

Abstract
In the title compound, C7H7N5, the non-H atoms are almost coplanar (r.m.s. deviation = 0.050 Å), with the N atom of pyridine ring oriented to the N—N(H) side of the 1,2,4-triazole ring. The mean planes of the pyridine and 1,2,4-triazole rings form a dihedral angle of 5.58 (7)°. The N atom of the amino group adopts a pyramidal configuration. The molecules are linked into a two-dimensional network parallel to (10 ) by N—H⋯N hydrogen bonds.
) by N—H⋯N hydrogen bonds.
Related literature
For 1,2,4-triazol-5-amines as building blocks in the synthesis of fused heterocyclic systems, see: Dolzhenko et al. (2006 ▶, 2007a
▶,b
▶); Fischer, (2007 ▶). For a summary of structural data for 1,2,4-triazoles, see: Buzykin et al. (2006 ▶). For crystal structures of CuII complexes with 3-pyridin-2-yl-1,2,4-triazol-5-amine, see: Ferrer et al. (2004 ▶).
Experimental
Crystal data
C7H7N5
M r = 161.18
Monoclinic,
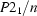
a = 7.3863 (6) Å
b = 7.9096 (6) Å
c = 13.2157 (11) Å
β = 91.832 (2)°
V = 771.70 (11) Å3
Z = 4
Mo Kα radiation
μ = 0.10 mm−1
T = 223 (2) K
0.36 × 0.16 × 0.12 mm
Data collection
Bruker SMART APEX CCD diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 2001 ▶) T min = 0.967, T max = 0.989
5336 measured reflections
1772 independent reflections
1519 reflections with I > 2σ(I)
R int = 0.026
Refinement
R[F 2 > 2σ(F 2)] = 0.042
wR(F 2) = 0.110
S = 1.05
1772 reflections
121 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.21 e Å−3
Δρmin = −0.20 e Å−3
Data collection: SMART (Bruker, 2001 ▶); cell refinement: SAINT (Bruker, 2001 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808042177/ci2719sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808042177/ci2719Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H2N⋯N5i | 0.90 (2) | 2.01 (2) | 2.9010 (16) | 171 (1) |
| N4—H4A⋯N3ii | 0.90 (2) | 2.11 (2) | 2.9971 (16) | 172 (1) |
| N4—H4B⋯N1i | 0.93 (2) | 2.19 (2) | 3.0264 (16) | 151 (1) |
Symmetry codes: (i)  ; (ii)
; (ii) 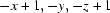 .
.
Acknowledgments
This work was supported by the National Medical Research Council, Singapore (grant Nos. NMRC/NIG/0019/2008 and NMRC/NIG/0020/2008).
supplementary crystallographic information
Comment
1,2,4-Triazol-5-amines have been recognized as valuable synthons for the construction of fused heterocyclic systems, e.g. 1,2,4-triazolo[1,5-a]pyrimidines (Fischer, 2007) and 1,2,4-triazolo[1,5-a][1,3,5]triazines (Dolzhenko et al., 2006). It also should be mentioned that 1,2,4-triazol-5-amines are widely used as ligands and crystallographic data on three different mononuclear complexes of 3-pyridin-2-yl-1,2,4-triazol-5-amine with CuII have been reported by Ferrer et al. (2004). However, no crystallographic study has been performed on the ligand.
In continuation of our investigations on using 1,2,4-triazol-5-amines in the synthesis of fused heterocyclic systems (Dolzhenko et al., 2007a,b), we report herein the crystal structure of a synthetically important building block viz. 3-pyridin-2-yl-1,2,4-triazol-5-amine.
Due to annular tautomerism, 3-pyridin-2-yl-1,2,4-triazol-5-amine may theoretically exist in three tautomeric forms (A, B and C) and for each of them, rotameric structures A', B' and C' are possible (Fig.1). As observed in reported CuII complexes (Ferrer et al., 2004), 3-pyridin-2-yl-1,2,4-triazol-5-amine was the only tautomeric form found in the crystal (Fig. 2). However, the molecule exists in the crystal as rotamer A in contrast to rotamer A' found in CuII complexes.
Bond lengths and angles in the molecule of 3-pyridin-2-yl-1,2,4-triazol-5-amine are within normal ranges, and comparable with values summarized for 1,2,4-triazoles by Buzykin et al. (2006). 3-Pyridin-2-yl-1,2,4-triazol-5-amine has practically planar geometry with slight deviation of the pyridyl moiety, which makes a dihedral angle of 5.58 (7)° with mean plane of the 1,2,4-triazole ring. The nitrogen atom (N4) of the amino group adopts a pyramidal configuration with 0.26 (2) Å deviation of the nitrogen atom from the C2/H4A/H4B plane.
The molecules are linked into a two-dimensional network parallel to the (101) by N—H···N hydrogen bonds (Table 1 and Fig.3).
Experimental
3-Pyridin-2-yl-1,2,4-triazol-5-amine was prepared according to general method reported by Dolzhenko et al. (2007a,b). Single crystals suitable for crystallographic analysis were grown by recrystallization from ethanol.
