

Abstract
The molecular structure of the title compound, C6H5BF2O2, is essentially planar (mean deviation = 0.019 Å), indicating electronic delocalization between the dihydroxyboryl group and the aromatic ring. In the crystal structure, inversion dimers linked by two O—H⋯O hydrogen bonds arise. An intramolecular O—H⋯F hydrogen bond reinforces the conformation and the same H atom is also involved in an intermolecular O—H⋯F link, leading to molecular sheets in the crystal.
Related literature
For general backround to boronic acids, see: Hall (2005 ▶); Höpfl (2002 ▶); Fujita et al. (2008 ▶); Soloway et al. (1998 ▶). For hydrogen-bond motifs, see: Bernstein et al. (1995 ▶); Desiraju (2002 ▶). For related structures, see: Wu et al. (2006 ▶); Bradley et al. (1996 ▶); Horton et al. (2004 ▶). For crystal engineering, see: Fournier et al. (2003 ▶); Rodríguez-Cuamatzi et al. (2004 ▶, 2005 ▶).
Experimental
Crystal data
C6H5BF2O2
M r = 157.91
Monoclinic,
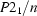
a = 3.7617 (11) Å
b = 12.347 (4) Å
c = 14.620 (4) Å
β = 95.450 (5)°
V = 676.0 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.15 mm−1
T = 293 (2) K
0.37 × 0.35 × 0.22 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.947, T max = 0.968
3196 measured reflections
1190 independent reflections
1012 reflections with I > 2σ(I)
R int = 0.028
Refinement
R[F 2 > 2σ(F 2)] = 0.056
wR(F 2) = 0.127
S = 1.15
1190 reflections
106 parameters
2 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.14 e Å−3
Δρmin = −0.18 e Å−3
Data collection: SMART (Bruker, 2000 ▶); cell refinement: SAINT-Plus-NT (Bruker, 2001 ▶); data reduction: SAINT-Plus-NT; program(s) used to solve structure: SHELXTL-NT (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL-NT; molecular graphics: CAMERON (Watkin et al., 1996 ▶); software used to prepare material for publication: PLATON (Spek, 2003 ▶) and publCIF (Westrip, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808040646/hb2865sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808040646/hb2865Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯F1 | 0.841 (15) | 2.16 (3) | 2.799 (3) | 133 (2) |
| O1—H1⋯F2i | 0.841 (15) | 2.39 (2) | 3.086 (3) | 140 (3) |
| O2—H2⋯O1ii | 0.841 (19) | 1.97 (2) | 2.809 (3) | 174 (3) |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Acknowledgments
This work was supported by Consejo Nacional de Ciencia y Tecnología (CIAM-59213 for HH).
supplementary crystallographic information
Comment
Boronic acids, RB(OH)2 with R = alkyl and aryl, have applications in organic synthesis (Hall, 2005), host–guest chemistry (Höpfl, 2002), the molecular recognition of biochemically active molecules (Fujita et al., 2008) and in medicine as antibiotics, inhibitors and for the treatment of tumors (Soloway et al., 1998). Similar to carboxylic acids they are capable to form hydrogen-bonded dimeric units and, therefore, boronic acids have been used recently as new building blocks in crystal engineering (Fournier et al., 2003; Rodríguez-Cuamatzi et al., 2004; Rodríguez-Cuamatzi et al., 2005). Previously, the structures of 3-fluorophenylboronic acid (Wu et al., 2006), 2,6-difluoroboronic acid (Bradley et al., 1996) and pentafluoroboronic acid (Horton et al., 2004) had been reported. We now present the crystal structure of (I).
