivity of RCO2Hc | pKa |
| 1 | adamantyl | 1:3.6 | 26 | 6.8 |
| 2 | t-Bu | 1:7.8 | 40 | 5.0 |
| 3 | p-MeC6H5 | 1:4.6 | 67 | 4.4 |
| 4 | Ph | 1:10.8 | 84 | 4.2 |
| 5 | o-ClC6H4CH2 | 1:3.1 | 92 | 4.1 |
| 6 | ClCH2CH2 | 1.2:1 | 40 | 4.0 |
| 7 | MeOCH2 | 1:5.0 | 38 | 3.6 |
| 8 | Cl2CH | 0:>95 | 14 | 1.3 |
[4] = 9.3 mM, [RCO2H] = 18.6 mM, CD2Cl2.
Measured as the sum of all dimeric or heterodimeric species that are not homodimeric 4. See Supplementary Information for more details.
% Conversion after 24 h: [imidate 1 (R = Bn)] = 0.5 M, 3 equiv RCO2H, 1 mol % 4, in CD2Cl2, 23 °C.
% ee as a Function of Time
Enantiomeric excess of the allylic ester product 2 (R = Bn) produced from the [COP-OAc]2-catalyzed reaction of (Z)-allylic imidate 1 (R = Bn) with acetic acid was monitored during the course of the reaction (Table 3). The enantiomeric excess of the product formed during the reaction was found to decrease only slightly over 25 h (96–91%). These results show that the distribution of [COP-OAc]2 (4), [COP-HNCOCCl3]2 (6) and unsymmetrical dimers has no, or only a minor, influence on stereoinduction. The observed small decrease in enantiomeric excess of 2 (R = Bn) could well result from slight catalyst decomposition at extended reaction times. In a control experiment, ester 2 (R = Bn) was found to be configurationally stable when exposed to 1 mol % of catalyst 4, 1 equiv of trichloroacetamide, and 2 equiv of acetic acid in dichloromethane at room temperature for 24 h.
Table 3.
Product Enantiomeric Excess vs. Conversion
Determined by enantioselective HPLC analysis.
Determined by HPLC analysis against an internal standard.
Non-Linear Effects
Because nonracemic dimeric enantioselective catalysts can exhibit non-linear effects,12 we examined the [COP-OAc]2-catalyzed reaction of (Z)-allylic imidate 1 (R = Bn) with acetic acid by using catalysts of varying enantiomeric purity. Non-linear effects were not observed (Table 4). This result is consistent with an active catalyst monomer where neither the (R/S) nor (R/R) dimeric resting state is thermodynamically favored.
Table 4.
Test for Non-linear Effects in the [COP-OAc]2-Catalyzed Allylic Esterificationa
| entry | cat. ee (%)b | product ee (%)c | eecalc (%)d |
|---|---|---|---|
| 1 | 0 | −2 | 0 |
| 2 | 20 | 15 | 16 |
| 3 | 40 | 34 | 34 |
| 4 | 60 | 52 | 53 |
| 5 | 80 | 75 | 71 |
| 6 | >99 | 90 | 90 |
1 equiv 1 (R = Bn), 3 equiv PhCO2H, 1 mol % 4, substrate concentration = 0.5 M.
Produced by mixing solutions of opposite enantiomer catalyst of >99% ee.
Determined by enantioselective SFC.
Calculated as straight line between 0% ee catalyst and 100% ee catalyst (eecalc = ee0·eecat, where ee0 is product ee when eecat = 100%, i.e. 90 %)
Steric Course of the SN2′ Reaction
In order to determine the geometry of the [COP-OAc]2-catalyzed allylic esterification reaction, deuterium-labeled primary allylic imidates 13 and 14 were prepared (Scheme 2). Deuterium incorporation was achieved by lithium aluminum deuteride reduction of ynoate 1513 to yield propargyl alcohol 16. Reduction of the triple bond using Lindlar’s catalyst provided (Z)-allylic alcohol 17 (Z:E > 95:5). Alternatively, sodium bis(2-methoxyethoxy)aluminumhydride (Red-Al) reduction of alkynol 16 yielded (E)-allylic alcohol 18 (E:Z > 95:5). Oxidation of these isomeric allylic alcohols with TPAP/NMO14 provided the corresponding aldehydes 19 and 20. Enantioselective Keck reduction of these aldehydes gave alcohols 21 and 22 in enantiomeric excesses of 85% and 91%, respectively.15 Standard base-promoted condensation of these alcohols with trichloroacetonitrile delivered allylic imidates 13 and 14.16 Quantitative NMR analyses of imidates 13 and 14 found them to have E:Z ratios of 3:97 and 99:1, respectively.
Scheme 2.