Refinement
N-bound H-atoms were located in a difference map and refined freely [N—H = 0.90 (2)–0.92 (2) Å]. C-bound H atoms were positioned geometrically (C—H = 0.94 Å) and were constrained in a riding motion approximation with Uiso(H) = 1.2Ueq(C).
Figures
Fig. 1.

Possible tautomers and rotamers of 3-pyridin-2-yl-1,2,4-triazol-5-amine.
Fig. 2.

The molecular structure of the title compound, with the atomic numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 3.

Molecular packing of the title compound, viewed along the c axis.
Crystal data
| C7H7N5 | F(000) = 336 |
| Mr = 161.18 | Dx = 1.387 Mg m−3 |
| Monoclinic, P21/n | Melting point: 493 K |
| Hall symbol: -P 2yn | Mo Kα radiation, λ = 0.71073 Å |
| a = 7.3863 (6) Å | Cell parameters from 1841 reflections |
| b = 7.9096 (6) Å | θ = 3.0–26.6° |
| c = 13.2157 (11) Å | µ = 0.10 mm−1 |
| β = 91.832 (2)° | T = 223 K |
| V = 771.70 (11) Å3 | Block, colourless |
| Z = 4 | 0.36 × 0.16 × 0.12 mm |
Data collection
| Bruker SMART APEX CCD diffractometer | 1772 independent reflections |
| Radiation source: fine-focus sealed tube | 1519 reflections with I > 2σ(I) |
| graphite | Rint = 0.026 |
| φ and ω scans | θmax = 27.5°, θmin = 3.0° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 2001) | h = −9→9 |
| Tmin = 0.967, Tmax = 0.989 | k = −8→10 |
| 5336 measured reflections | l = −14→17 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.110 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0539P)2 + 0.1487P] where P = (Fo2 + 2Fc2)/3 |
| 1772 reflections | (Δ/σ)max = 0.001 |
| 121 parameters | Δρmax = 0.21 e Å−3 |
| 0 restraints | Δρmin = −0.20 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.72741 (16) | 0.34999 (14) | 0.68693 (8) | 0.0383 (3) | |
| N2 | 0.66107 (16) | 0.19639 (15) | 0.71630 (8) | 0.0393 (3) | |
| H2N | 0.669 (2) | 0.164 (2) | 0.7817 (14) | 0.052 (5)* | |
| N3 | 0.61324 (14) | 0.20352 (14) | 0.55193 (8) | 0.0345 (3) | |
| N4 | 0.51489 (16) | −0.04181 (15) | 0.64256 (9) | 0.0398 (3) | |
| H4A | 0.488 (2) | −0.095 (2) | 0.5842 (13) | 0.049 (4)* | |
| H4B | 0.564 (2) | −0.106 (2) | 0.6948 (13) | 0.050 (4)* | |
| N5 | 0.80901 (15) | 0.62905 (15) | 0.56803 (8) | 0.0388 (3) | |
| C1 | 0.69411 (16) | 0.34759 (16) | 0.58805 (9) | 0.0331 (3) | |
| C2 | 0.59494 (16) | 0.11150 (17) | 0.63532 (9) | 0.0343 (3) | |
| C3 | 0.74353 (16) | 0.48955 (17) | 0.52229 (9) | 0.0343 (3) | |
| C4 | 0.72299 (19) | 0.47748 (19) | 0.41755 (10) | 0.0425 (3) | |
| H4 | 0.6758 | 0.3786 | 0.3873 | 0.051* | |
| C5 | 0.77288 (19) | 0.6128 (2) | 0.35870 (11) | 0.0491 (4) | |
| H5 | 0.7598 | 0.6076 | 0.2878 | 0.059* | |
| C6 | 0.8419 (2) | 0.7554 (2) | 0.40516 (12) | 0.0486 (4) | |
| H6 | 0.8781 | 0.8489 | 0.3668 | 0.058* | |
| C7 | 0.85663 (19) | 0.75796 (19) | 0.50930 (12) | 0.0455 (4) | |
| H7 | 0.9029 | 0.8561 | 0.5408 | 0.055* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0476 (6) | 0.0387 (6) | 0.0282 (5) | −0.0007 (5) | −0.0076 (4) | 0.0006 (4) |
| N2 | 0.0518 (7) | 0.0392 (6) | 0.0261 (6) | −0.0013 (5) | −0.0092 (5) | 0.0000 (5) |
| N3 | 0.0362 (5) | 0.0396 (6) | 0.0273 (5) | 0.0042 (4) | −0.0059 (4) | −0.0028 (4) |
| N4 | 0.0498 (7) | 0.0397 (6) | 0.0291 (6) | −0.0021 (5) | −0.0110 (5) | −0.0001 (5) |