The molecular structure is essentially planar, O1—B1—C1—C2 = 4.4 (4)°, indicating that there is a π···π interaction between the dihydroxyboryl group and the aromatic ring, to which it is attached (Fig. 1). This interaction is also evidenced by the B—C bond length of 1.566 (3) Å, which is significantly shorter than that observed in boronates containing tetra-coordinate boron atoms (Höpfl, 2002). The crystal structure is stabilized by strong O2—H2···O1 hydrogen-bonding interactions, forming R22(8) motifs (Bernstein et al., 1995), as well as, O1—H1···F1 and O1—H1···F2 bifurcated hydrogen bonds (Fig. 2; Table 1) (Desiraju, 2002). Due to these interactions each boronic acid homodimer is linked to two neighboring homodimeric units, thus creating a two-dimensional hydrogen-bonded network, in which fluorine is therefore an essential structural component.
Experimental
2,4-Difluorophenylboronic acid was purchased from Aldrich and crystallized from water to yield colourless blocks of (I).
Refinement
The aromatic H atoms were positioned geometrically (C—H = 0.93Å) and refined as riding with Uiso(H) = 1.2Ueq(C). The O—H hydrogen atoms were localized in a difference map and their coordinates were refined with O—H = 0.84+/0.01Å and Uiso(H) = 1.5 Ueq(O).
Figures
Fig. 1.

The molecular structure of (I) with displacement ellipsoids drawn at the 30% probability level and H atoms shown as small spheres of arbitrary radius.
Fig. 2.

View of the packing arrangement of the two-dimensional network of (I)(I).
Crystal data
| C6H5BF2O2 | F(000) = 320 |
| Mr = 157.91 | Dx = 1.552 Mg m−3 |
| Monoclinic, P21/n | Melting point = 521–522 K |
| Hall symbol: -P 2yn | Mo Kα radiation, λ = 0.71073 Å |
| a = 3.7617 (11) Å | Cell parameters from 1052 reflections |
| b = 12.347 (4) Å | θ = 2.3–26.2° |
| c = 14.620 (4) Å | µ = 0.15 mm−1 |
| β = 95.450 (5)° | T = 293 K |
| V = 676.0 (3) Å3 | Block, colorless |
| Z = 4 | 0.37 × 0.35 × 0.22 mm |
Data collection
| Bruker SMART APEX CCD area-detector diffractometer | 1190 independent reflections |
| Radiation source: fine-focus sealed tube | 1012 reflections with I > 2σ(I) |
| graphite | Rint = 0.028 |
| Detector resolution: 8.3 pixels mm-1 | θmax = 25.0°, θmin = 2.2° |
| φ and ω scans | h = −3→4 |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | k = −14→12 |
| Tmin = 0.947, Tmax = 0.968 | l = −17→17 |
| 3196 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.056 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.127 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.15 | w = 1/[σ2(Fo2) + (0.0442P)2 + 0.2673P] where P = (Fo2 + 2Fc2)/3 |
| 1190 reflections | (Δ/σ)max < 0.001 |
| 106 parameters | Δρmax = 0.14 e Å−3 |
| 2 restraints | Δρmin = −0.18 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| B1 | 0.7704 (8) | 0.4548 (2) | 0.62567 (18) | 0.0452 (7) | |