Synthesis of Enantioenriched Chiral Primary Imidates 13 and 14
As enantioselective reduction of only one α,β-unsaturated aldehyde, (E)-cinnamaldehyde, was reported by Keck,15 we felt it was essential to establish the absolute configuration of chiral allylic alcohols 21 and 22 by an independent means (Figure 1). Therefore, alcohols 21 and 22 were converted into their corresponding R and S Mosher esters, which were analyzed by 1H and 2H NMR spectroscopy.17,18 Mosher has shown that shielding by the phenyl group of an MTPA ester causes a distinctive upfield shift of groups oriented in-plane with the π-electrons of the phenyl ring (Figure 1a). However, because of the similar size of 1H and 2H, no distinct preference for 60° rotation about the allylic C–O bond (A or B, A′ or B′) would be expected. A modification of the Mosher model was developed in order to address this issue. For the purposes of this model, discussion is centered on the S enantiomers of primary alcohols 21 and 22. In the R,S MTPA ester (A or B), the deuterium atom would never experience the in-plane shielding effects of the phenyl ring in either conformation A or B. Instead, it would only be deshielded by interaction with the methoxy group in conformation A. In contrast, the proton would be shielded by interaction with the phenyl ring in conformation B (Figure 1, b). The opposite trends would be expected for the S,S diastereomer (Figure 1, c).19 Therefore, the α-hydrogen of the R,S diastereomer should be shifted upfield relative to that of the S,S diastereomer. Accordingly, the α-deuterium atom of the R,S diastereomer should be shifted downfield relative to that of the S,S diastereomer. The opposite effects (relative deshielding of 1H and shielding of 2H) would be expected for an R configuration. As summarized in Table 5, the observed 1H- and 2H-NMR shifts of the methylene hydrogen and deuterium of the Mosher esters derived from allylic alcohols 21 and 22 are consistent with an S configuration. These findings are in agreement with the absolute configurations derived from the expected stereoselectivity of the Keck reduction.15
Figure 1.

Mosher’s Conformational Model for MTPA Esters Derived from Chiral Primary Alcohols 21 and 22
Table 5.
1H and 2H NMR Chemical shifts of MTPA adducts of 21 and 22
| entry | compound | H δ (ppm) | D δ (ppm) |
|---|---|---|---|
| 1 | 21-(R)-MTPA ester (A/B) | 4.82 | 4.90 |
| 2 | 21-(S)-MTPA ester (A′/B′) | 4.86 | 4.85 |
| 3 | 22-(R)-MTPA ester (A/B) | 4.69 | 4.76 |
| 4 | 22-(S)-MTPA ester (A′/B′) | 4.73 | 4.72 |
Enantioenriched (1S,2Z)-allylic trichloroacetimidate 13 was allowed to react with benzoic acid and phenol in the presence of 1 mol % of [(Rp,S)-COP-OAc]2 under standard conditions (Figure 2). Within experimental uncertainty, chirality transfer was complete, with the Rp,S enantiomer of the catalyst giving respectively (1E,3R)-allylic benzoate 23 and (1E,3R)-allylic phenyl ether 24.20 In accord with these observations, reaction of imidate 13 with benzoic acid in the presence of the Sp,R catalyst enantiomer yielded (1Z,3S)-allylic benzoate 25. These results establish that the SN2′ substitution reactions of (Z)-allylic imidates with carboxylic acids and phenols cleanly occur with antarafacial stereospecificity.
Figure 2.

SN2′ Substitution with Enantioenriched Allylic Imidate Precursors21
Chirality transfer in the reaction of (1S,2E)-allylic trichloroacetimidate 14 with phenol catalyzed by the Rp,S enantiomer of di-μ-trichloroacetamidate dipalladium complex 6 also proceeded with complete transfer of chirality to produce (1E,3S)-allylic phenyl ether ent-24 (eq 5). This result shows that the [COP-NHCOCCl3]2-catalyzed asymmetric SN2′ etherification in the E allylic imidate series also takes place by clean antarafacial stereospecificity.
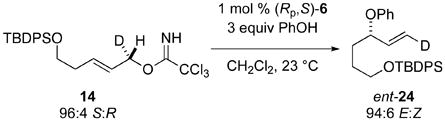 |
(5) |
As expected, allylic rearrangement of trichloroacetimidate 14 with [(Rp,S)-COP-NHCOCCl3]2 yielded (1Z,3S)-allylic trichloroacetamide 26 (eq 6). This result is consistent with the established suprafacial nature of this transformation.7
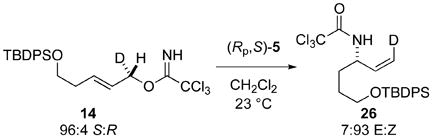 |
(6) |
Secondary Kinetic Isotope Effects
To gain some insight into the nature of the rate-limiting step of the catalytic cycle, hydrogen secondary kinetic isotope effects were measured. The straightforward synthesis and isotopic analysis of the deuterium-labeled substrates 1 (R = CH2CH2OTBDPS) having deuterium at either carbons 1, 2 or 3 are documented in the Supporting Information. Kinetic isotope effect studies were carried out as competition experiments using mixtures of unlabeled and deuterium-labeled 1, with the ratios of unlabeled and deuterium-labeled starting allylic imidate 1 before addition of catalyst and allylic ester products 27 being measured by 1H NMR. Kinetic isotope effects were calculated as the ratio of these two ratios [(27-1H:27-2H)/(1-1H:1-2H)]. p-Methoxybenzoic acid was employed as the nucleophile, because the 1H NMR signals of 27 corresponding to Ha, Hb, and Hc were fully resolved. An inverse hydrogen secondary kinetic isotope effect at C3 of 0.79, and a normal secondary kinetic isotope effect at C1 of 1.16, were observed (Figure 3). No kinetic isotope effect at the internal allylic carbon was observed.
Figure 3.

Secondary Kinetic Isotope Effect Studies (R = CH2CH2OTBDPS, Ar = p-MeOC6H4)
Computational Modeling of Potential Mechanisms
The results of the chirality-transfer experiments indicate that the [COP-OAc]2-catalyzed synthesis of allylic esters (eq 1) and allylic aryl ethers (eq 2) occur by an antarafacial process wherein the imidate leaving group is eliminated from the opposite face of the allyl system as carboxylate is added. In the context of an oxypalladation/deoxypalladation mechanism, this result requires that the oxypalladation and deoxypalladation steps occur with opposite geometries. The two possibilities are depicted in Scheme 3 for the [(Rp,S)-COP-OAc]2-catalyzed reaction of a (1S,2Z)-allylic trichloroacetimidate with a carboxylate nucleophile. In pathway A, the palladium catalyst binds to the Si face of the C–C π-bond activating it for anti oxypalladation to generate the observed 3R configuration of the allylic ester product. This step is followed by syn deoxypalladation to provide the 1E alkene. Alternatively, catalyst binding to the Re face of the C–C π-bond followed by syn oxypalladation and anti deoxypalladation would provide the same (1E,3R)-allylic ester product.