| N5 | 0.0420 (6) | 0.0409 (6) | 0.0332 (6) | 0.0030 (5) | −0.0043 (5) | 0.0012 (5) |
| C1 | 0.0330 (6) | 0.0385 (7) | 0.0273 (6) | 0.0057 (5) | −0.0056 (5) | −0.0035 (5) |
| C2 | 0.0357 (6) | 0.0391 (7) | 0.0274 (6) | 0.0053 (5) | −0.0068 (5) | −0.0030 (5) |
| C3 | 0.0305 (6) | 0.0419 (7) | 0.0301 (6) | 0.0076 (5) | −0.0028 (5) | −0.0010 (5) |
| C4 | 0.0442 (7) | 0.0522 (8) | 0.0310 (7) | 0.0046 (6) | −0.0016 (5) | −0.0025 (6) |
| C5 | 0.0488 (8) | 0.0683 (10) | 0.0304 (7) | 0.0080 (7) | 0.0032 (6) | 0.0071 (7) |
| C6 | 0.0444 (8) | 0.0558 (9) | 0.0457 (8) | 0.0057 (7) | 0.0046 (6) | 0.0150 (7) |
| C7 | 0.0454 (8) | 0.0447 (8) | 0.0460 (8) | 0.0013 (6) | −0.0035 (6) | 0.0050 (6) |
Geometric parameters (Å, °)
| N1—C1 | 1.3221 (16) | N5—C3 | 1.3410 (17) |
| N1—N2 | 1.3708 (16) | C1—C3 | 1.4729 (18) |
| N2—C2 | 1.3422 (16) | C3—C4 | 1.3909 (18) |
| N2—H2N | 0.902 (19) | C4—C5 | 1.380 (2) |
| N3—C2 | 1.3312 (17) | C4—H4 | 0.94 |
| N3—C1 | 1.3656 (16) | C5—C6 | 1.374 (2) |
| N4—C2 | 1.3538 (18) | C5—H5 | 0.94 |
| N4—H4A | 0.896 (18) | C6—C7 | 1.377 (2) |
| N4—H4B | 0.925 (17) | C6—H6 | 0.94 |
| N5—C7 | 1.3354 (18) | C7—H7 | 0.94 |
| C1—N1—N2 | 102.14 (10) | N5—C3—C4 | 122.10 (13) |
| C2—N2—N1 | 109.99 (11) | N5—C3—C1 | 116.99 (11) |
| C2—N2—H2N | 129.2 (11) | C4—C3—C1 | 120.91 (12) |
| N1—N2—H2N | 120.8 (11) | C5—C4—C3 | 119.01 (14) |
| C2—N3—C1 | 102.81 (10) | C5—C4—H4 | 120.5 |
| C2—N4—H4A | 116.4 (10) | C3—C4—H4 | 120.5 |
| C2—N4—H4B | 112.7 (10) | C6—C5—C4 | 119.12 (14) |
| H4A—N4—H4B | 117.2 (14) | C6—C5—H5 | 120.4 |
| C7—N5—C3 | 117.65 (12) | C4—C5—H5 | 120.4 |
| N1—C1—N3 | 115.02 (12) | C5—C6—C7 | 118.36 (14) |
| N1—C1—C3 | 122.04 (12) | C5—C6—H6 | 120.8 |
| N3—C1—C3 | 122.94 (11) | C7—C6—H6 | 120.8 |
| N3—C2—N2 | 110.03 (12) | N5—C7—C6 | 123.76 (15) |
| N3—C2—N4 | 127.19 (11) | N5—C7—H7 | 118.1 |
| N2—C2—N4 | 122.71 (12) | C6—C7—H7 | 118.1 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2N···N5i | 0.90 (2) | 2.01 (2) | 2.9010 (16) | 171 (1) |
| N4—H4A···N3ii | 0.90 (2) | 2.11 (2) | 2.9971 (16) | 172 (1) |
| N4—H4B···N1i | 0.93 (2) | 2.19 (2) | 3.0264 (16) | 151 (1) |
Symmetry codes: (i) −x+3/2, y−1/2, −z+3/2; (ii) −x+1, −y, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: CI2719).
References
- Bruker (2001). SMART and SAINT Bruker AXS GmbH, Karlsruhe, Germany.
- Buzykin, B. I., Mironova, E. V., Nabiullin, V. N., Gubaidullin, A. T. & Litvinov, I. A. (2006). Russ. J. Gen. Chem.76, 1471–1486.
- Dolzhenko, A. V., Dolzhenko, A. V. & Chui, W. K. (2006). Heterocycles, 68, 1723–1759.
- Dolzhenko, A. V., Dolzhenko, A. V. & Chui, W. K. (2007a). Heterocycles, 71, 429–436.
- Dolzhenko, A. V., Dolzhenko, A. V. & Chui, W. K. (2007b). Tetrahedron, 63, 12888–12895.
- Ferrer, S., Ballesteros, R., Sambartolome, A., Gonzalez, M., Alzuet, G., Borras, J. & Liu, M. (2004). J. Inorg. Biochem.98, 1436–1446. [DOI] [PubMed]
- Fischer, G. (2007). Adv. Heterocycl. Chem.95, 143–219.
- Sheldrick, G. M. (2001). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808042177/ci2719sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808042177/ci2719Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report