| O1 | 0.6825 (6) | 0.38623 (15) | 0.55419 (13) | 0.0695 (6) | |
| H1 | 0.748 (9) | 0.3215 (9) | 0.562 (2) | 0.104* | |
| O2 | 0.6880 (6) | 0.55977 (14) | 0.61557 (12) | 0.0630 (6) | |
| H2 | 0.593 (8) | 0.577 (3) | 0.5632 (10) | 0.094* | |
| F1 | 1.0385 (5) | 0.23728 (11) | 0.67473 (11) | 0.0768 (6) | |
| F2 | 1.4329 (5) | 0.33016 (14) | 0.97634 (10) | 0.0822 (6) | |
| C1 | 0.9591 (6) | 0.41789 (18) | 0.72069 (15) | 0.0424 (6) | |
| C2 | 1.0796 (7) | 0.31430 (18) | 0.74175 (16) | 0.0470 (6) | |
| C3 | 1.2380 (7) | 0.2819 (2) | 0.82553 (17) | 0.0539 (7) | |
| H3 | 1.3138 | 0.2109 | 0.8363 | 0.065* | |
| C4 | 1.2785 (7) | 0.3593 (2) | 0.89223 (17) | 0.0547 (7) | |
| C5 | 1.1696 (8) | 0.4640 (2) | 0.87828 (17) | 0.0586 (7) | |
| H5 | 1.2013 | 0.5150 | 0.9251 | 0.070* | |
| C6 | 1.0119 (7) | 0.49169 (19) | 0.79282 (16) | 0.0498 (6) | |
| H6 | 0.9371 | 0.5628 | 0.7827 | 0.060* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| B1 | 0.0451 (16) | 0.0425 (15) | 0.0471 (16) | −0.0009 (12) | −0.0005 (12) | 0.0033 (12) |
| O1 | 0.1000 (17) | 0.0484 (11) | 0.0540 (11) | 0.0158 (10) | −0.0241 (10) | −0.0009 (9) |
| O2 | 0.0850 (15) | 0.0441 (10) | 0.0552 (11) | 0.0087 (9) | −0.0172 (10) | 0.0048 (8) |
| F1 | 0.1192 (15) | 0.0456 (9) | 0.0602 (10) | 0.0151 (9) | −0.0187 (9) | −0.0046 (7) |
| F2 | 0.1070 (14) | 0.0804 (12) | 0.0527 (10) | −0.0120 (10) | −0.0262 (9) | 0.0181 (8) |
| C1 | 0.0383 (13) | 0.0412 (13) | 0.0473 (13) | −0.0039 (10) | 0.0019 (10) | 0.0048 (10) |
| C2 | 0.0523 (16) | 0.0410 (13) | 0.0467 (13) | −0.0017 (11) | 0.0001 (11) | 0.0008 (10) |
| C3 | 0.0584 (17) | 0.0449 (14) | 0.0564 (15) | 0.0002 (12) | −0.0053 (13) | 0.0124 (12) |
| C4 | 0.0579 (17) | 0.0613 (17) | 0.0426 (13) | −0.0099 (13) | −0.0075 (12) | 0.0135 (12) |
| C5 | 0.0696 (19) | 0.0552 (16) | 0.0489 (14) | −0.0109 (13) | −0.0058 (13) | −0.0020 (12) |
| C6 | 0.0559 (16) | 0.0399 (13) | 0.0522 (14) | −0.0011 (11) | −0.0011 (12) | 0.0029 (11) |
Geometric parameters (Å, °)
| B1—O2 | 1.338 (3) | C1—C6 | 1.394 (3) |
| B1—O1 | 1.361 (3) | C2—C3 | 1.370 (3) |
| B1—C1 | 1.566 (3) | C3—C4 | 1.363 (4) |
| O1—H1 | 0.841 (15) | C3—H3 | 0.93 |
| O2—H2 | 0.841 (15) | C4—C5 | 1.366 (4) |
| F1—C2 | 1.364 (3) | C5—C6 | 1.374 (3) |
| F2—C4 | 1.358 (3) | C5—H5 | 0.93 |
| C1—C2 | 1.382 (3) | C6—H6 | 0.93 |
| O2—B1—O1 | 118.7 (2) | C4—C3—H3 | 121.8 |
| O2—B1—C1 | 117.4 (2) | C2—C3—H3 | 121.8 |
| O1—B1—C1 | 123.8 (2) | F2—C4—C3 | 118.1 (2) |
| B1—O1—H1 | 116 (2) | F2—C4—C5 | 118.8 (2) |
| B1—O2—H2 | 115 (2) | C3—C4—C5 | 123.0 (2) |
| C2—C1—C6 | 114.6 (2) | C4—C5—C6 | 117.9 (2) |
| C2—C1—B1 | 125.3 (2) | C4—C5—H5 | 121.0 |
| C6—C1—B1 | 120.1 (2) | C6—C5—H5 | 121.0 |
| F1—C2—C3 | 116.7 (2) | C5—C6—C1 | 122.9 (2) |