Scheme 3.

The Two Possible Stereochemical Courses of Antarafacial-SN2′ Displacement
Oxypalladation reactions are known that take place in both anti and syn geometries.22 As the two reaction pathways depicted in Scheme 3 can not be unambiguously distinguished by existing precedent, we turned to DFT calculations to examine which of the allowable reaction pathways is most plausible by investigating computationally derived reaction coordinate diagrams.
The TURBOMOLE V6.223 quantum chemistry program using the B3-LYP24 hybrid functional was utilized throughout this study. All atoms were represented by the triple-ζ def2-TZVP basis set.25 A polarizable continuum solvation model (COSMO) was implemented to model the bulk effects of the dichloromethane solvent.26 Full geometry optimization and numerical frequency calculations were performed on all structures. Transition-state structures were characterized by exactly one imaginary vibrational mode along the elemental step studied; intermediates were confirmed by the absence of imaginary vibrational modes.27 All energies are reported as electronic energies plus unscaled zero-point corrections.
For ease of calculation, palladacyclic fragment 28 was chosen as a model for the COP ligand. Natural population analysis (NPA)28 indicated that 28 was a reasonable electronic substitute for the COP fragment (4). Furthermore, a geometric comparison of bond lengths and angles confirmed 28 as a reasonable surrogate of crystallographically characterized COP complexes.8,29,30
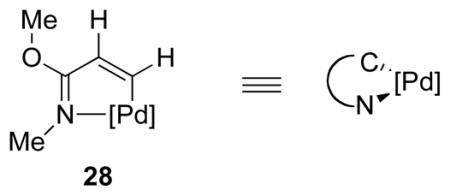
Three possible reaction pathways were investigated (Figure 4). The first involved an anti-oxypalladation pathway, wherein external acetate attacks a COP-alkene complex having an acetate ligand (I, Figure 4). In this reaction pathway, the imidate nitrogen is protonated. The second anti-oxypalladation mechanism investigated involved external acetate attack on a cationic palladium complex in which the imidate nitrogen is bonded to the palladium center (II). The third reaction simulation involved syn oxypalladation of a palladium complex wherein the acetate nucleophile is delivered internally (III). Although the possibility of a concerted oxypalladation/deoxypalladation pathway was initially pursued, transition states corresponding to this reaction mode were not found.31 We reasoned that syn-oxypalladation processes in which the nucleophile is not bound to palladium would be sterically improbable because of the necessity of the nucleophile to pass by either the tetraphenylcyclobutadiene or oxazoline fragments of the COP framework to interact with the activated π-bond.
Figure 4.

Three Reaction Pathways Investigated
Coordination Geometry of Palladium in a COP Complex
The coordination geometry at the palladium center was examined first (Figure 5). Each of the three pathways required slightly different coordination geometries of the starting complex. For the anti-oxypalladation pathway I, monodentate complexes, in which the palladium was bound to the alkene Si-face, were investigated (29–32). Monodentate complexes (33–36) with palladium bound to the Re face of the C–C π-bond were investigated as the starting complex for syn-oxypalladation mechanism III. In these two model complexes, the allylic imidate substrate could bind in one of four possible configurations, either trans or cis to the oxazoline ligand with the alkene substituents either pointed towards or away from the acetate ligand. Because of geometric constraints, only the cis and trans isomers of the bidentate allylic imidate ligand were calculated (37 and 38).
Figure 5.

Relative Energies of Imidate Binding Geometries
A universal preference for the alkene π-bond to bind trans to the oxazoline ligand (29, 31, 33, 35, and 37) was found. This coordination geometry was favored by 2–5 kcal/mol. This observation agrees with earlier investigations of the palladium-catalyzed [3,3]-sigmatropic rearrangement of allylic imidates,7 and with the trans influence of ligands in palladium(II) square-planar complexes.32 In these complexes, the preference for orientation of the alkene substituents towards or away from the acetate ligand was small. In the series of Si-bound complexes, orientation towards the acetate was slightly favored, whereas essentially no preference was found in the Re-bound complexes. This trend held also for complexes in which the alkene binds cis to the nitrogen ligand. These small energy differences in the conformation of the alkene were well within the error of the computational methods employed.33 Complexes 31, 33, and 37 were chosen as the starting geometries for the three reaction pathways.
Energies of Imidate-Bound Palladium Complexes
In order to gain a better understanding of various starting complexes that could be present under catalysis conditions, several additional complexes were evaluated computationally (Figure 6). Bidentate complex 39 would originate from N-bound imidate complex 40 or alkene-bound complex 41. These latter two neutral complexes were calculated to be 16.1 and 9.0 kcal/mol respectively lower in energy than cationic complex 39. The Re-bound η2-alkenepalladium complex 33 was found to be 1.4 kcal/mol lower in energy than the Si-bound complex 41. Protonation of 41 to generate complex 42 costs 23.9 kcal/mol. Because of the expected low basicity of a trichloroacetimidate,34 protonation of neutral complex 41 by acetic acid would not be expected to be favorable in CH2Cl2, but would be necessary in order to achieve a sufficiently nucleophilic acetate species while maintaining charge balance.
Figure 6.

