


Abstract
The mechanisms that underlie the maintenance of and increase in mutant mitochondrial DNA (mtDNA) are central to our understanding of mitochondrial disease. We have therefore developed a technique based on saponin permeabilisation that allows the study of mtDNA synthesis in intact cells. Permeabilisation of cells has been extensively used in an established method both for studying transcription and DNA replication in the nucleus and for measuring respiratory chain activities in mitochondria. We have quantitatively studied incorporation of radiolabelled DNA precursors into mtDNA in human cell lines derived from controls and from patients with mitochondrial DNA disease. Total cell DNA is extracted, restriction digested and Southern blotted, newly synthesised mtDNA being proportional to the label incorporated in each restriction band. A rate of synthesis can then be derived by estimating the relative steady-state mtDNA after probing with full-length mtDNA. Where co-existing mutant and wild-type mtDNA (heteroplasmy) can be distinguished using restriction digestion, their rates of synthesis can be compared within a single cell line. This will be particularly useful in elucidating the pathophysiology of mtDNA diseases in which the distribution of mutant and wild-type mtDNA in cell lines in patient tissues may evolve with time.
INTRODUCTION
Understanding mammalian mitochondrial DNA (mtDNA) replication is important in studies of mtDNA disease, caused by mutations in either the nuclear or mitochondrial genomes (1). Mutations in two nuclear genes of mitochondrial biogenesis, encoding thymidine phosphorylase (2) and the adenine nucleotide transporter (3), are associated with Mendelian disorders of mtDNA. In patients with maternally inherited mitochondrial diseases pathogenic mtDNA mutations frequently co-exist with wild-type mtDNA (heteroplasmy) (4). Variation in the proportion of mutant mtDNA between tissues is common in these patients and preferential accumulation of mutant mtDNA in affected tissues may account for the progressive and tissue-specific nature of these disorders. Hence, studying synthesis of wild-type and mutant mtDNA, both replication and repair, is central to elucidating the pathogensis of mtDNA disease.
Several methods have been used to study mtDNA replication, which continues throughout the cell cycle (5). These methods include whole cell techniques with [3H]thymidine (6,7) and in organellar techniques (8). To our knowledge, there have been no previous studies investigating incorporation of radionucleotides into mtDNA in permeabilised cells. We have adapted an established technique, which uses permeabilised cells to investigate nuclear DNA replication, to mitochondria. The method (Fig. 1) (henceforth the permeabilised cell assay) has been applied to an analysis of cell lines derived from patients heteroplasmic for the A3243G mutation associated with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS). The rates of incorporation of labelled deoxynucleotides into the A3243G mutant and wild-type mtDNA can be compared in these cell lines.
Figure 1.

The permeabilised cell assay: a schematic diagram showing the final protocol used for quantitating incorporation of [α-32P]dTTP into mtDNA. In outline, mtDNA was cut with a restriction enzyme such as ApaI, labelled with [α-32P]dTTP, detected by Southern blotting and the amount of radiolabel incorporated into each fragment determined (I). The steady-state amount of radiolabel bound to each mtDNA fragment (S) was determined by subsequent hybridisation of the filter with a 32P-labelled mtDNA-specific probe. As the I signal at time 0 was negligible in all experiments, relative rates of incorporation into each mtDNA fragment can be estimated from the ratio I/S, which represents incorporated label relative to steady-state amount of mtDNA for each fragment.
MATERIALS AND METHODS
General principles
Permeabilised cells have successfully been used to study the activity of both nuclear DNA polymerases, where they sustain rates of replication that are >85% of those measured in vivo (9), and RNA polymerases (10). Permeabilisation of the plasma membrane allows controlled manipulation of the cell interior by introduction of exogenous substances such as radionucleotides, which would otherwise not permeate the cell membrane. This enables nuclear but not mitochondrial matrix access to polar probes. Saponin permeabilisation is particularly useful for studying processes such as respiratory chain activities, as it leaves mitochondria and other organelles intact and functional (11). At low concentrations it maintains cytoplasmic integrity, hence minimising artifacts (12), but at high concentrations cells are disrupted. Saponin complexes with membrane cholesterol and forms a hexagonal pore allowing passage of molecules with molecular weights of up to 200 kDa. Plasma membranes that are rich in cholesterol are readily permeabilised by saponin, but mitochondrial membranes are low in cholesterol and are less readily permeabilised. Hence, we used saponin to allow access of labelled nucleotide precursors to the cytosol. Cold shock has also been used to permeabilise mitochondria (13). As the effects of cold shock on cell growth are reversible, we attempted to allow radiolabelled precursors in the cytosol access to mitochondria by cold shock.