| F1—C2—C1 | 118.2 (2) | C5—C6—H6 | 118.5 |
| C3—C2—C1 | 125.1 (2) | C1—C6—H6 | 118.5 |
| C4—C3—C2 | 116.4 (2) | ||
| O2—B1—C1—C2 | −176.5 (2) | C1—C2—C3—C4 | −0.3 (4) |
| O1—B1—C1—C2 | 4.5 (4) | C2—C3—C4—F2 | 179.7 (2) |
| O2—B1—C1—C6 | 4.6 (4) | C2—C3—C4—C5 | 0.0 (4) |
| O1—B1—C1—C6 | −174.5 (2) | F2—C4—C5—C6 | −179.6 (2) |
| C6—C1—C2—F1 | −179.9 (2) | C3—C4—C5—C6 | 0.1 (4) |
| B1—C1—C2—F1 | 1.1 (4) | C4—C5—C6—C1 | 0.0 (4) |
| C6—C1—C2—C3 | 0.4 (4) | C2—C1—C6—C5 | −0.3 (4) |
| B1—C1—C2—C3 | −178.6 (2) | B1—C1—C6—C5 | 178.8 (2) |
| F1—C2—C3—C4 | 180.0 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···F1 | 0.84 (2) | 2.16 (3) | 2.799 (3) | 133 (2) |
| O1—H1···F2i | 0.84 (2) | 2.39 (2) | 3.086 (3) | 140 (3) |
| O2—H2···O1ii | 0.84 (2) | 1.97 (2) | 2.809 (3) | 174 (3) |
Symmetry codes: (i) x−1/2, −y+1/2, z−1/2; (ii) −x+1, −y+1, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB2865).
References
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Bradley, D. C., Harding, I. S., Keefe, A. D., Motevalli, M. & Zheng, D. H. (1996). J. Chem. Soc. Dalton Trans. pp. 3931–3936.
- Bruker (2000). SMART. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2001). SAINT-Plus NT Bruker AXS Inc., Madison, Wisconsin, USA.
- Desiraju, G. R. (2002). Acc. Chem. Res.35, 565–573. [DOI] [PubMed]
- Fournier, J.-H., Maris, T., Wuest, J. D., Guo, W. & Galoppini, E. (2003). J. Am. Chem. Soc.125, 1002–1006. [DOI] [PubMed]
- Fujita, N., Shinkai, S. & James, T. D. (2008). Chem. Asian J.3, 1076–1091. [DOI] [PubMed]
- Hall, D. G. (2005). Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine Weinheim: Wiley-VCH.
- Höpfl, H. (2002). Structure and Bonding, edited by H. W. Roesky & D. A. Atwood, Vol. 103, pp. 1-56. Berlin: Springer Verlag.
- Horton, P. N., Hursthouse, M. B., Beckett, M. A. & Rugen-Hankey, M. P. (2004). Acta Cryst. E60, o2204–o2206.
- Rodríguez-Cuamatzi, P., Arillo-Flores, O. I., Bernal-Uruchurtu, M. I. & Höpfl, H. (2005). Cryst. Growth Des.5, 167–175.
- Rodríguez-Cuamatzi, P., Vargas-Díaz, G. & Höpfl, H. (2004). Angew. Chem. Int. Ed.43, 3041–3044. [DOI] [PubMed]
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Soloway, A. H., Tjarks, W., Barnum, B. A., Rong, R.-A., Barth, R. F., Codogni, I. M. & Wilson, J. G. (1998). Chem. Rev.98, 1515–1562. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Watkin, D. J., Prout, C. K. & Pearce, L. J. (1996). CAMERON Chemical Crystallography Laboratory Oxford, Oxford, England.
- Westrip, S. P. (2009). publCIF In preparation.
- Wu, Y.-M., Dong, C.-C., Liu, S., Zhu, H.-J. & Wu, Y.-Z. (2006). Acta Cryst. E62, o4236–o4237.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808040646/hb2865sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808040646/hb2865Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report