Relative Energies of Various Imidate-Bound Palladium Complexes
The [COP-OAc]2-Catalyzed SN2′ Displacement Reaction
Reaction coordinate diagrams were calculated for the three pathways illustrated in Figure 4 based on these optimized starting geometries (Figure 7).35 The monodentate anti-oxypalladation pathway I begins at high-energy iminium complex 42, which requires 8.7 kcal/mol to reach oxypalladation transition-state structure 43. The resulting zwitterionic oxypalladate 44 is calculated to possess similar energy to starting complex 42. Deoxypalladation has a small activation barrier (<1 kcal/mol), via transition-state structure 45, to generate neutral alkene complex 46. Overall pathway I is calculated to be exothermic by 41.6 kcal/mol.
Figure 7.

Calculated Reaction Coordinate Diagrams for Three Palladium-Catalyzed Antarafacial SN2′ Reactions
Pathway II begins with cationic bidentate palladium alkene complex 39. The activation energy for CO bond formation via transition-state structure 47 is calculated to be 4.4 kcal/mol. This oxypalladation step produces neutral alkylpalladium complex 48, which is calculated to be 9.2 kcal/mol lower in energy than starting complex 39. Deoxypalladation of this intermediate via transition-state structure 49 is found to have an 11.0 kcal/mol activation barrier, with 12.5 kcal/mol being released in the formation of amidate-alkene complex 50. Pathway II to generate complex 50 is calculated to be highly favorable: 4.4 kcal/mol activation barrier and exothermic by 21.7 kcal/mol.
The syn-oxypalladation process (pathway III) begins with acetate-alkene complex 33, which lies 10.4 kcal/mol lower in energy than cationic bidentate complex 39. Intramolecular oxypalladation of 33 requires 21.0 kcal/mol to reach transition-state structure 51. The product of this syn oxypalladation, intermediate 52, is calculated to be 9.7 kcal/mol higher in energy than the starting complex 33. An energy barrier of 9.9 kcal/mol (via 53) separates this intermediate from the cationic ester complex 54, which is 2.4 kcal/mol higher in energy than intermediate complex 52. It is no surprise that this SN2′ pathway, which transforms a cationic alkene complex having an acetate counter ion to one having a more basic trichloroacetamidate counter ion, is calculated to be endothermic (by 12.1 kcal/mol).
The results of this computational study indicate that pathway II is the most likely mechanism. Although the chelated ion-pair intermediate 39 is not the lowest energy imidate-bound palladium complex, the transition-state structure for oxypalladation of this intermediate, which in the three mechanisms investigated is found to be the rate-limiting step, is the lowest energy oxypalladation transition state by 6.2 kcal/mol.
Transition-State Structures for [COP-OAc]2-Catalyzed Oxypalladation
Based on the results from the model studies, preferred antarafacial SN2′ pathway II was investigated computationally using the entire COP catalyst structure (Figure 8).36 Enantiodetermining transition-state structure 47 was used as the starting geometry for calculations of isomeric transition-state structures 55–58. Transition structures 55–58 were confirmed by the presence of a single imaginary vibrational mode along the formation of the C–O bond. These complexes would be derived from reaction of imidate 1 (R = H) with acetic acid in the presence of [(Rp,S)-COP-OAc]2 (4).37 Imidate bound starting-complexes based on structure 39 were also calculated as a comparison. In transition-state structures 55 and 56, the reacting C–C π-bond is positioned trans to the oxazoline ligand, whereas the alkene is cis to the oxazoline in transition-state structures 57 and 58. Transition-state structures 55 and 57 would produce the observed R enantiomer of the allylic ester product; the minor S enantiomer would be generated from transition-state structures 56 and 58. Transition-state structure 55 was found to be the lowest energy of the four complexes investigated when considering both the relative energies of the transition states (ΔΔG‡) and the activation energy from the corresponding precursor imidate complex (ΔG‡, e.g. 39 to 47, Figure 7). Complexes 57 and 58 having the coordinated nitrogen atoms trans are calculated to be 3.5 and 4.9 kcal/mol higher in energy than the corresponding cis complexes. Transition-state structure 56, which leads to the minor enantiomer, is found to be 1.5 kcal/mol higher in energy than the lowest-energy transition-state structure 55.
Figure 8.


Relative Energies of Four Isomeric Transition-State Structures Based on 4738
To gain an appreciation of the non-bonded interactions that are likely responsible for the differences in energy of the isomeric transition-state structures 55–58, the closest contacts between the imidate substrate and COP ligand were examined (Table 6). In low energy transition-state complex 55, the closest contact between the imidate and the cyclopentadiene ligand is the coordinated alkene hydrogen H59–cyclopentadiene hydrogen H75 distance of 2.37 Å (entry 1). This computed structure also exhibits a 2.66 Å contact between the coordinated-alkene hydrogen H62 and H92 of the tetraphenylcyclobutadiene fragment. The closest imidate-to-cyclopentadiene contact in transition-state complex 56 is 2.33 Å (H56–H95), also between a coordinated-alkene hydrogen and a hydrogen of the cyclopentadiene fragment (entry 2). Two additional close contacts of the imidate fragment occur between coordinated alkene hydrogen H95 and H73 of the tetraphenylcyclobutadiene fragment (2.57 Å), and between methyl hydrogen H93 and carbon C31 tetraphenylcyclobutadiene unit (2.85 Å).
Table 6.