Because mtDNA comprises <1% of total cell DNA, we used aphidicolin to inhibit incorporation of radionucleotides into nuclear DNA. Aphidicolin does not inhibit DNA-dependent DNA polymerisation by mitochondrial polymerase γ (14).
Cells and cell culture maintenance
Cells were cultured from both controls and patients with mtDNA disease. Control fibroblast cultures were derived from patients with non-mitochondrial diseases such as glycogen storage disease type 1a (control fib 1). Immortalised lines containing 100% wild-type mtDNA derived from rhabdomyosarcoma, osteosarcoma and lung adenocarcinoma were used (RMS, 143B and A549, respectively). Three heteroplasmic cybrid lines derived from two patients with MELAS due to a heteroplasmic A3243G transition (15) were used: P1 cyb was derived from fibroblasts and the ρ0 line 206 (16) and P2 cyb and P3 cyb from myoblasts from a second patient and the ρ0 line B2 (17), courtesy of Dr I. Holt. They contained ∼33, 81 and 0% A3243G mutant mtDNA, respectively. The mutant mtDNA can readily be distinguished from wild-type by the creation of a new ApaI site at base pair 3243, so that the 2965 bp wild-type band flanked by restriction sites at base pairs 1472 and 4432 is replaced by two bands of 1776 and 1189 bp (see Fig. 2A and B). Cell line P1 cyb has an additional polymorphic ApaI site due to an A→G transition at base pair 10 289, so that the 1 kb ApaI band represents two co-migrating fragments and a 6.2 kb fragment replaces the 7.2 kb fragment. Each of these cybrid lines was clonal. The mtDNA-free (ρ0) line used to exclude involvement of pseudogenes was the 206 line (courtesy of Dr M. King) (18). These cell lines were grown in Dulbecco’s modified Eagle’s medium with 0.1 M pyruvate (Sigma D6546) and 10% foetal calf serum, supplemented with uridine to 50 µg/ml.