Key Interactions Between the Allylic Imidate Fragment and the COP-liganda
| entry | structure | ΔΔG‡ (kcal/mol) | imidate to Cp or imidate to tetraphenylcyclobutadiene oxazoline |
|||||
|---|---|---|---|---|---|---|---|---|
| atoms | distance (Å) | atoms | distance (Å) | atoms | distance (Å) | |||
| 1 | 55 | 0.0 | H59–H75 | 2.374 | H62–H92 | 2.662 | H61–C33 | 3.298 |
| 2 | 56 | +1.5 | H95–H56 | 2.327 | H95–H73 | 2.572 | H93–C31 | 2.852 |
| 3 | 57 | +3.5 | H91–H47 | 1.979 | H95–H58 | 2.508 | H94–C33 | 3.213 |
| 4 | 58 | +6.4 | H95–H47 | 1.997 | H91–H57 | 2.398 | H89–C33 | 3.063 |
Distances that are less than the sum of the Van Der Waals radii of the atoms involved are shown in bold font.
A similar analysis of transition-state complexes 57 and 58 identifies several close contacts (Table 6, entries 3 and 4). Coordinated-alkene hydrogen H91 of transition-state structure 57 is positioned only 1.98 Å away from H47 of the isopropyl group. Coordinated-alkene hydrogen H95 of transition-state structure 57 and methyl hydrogen H94 have contacts with H58 and C33 of the nearest phenyl ring of the tetraphenylcyclobutadiene moeity of 2.51 Å and 3.21 Å, respectively. Transition-state complex 58 also has a similar highly destabilizing interaction between coordinated-alkene hydrogen H95 and H47 of the isopropyl group (2.00 Å), and has 2.40 Å and 3.06 Å contacts between H91 and H89 with a hydrogen (H57) and carbon (C33) of the tetraphenylcyclobutadiene floor, respectively.
Discussion
A Monomeric Palladium(II) Catalyst in Equilibrium with Carboxylic Acids and trichloroacetamide
The studies summarized in Table 1 show that in the presence of trichloroacetamide, [COP-OAc]2 (4) is in a statistical equilibrium with [COP-NHCOCCl3]2 (6) and an unsymmetrical dimer (12). When carboxylic acid nucleophiles other than acetic acid are present, catalyst dimers incorporating these nucleophiles are formed rapidly. However, the propensity of a carboxylic acid to displace acetic acid from [COP-OAc]2 (4) does not correlate with the relative reactivity of the acid towards imidate electrophiles (Table 2). Furthermore, analysis of the % ee of allylic ester products at different extents of conversion show negligible change as trichloroacetamide is produced and the relative abundance of dimeric-amidate catalysts 6 and 12 increases (Table 3). In addition, non-linear effects are not observed (Table 4). Although the resting state of the catalyst is a mixture of various COP complexes, these combined observations suggest that in the presence of an allylic trichloroacetimidate substrate these dipalladium complexes generate a monomeric complex, most likely imidate-nitrogen-coordinated complex 59 (Scheme 4).7
Scheme 4.

Proposed Catalytic Cycle and Off-Cycle Catalyst Equilibria
Allylic Imidate Chelation Followed by Anti Oxypalladation/Syn Deoxypalladation
Deuterium labeling studies showed that the [COP-OAc]2-catalyzed SN2′ reaction of (Z)- and (E)-allylic trichloroacetimidates with carboxylic acids proceeds in an overall antarafacial fashion (Figure 2), requiring that the steric course of the oxypalladation and deoxypalladation steps are different. The inverse secondary kinetic isotope effect observed for the C3 vinylic hydrogen of the allylic imidate substrate indicates rehybridization from sp2 to sp3 occurs during the rate-limiting step (Figure 3). Conversely, the smaller normal secondary kinetic hydrogen isotope effect at C1 suggests that some rehybridization from sp3 to sp2 also occurs during the rate-limiting step. These kinetic isotope effect studies would be consistent with either a concerted oxypalladation-deoxypalladation process,39 a stepwise mechanism wherein deoxypalladation is turnover-limiting,40–42 or a stepwise non-steady-state mechanism wherein the transition states for oxypalladation and deoxypalladation are similar in energy.
To pursue whether the antarafacial selectivity of this catalytic enantioselective synthesis of allylic esters results from anti acyloxypalladation followed by syn-deoxypalladation or the inverse (syn acyloxypalladation followed by anti deoxypalladation), as well as which step in the catalytic cycle is rate-limiting, three possible mechanisms were evaluated by DFT calculations (Figure 7). This computational investigation suggests that pathway II is the most likely mechanism. In this mechanism, palladium-imidate coordination to form complex 59 is followed by alkene coordination to form chelated-cationic palladium(II) intermediate 60 (Scheme 4). Anti acyloxypalladation by external nucleophilic attack of a carboxylic acid at C3 of the palladium-imidate complex 60 generates palladacyclic intermediate 61. Subsequent syn-deoxypalladation forms the alkene trichloroacetamidate complex 62. Finally, reaction of this complex with allylic trichloroacetimidate 1 liberates allylic ester product 2 and trichloroacetamide and regenerates intermediate 59.
The DFT calculations support our original conjecture4b that chelation of the allylic trichloroacetimidate substrate to generate a cationic palladium(II)-alkene complex (60) significantly lowers the activation energy for oxypalladation (see the relative energies of 33, 39, and 42, Figure 7). Intramolecular syn oxypalladation is further made unlikely because of the lack of a correlation between reactivity and the ability of a carboxylic acid to displace acetic acid from [COP-OAc]2 (mechanism III of Figure 7). The expectation would be that a higher relative concentration of reacting complex 33 would increase the overall rate of reaction in this pathway.
Our kinetic isotope effect investigations are consistent with the calculated results. The transition-state structures for both the oxypalladation, 47, and deoxypalladation, 49, steps of mechanism II are sufficiently similar in energy to be consistent with a non-steady-state kinetics scenario: the activation barrier for the reverse reaction from intermediate 48 to complex 39 is calculated to be only slightly higher (by 2.6 kcal/mol) than the activation barrier for the forward reaction from 48 to 50. This small energy difference would allow for secondary kinetic isotope effects at the sites of both oxypalladation and deoxypalladation.
A Model for Enantioselection
An important outcome of the computational study was the development of a model for the high degree of diastereoselection induced by the COP catalyst framework (Figure 8). In this model, bidentate coordination of the allylic imidate substrate is determined by a combination of steric and stereoelectronic effects. The two nitrogen (L-type) ligands prefer a cis orientation because of an electronic preference for the strong σ-donating cyclopentadienide ligand to be trans to the weak σ-donating nitrogen atom of the imidate. This mode of chelation is also favored sterically as the more bulky alkene ligand is positioned away from the isopropyl substituent of the oxazoline ligand. The DFT calculations of the four potential transition-state structures for anti acyloxypalladation suggest that these effects are also manifest in this enantiodetermining step, as the calculated energies of transition-state structures 55 and 56, in which the nitrogen ligands are cis, are lower than structures 57 and 58, where these ligands are trans (Figure 8). The destabilizing steric interactions between the isopropyl substituent of the oxazoline ligand and C3 of the imidate substrate in transition-state complexes 57 and 58 are highlighted in red in Figure 9.
Figure 9.

Model for Enantioselection
However, it is the subtle difference in steric interactions between the substrate and the tetraphenylcyclobutadiene floor that likely are decisive in favoring transition-state structure 55, which leads to the observed R enantiomer of the allylic ester product, over transition-state structure 56 leading to the minor S enantiomer. In these two cis complexes, the alkene hydrogen H95 of complex 56 is calculated to be 0.1 Å closer to the proximal phenyl rings of the tetraphenylcyclobutadiene fragment than the corresponding hydrogen H62 of complex 55 (Figure 8 and Table 6). This trend is more significant when comparing the next-closest groups: the methyl group of transition-state structure 56 is positioned 0.45 Å closer tetraphenylcyclobutadiene floor (Table 6, H93–C31) than the C1 methylene group of low energy transition-state conformation 55 (Table 6, H61–C33). In transition-state structure 55, the methyl group of the substrate is positioned away from the tetraphenylcyclobutadiene floor and is pointed away from the COP ligand entirely. Whereas in transition-state structure 56, the methyl substituent projects towards the large tetraphenylcyclobutadiene fragment.43
Conclusion
The experimental and computational data acquired during this study are consistent with a novel mechanism for the [COP-OAc]2-catalyzed SN2′ reaction of allylic trichloroacetimidates with carboxylic acids to form 3-acyloxy-1-alkenes of high enantiomeric purity.4 Experimental studies establish that a variety of bridged-dipalladium complexes are present and constitute resting states of the COP catalyst; however, monomeric palladium (II) complexes are undoubtedly involved in the catalytic cycle. Deuterium-labeling experiments establish that that the [COP-OAc]2-catalyzed SN2′ reaction of (Z)- and (E)-allylic trichloroacetimidates with carboxylic acids proceeds in an overall antarafacial fashion.
Computational studies indicate that this antarafacial selectivity results from an anti acyloxypalladation/syn deoxypalladation reaction sequence (Scheme 4). In this mechanism, chelation of the allylic imidate substrate to the catalyst to form a cationic Pd(II) intermediate (complex 60 of Scheme 4) activates the alkene for external attack by a carboxylate nucleophile. This step is enantiodetermining. Computational studies also led to a model for enantioselection. In this model, the tetraphenylcyclobutadiene floor and not the isopropyl group of the oxazoline ligand is the significant structural feature of the catalyst determining enantioselection.43 Positioning of the terminal alkyl groups of the allylic imidate substrate away from tetraphenylcyclobutadiene moeity of the Rp,S enantiomer of [COP-OAc]2 activates the Re-face of the olefin for nucleophilic attack. Additionally, steric interactions between the imidate and the isopropyl group and the trans effect of the ligands direct coordination of the C–C π-bond of the substrate trans to the oxazoline ligand. As the [COP-OAc]2-catalyzed SN2′ reaction of (Z)-allylic trichloroacetimidates with phenols takes place in an identical antarafacial fashion, it is reasonable to propose that a similar transition-state model rationalizes stereoselection in this reaction also.
By applying the lessons learned from these mechanistic findings, we hope to develop more effective and more practical catalysts for SN2′ allylic substitution reactions.
Supplementary Material
Acknowledgments
We thank Professor Filipp Furche and Dr. Nathan Crawford for assistance with the computational studies as well as helpful discussion. This research was supported by NSF (CHE-9726471) and postdoctoral fellowships for S.F.K. from the Alexander von Humboldt Foundation (Feodor Lynen Fellowship) and for H.F.S. from the Royal Commission for the Exhibition of 1851. Additional unrestricted support from Amgen and Merck and Pfizer is also gratefully acknowledged. Computational studies were performed on hardware purchased with funding from CRIF (CHE-0840513), and NMR and mass spectra with instruments acquired with the assistance of NSF and NIH shared instrumentation grants.
Footnotes
Supporting Information Available: General methods, experimental procedures, NMR, and HPLC data of new compounds, computational data and XYZ coordinates. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Enantioenriched allylic esters are a ubiquitous intermediate for the synthesis of complex molecules. Selected recent examples include: Shimizu Y, Shi SL, Usuda H, Kanai M, Shibasaki M. Angew Chem, Int Ed. 2010;49:1103–1106. doi: 10.1002/anie.200906678.Crimmins MT, Jacobs DL. Org Lett. 2009;11:2695–2698. doi: 10.1021/ol900814w.Stivala CE, Zakarian A. J Am Chem Soc. 2008;130:3774–3776. doi: 10.1021/ja800435j.