Figure 2.
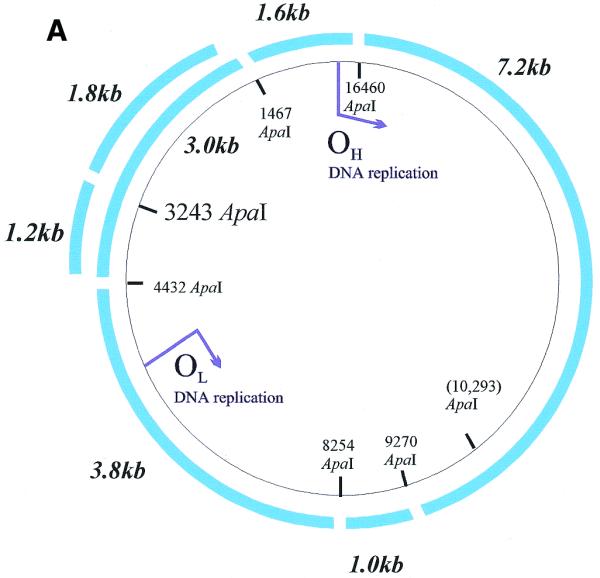
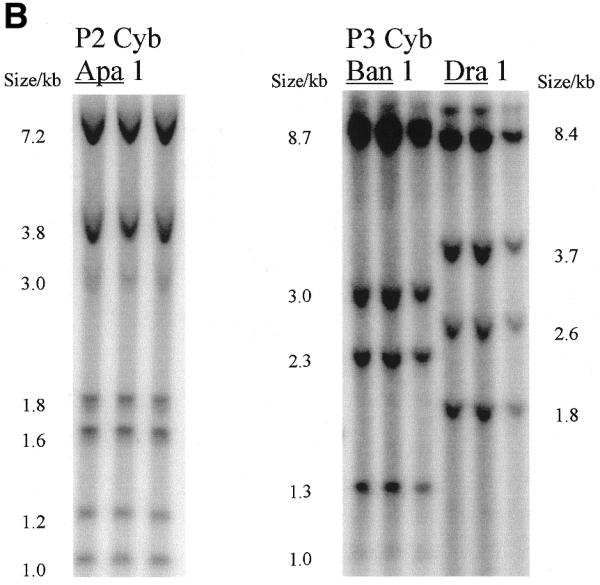
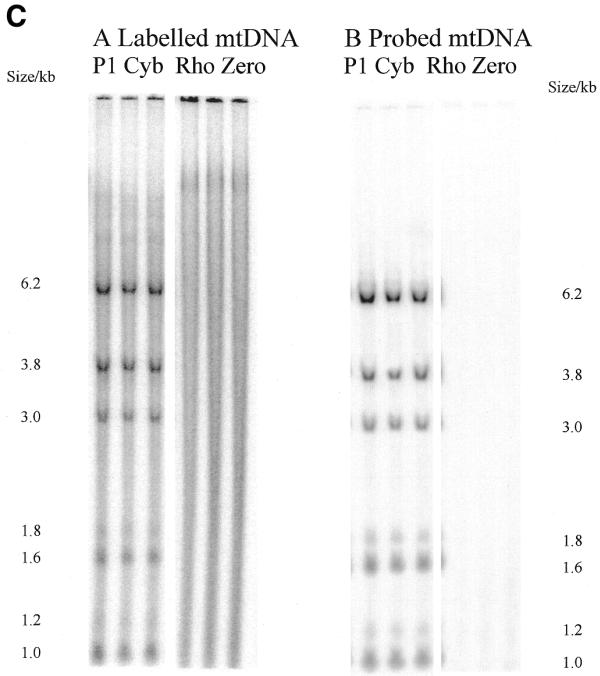
Radioactivity is incorporated into wild-type and A3243G mutant mtDNA and corresponding steady-state mtDNA. (A) Map of human mtDNA to show ApaI restriction sites. ApaI restriction sites and sizes of the fragments generated are indicated. The polymorphic site in line P1 cyb is in brackets. The additional fragments generated when the heteroplasmic 3243 mutation is present are shown outside the wild-type fragments. Origins of DNA replication OH and OL are marked with arrows. (B) Southern blot of labelled mtDNA digested with various restriction enzymes. DNA from patient-derived cell lines P2 cyb and P3 cyb was extracted after pulse labelling in the presence of 5 mg/ml aphidicolin to inhibit nuclear replication. Restriction by ApaI, BanI and DraI all resulted in the predicted restriction fragments for mtDNA. In addition, the A3243G mutation introduces an ApaI restriction site so that the 3 kb band is replaced by bands of 1.2 and 1.8 kb. (C) Comparison of Southern blotted, labelled and probed mtDNA from a patient-derived cybrid and a mtDNA-free cell line. (Left) Phosphorimage of a filter with Southern blotted labelled DNA from a permeabilised cell assay using DNA from patient-derived cell line P1 cyb and a ρ0 line cut with restriction enzyme ApaI. (Right) The same filter probed with a full-length mtDNA probe. Both resulted in the predicted restriction fragments for mtDNA in the patient-derived line which are absent from the ρ0 line. Cell line P1 cyb has an additional ApaI site which maps to base pair 10 293, so that a 6.2 kb band replaces the 7.2 kb band and the 1 kb ApaI band represents two co-migrating fragments.