- 2.Hodgson DM, Humphreys PG. In: Science of Synthesis. Clayden JP, editor. Vol. 36. Thieme; Stuttgart: 2007. pp. 583–665. [Google Scholar]
- 3.(a) Anderson CE, Kirsch SF, Overman LE, Richards CJ, Watson MP. Org Synth. 2007;84:148–155. [Google Scholar]; (b) Anderson CE, Overman LE, Richards CJ, Watson MP, White NS. Org Synth. 2007;84:139–147. [Google Scholar]; (c) Stevens AM, Richards CJ. Organometallics. 1999;18:1346–1348. [Google Scholar]
- 4.(a) Cannon JS, Kirsch SF, Overman LE. previous paper in this issue. [Google Scholar]; (b) Kirsch SF, Overman LE. J Am Chem Soc. 2005;127:2866–2867. doi: 10.1021/ja0425583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirsch SF, Overman LE, White NS. Org Lett. 2007;9:911–913. doi: 10.1021/ol070110b. [DOI] [PubMed] [Google Scholar]
- 6.Anderson CE, Overman LE. J Am Chem Soc. 2003;125:12412–12413. doi: 10.1021/ja037086r. [DOI] [PubMed] [Google Scholar]
- 7.Watson MP, Overman LE, Bergman RG. J Am Chem Soc. 2007;129:5031–5044. doi: 10.1021/ja0676962. [DOI] [PubMed] [Google Scholar]
- 8.Olson AC, Overman LE, Sneddon HF, Ziller JW. Adv Synth Catal. 2009;351:3186–3192. doi: 10.1002/adsc.200900678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Although anti oxypalladation was originally suggested, and is depicted in Scheme 1, no evidence concerning this stereochemical issue was available at the time.
- 10.(a) Onitsuka K. J Synth Org Chem Jpn. 2009;67:584–594. [Google Scholar]; (b) Trost BM. J Org Chem. 2004;69:5813–5837. doi: 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Crawley ML. Chem Rev. 2003;103:2921–2943. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (d) Szabó KJ. J Am Chem Soc. 1996;118:7818–7826. [Google Scholar]
- 11.The structure shown for complexes 4 and 6 is consistent with single-crystal X-ray analyses; see ref 8 for a detailed discussion and leading references.
- 12.Guillaneux D, Zhao SH, Samuel O, Rainford D, Kagan HB. J Am Chem Soc. 1994;116:9430–9439. [Google Scholar]
- 13.Hall DG, Deslongchamps P. J Org Chem. 1995;60:7796–7814. [Google Scholar]
- 14.TPAP = Tetrapropylammonium perruthenate (Pr4N+RuO4−): Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639–666.
- 15.Keck GE, Krishnamurthy D. J Org Chem. 1996;61:7638–7639. doi: 10.1021/jo961593s. [DOI] [PubMed] [Google Scholar]
- 16.(a) Nishikawa T, Asai M, Ohyabu N, Yamamoto N, Fukuda Y, Isobe M. Tetrahedron. 2001;57:3875–3883. [Google Scholar]; (b) Numata M, Sugimoto M, Koike K, Ogawa T. Carbohyd Res. 1987;163:209–225. doi: 10.1016/0008-6215(87)80182-9. [DOI] [PubMed] [Google Scholar]
- 17.Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519. [Google Scholar]
- 18.(a) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]; (b) Seco JM, Quinoa E, Riguera R. Tetrahedron: Asymmetry. 2000;11:2781–2791. [Google Scholar]; (c) Kusumi T, Ooi T, Ohkubo Y, Yabuuchi T. Bull Chem Soc Jpn. 2006;79:965–980. [Google Scholar]
- 19.Because the C2 vinylic hydrogen would experience both the shielding phenyl and the deshielding methoxy, a small difference in chemical shift between the two diastereomers would be expected. In fact, the chemical shift for the C2 proton was nearly identical in both diastereomers.
- 20.The E:Z ratio of purified products was determined by integration of the terminal vinyl signal and are estimated to be accurate within 2%. The E and Z protons were differentiated by analyzing the magnitude of the vicinal vinyl H–H coupling constant JHH (E: JHH = 17.2 Hz; Z: JHH = 10.6 Hz).
- 21.Different batches of 21 provided different enantiomeric excesses. The enhancement of all batches was sufficient to determine the stereospecificity of the reactions depicted in Figure 2. The same level of stereospecificity was observed when different enantiomeric mixtures were exposed to the reaction conditions.