Incorporation of radiolabel using the permeabilised cell assay
Figure 1 summarises the protocol devised. In outline, the permeabilised cell assay involves transient cooling to 4°C and permeabilising cells with saponin. mtDNA is labelled with [α-32P]dTTP and specifically detected after Southern blotting from the sizes of the radiolabelled restriction fragments. We used aphidicolin to inhibit incorporation of radionucleotides into nuclear DNA.
Cultured cells were harvested with 0.25% porcine trypsin, 0.02% EDTA (Sigma) and then incubated in a 10-fold excess of tissue culture medium at 37°C for 30 min. The cells were resuspended in a physiological buffer (130 mM KCl, 10 mM Na2HPO4, 1 mM MgCl2, 1 mM Na2ATP, 1 mM dithiothreitol, buffered to pH 7.4 with 100 mM KH2PO4) and permeabilised with saponin and cold shock. Aphidicolin and NTPs and dNTPs for incorporation into mtDNA were added to final concentrations of 5 µg/ml, 100 µM and 250 µM, respectively, except for [α-32P]dTTP, which was at 13 µM (specific activity 10 µCi/µl or 3000 Ci/mmol, final concentration 0.04 µCi/µl).
DNA was extracted and digested with a restriction enzyme such as PvuII, which linearises mtDNA by cutting once, or with an enzyme making multiple cuts, like ApaI. The DNA fragments were run on a 1% agarose gel, blotted onto a Genescreen Plus nylon membrane and the radioactivity in each fragment quantitated using a Molecular Dynamics PhosphorImager with a Kodak Phosphorimager screen and ImageQuant software. The filter was then probed with 32P-labelled probe (labelled using Megaprime; Amersham), which was made from a 16 kb mtDNA PCR product including almost the complete mtDNA (19), to quantitate the steady-state level of each mtDNA fragment. The relative incorporation of [α-32P]dTTP into mtDNA at each time point (I) was derived from the radioactivity in mtDNA restriction fragments following incubation with radionucleotide compared with radioactivity of probe bound to mtDNA sequences (S) following hybridisation (representing incorporated and steady-state amounts of mutant and wild-type mtDNA, respectively). Blots were stored for ∼1 month, i.e. two half-lives of 32P, before hybridisation, by which time radioactivity incorporated into DNA was routinely two orders of magnitude less than the hybridisation signal. As the signal obtained at time 0 was negligible, the rate of synthesis of each mtDNA restriction fragment is effectively proportional to I/S (i.e. the radiolabel incorporated into newly labelled mtDNA compared with the activity of the mtDNA probe bound to that band following hybridisation).
In addition to providing an estimate of incorporation of 32P-labelled nucleotide into mtDNA in homoplasmic wild-type lines, this labelling technique can be used to measure the relative rate of incorporation into A3243G mutant and wild-type mtDNA in heteroplasmic cells as, fortuitously, the A3243G mutation introduces a new ApaI site (15). Digestion with ApaI (or its isoschizomer Bsp120I) thus enables a direct comparison of incorporation into fragments derived from either wild-type or mutant mtDNA within the same cybrid cell line.
RESULTS
Evidence that radiolabelled nucleotide is incorporated into mtDNA rather than nuclear pseudogenes
Figure 2B demonstrates that the radiolabelled nucleotide was incorporated into bands of the size predicted from the restriction map of circular mtDNA. In all cell lines used (including control, patient-derived and immortal lines RMS, 143B and A549) bands of appropriate sizes were obtained using seven restriction enzymes, three of which are shown in Figure 2B. The sub-stoichiometric bands in lanes 1–3 arise because the cell line P2 cyb is derived from a patient who is heteroplasmic for the A3243G mutation which introduces an ApaI restriction site: in mutant mtDNA the 3 kb band is replaced by bands of 1.2 and 1.8 kb. All of these labelled restriction fragments co-migrated with the corresponding bands detected by hybridisation with a radiolabelled mtDNA probe, as shown in Figure 2C. As all major bands were of the predicted size, it is unlikely that nuclear pseudogenes make a significant contribution to the signal.