- 22.(a) Bäckvall JE. Acc Chem Res. 1983;16:335–342. [Google Scholar]; (b) Francis JW, Henry PM. Organometallics. 1991;10:3498–3503. [Google Scholar]; (c) Francis JW, Henry PM. Organometallics. 1992;11:2832–2836. [Google Scholar]; (d) Zaw K, Henry PM. Organometallics. 1992;11:2008–2015. [Google Scholar]; (e) Hamed O, Henry PM. Organometallics. 1997;16:4903–4909. [Google Scholar]; (f) Hamed O, Thompson C, Henry PM. J Org Chem. 1997;62:7082–7083. doi: 10.1021/jo971051q. [DOI] [PubMed] [Google Scholar]; (g) Hamed O, Henry PM, Thompson C. J Org Chem. 1999;64:7745–7750. [Google Scholar]; (h) ten Brink G-J, Arends IWCW, Papadogianakis G, Sheldon RA. Appl Catal, A. 2000;194–195:435–442. [Google Scholar]; (i) Trend RM, Ramtohul YK, Stoltz BM. J Am Chem Soc. 2005;127:17778–17788. doi: 10.1021/ja055534k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) TURBOMOLE V62. Turbomole GmbH; Karlsruhe, Germany: 2009. http://www.turbomole.com. [Google Scholar]; (b) Treutler O, Ahlrichs R. J Chem Phys. 1995;102:346–354. [Google Scholar]
- 24.(a) Slater JC. The Self-Consistent Field for Molecules and Solids. Vol. 4. McGraw-Hill; New York: 1974. Quantum Theory of Molecules and Solids. [Google Scholar]; (b) Vosko SH, Wilk L, Nusair M. Can J Phys. 1980;58:1200–1211. [Google Scholar]; (c) Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (d) Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (e) Miehlich B, Savin A, Stoll H, Preuss H. Chem Phys Lett. 1989;157:200–206. [Google Scholar]; (f) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (g) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 25.(a) Eichkorn K, Weigend F, Treutler O, Ahlrichs R. Theor Chem Acc. 1997;97:119–124. [Google Scholar]; (b) Schäfer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100:5829–5835. [Google Scholar]; (c) Weigend F, Furche F, Ahlrichs R. J Chem Phys. 2003;119:12753–12762. [Google Scholar]
- 26.Klamt A, Schüürmann G. J Chem Soc, Perkin Trans. 1993;2:799–805. [Google Scholar]
- 27.Because the COSMO solvent model was used, analytical second derivatives were unavailable; as a result, numerical differentiation was performed (TURBOMOLE program NumForce) using a step size of 0.02 bohr on 3N coordinates.
- 28.Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735–746. [Google Scholar]
- 29.See the Supporting Information for details.
- 30.Kirsch SF, Overman LE, Watson MP. J Org Chem. 2004;69:8101–8104. doi: 10.1021/jo0487092.(b) The four Pd–C distances in cyclooctadienylpalladium(II) chloride were determined to be 2.200, 2.209, 2.211, and 2.259 Å by X-ray crystallography. These compare favorably to the Pd–C distances found in our computational studies. See: Rettig MF, Wing RM, Wiger GR. J Am Chem Soc. 1981;103:2980–2986.
- 31.All attempts to locate a concerted transition state converged on the transition structure 49 (Figure 7).
- 32.Hartley FR. Chem Soc Rev. 1973;2:163–179. [Google Scholar]
- 33.It was anticipated that for the purposes of these studies, this small energy difference would not effect the qualitative conclusions drawn from the computational results. Preliminary investigation of the small conformational preference of the alkene ligand using the full COP framework supported these findings.
- 34.(a) To estimate the pKa of the conjugate acid of trichloroacetimidates 1, mixtures of methyl trichloroacetimidate and various acids were analyzed by NMR. The pKa of 1 is estimated to be approximately 1.1 based on these studies. See the Supporting Information for further details. (b) Comparison of trichloroacetimidates to other imidates for which the pKa of their conjugate acids are known suggests that trichloroacetimidate salts should have a pKa in the range of 0–2: Granik VG, Pyatin BM, Persianova JV, Peresleni EM, Kostyuchenko NP, Glushkov RG, Sheinker YN. Tetrahedron. 1970;26:4367–4373.
- 35.Although ΔG and ΔS can be calculated for complexes 33, 39, and 42–54,23 there are so many assumptions involved in calculating the relative energies of structures of this complexity that we prefer to discuss ΔE. The analysis would be unchanged if entropy was included: see table of relative ΔS values in the Supporting Information. Additionally, the COSMO solvent model accounts for the entropy of solvation.
- 36.Zero-point energy corrections to obtain ΔG and ΔH values (298.15 K, 1 atm) were calculated using the program FreeH included in the TURBOMOLE V6.2 software package.
- 37.For ease of calculation, the carbon and hydrogen atoms of the four phenyl rings in complexes 55–58 were simplified using the single-ζ SZ.benzene basis set optimized for aromatics included with the TURBOMOLE V6.2 package. All other atoms were calculated with the def2-TZVP basis set.
- 38.An alternate view of structures 55–58 can be found in the Supporting Information.
- 39.The Bergman group has reported similar secondary kinetic isotope effects for a zirconium-promoted concerted syn-SN2′ substitution reaction of allylic chlorides, see: Fox RJ, Lalic G, Bergman RG. J Am Chem Soc. 2007;129:14144–14145. doi: 10.1021/ja075967i.
- 40.Small inverse deuterium equilibrium isotope effects would be expected at C2 and C3 for alkene complexation,41 and at C2 and C3 for reversible oxypalladation.42
- 41.Schröder D, Wesendrup R, Hertwig RH, Dargel TK, Grauel H, Koch W, Bender BR, Schwarz H. Organometallics. 2000;19:2608–2615. [Google Scholar]
- 42.To our knowledge, deuterium equilibrium isotope effects have not been reported for oxymetallation of alkenes. We assume that they would be similar, at least in direction, to those for addition of bromine to an alkene: Koerner T, Brown RS, Gainsforth JL, Klobukowski M. J Am Chem Soc. 1998;120:5628–5636.
- 43.The importance of interactions with the planar chiral tetraphenylcyclobutadiene moiety of the COP catalyst structure in dictating enantioselection is also found in allylic imidate rearrangements catalyzed by [COP-X]2 catalysts: see reference 7 and Prasad RS, Anderson CE, Richards CJ, Overman LE. Organometallics. 2005;24:77–81.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.