Similar experiments performed with the parent ρ0 206 cell line (18) and the ρ+ line P1 cyb, using an ApaI digest, are shown in Figure 2C. None of the mitochondrial restriction fragments detected by incorporation of radiolabelled nucleotide (left) or by hybridisation with the labelled mtDNA probe (right) in the ρ+ line P1 cyb were seen in the ρ0 cells. These experiments strongly suggest that the fragments shown in Figure 2B and C arose from incorporation of [α-32P]dTTP into mtDNA in intact mitochondria rather than into nuclear pseudogenes.
Optimisation of mtDNA replication in control and patient-derived cybrid lines
Experiments aimed at optimising the conditions for incorporation of [α-32P]dTTP into mtDNA were carried out. In different experiments the saponin concentration was varied from 0 to 130 µg/ml. At the highest concentrations mtDNA synthesis was not detectable, consistent with disruption of normal cell morphology. While the optimum concentration was not identical in all lines, low concentrations of 25–30 µg/ml were suitable in all the cell lines investigated. Trypan blue exclusion showed that >90% of HeLa cells are permeabilised at 30 µg/ml (M.Hollinshead and D.Vaux, unpublished results) and this concentration was therefore used in all subsequent experiments. Mitochondrial morphological changes suggest that higher concentrations of saponin may increasingly compromise normal cell physiology.
The duration of incorporation of radioalabelled nucleotide was varied from <30 s to 9 h, using both primary fibroblasts and cybrid lines derived from different patients and controls as well as immortalised lines. Figure 3A and B illustrates typical examples in which incorporation was virtually undetectable following a pulse of <30 s. The signal intensity in the smaller bands relative to each other varied little over the range 15–60 min (although the absolute signal was higher with longer periods of labelling). In all fragments <5 kb the incorporation of radiolabel (normalised for length and loading) into each band showed a roughly linear increase with time over the first hour with no detectable lag phase (Fig. 3B, r2 = 0.93–0.99). Figure 3A shows that when mtDNA was linearised there was a suggestion of a lag phase lasting 1–2 h. This is consistent with completion of the first full-length mtDNA molecules in this time period. Later time points (Fig. 3A) demonstrated that the linear phase continued for between 2 and 5 h, after which a plateau phase followed.
Figure 3.





Optimisaton of the conditions for incorporation of [α-32P]dTTP into mtDNA. (A) Effect of duration of pulse labelling. Control cell line fib1 was incubated with [α-32P]dTTP for <25 s and 1, 2, 3, 5 and 10 h and digested with PvuII. Error bars represent the standard errors of three loadings. (B) Time course of radiolabel incorporation into specific ApaI fragments. Radioactivity incorporated after various incubation times with [α-32P]dTTP (I) was normalised to DNA loading and the size of the fragment (derived from S, the signal obtained after probing the filter with the mtDNA probe) to derive I/S for the 3.8 (top left) and 1.0 kb (bottom left) ApaI fragments from cell line P1 cyb and the 3.8 (top right) and 1.0 kb (bottom right) ApaI fragments from cell line P2 cyb. Two or three experiments are shown for each condition and cell line. Each run has been standardised to an incorporation of 1.0 at 60 min. Each time point represents three loadings from a single experiment. (C) Incorporation of radiolabel into ApaI fragments of mtDNA in relation to fragment length. Incorporation of [α-32P]dTTP into mtDNA restriction fragments from an ApaI digest of cell line P3 cyb, which contains only wild-type mtDNA. The incorporation of [α-32P]dTTP over 30 min is expressed as a percentage of the total radioactivity incorporated into all the mtDNA restriction fragments in that lane (each point representing triplicate loadings of the same DNA sample). Signal intensity was roughly proportional to fragment size (up to 4–5 kb) or TA content (not shown) with a 30 min pulse (but see D). A similar relationship was evident with pulse durations of 15 and 45 min. (D) Time course of incorporation of dTTP into the 1.0 and 1.6 kb (D-loop-containing) bands relative to steady-state mtDNA. Radioactivity incorporated after various incubation times and normalised to DNA loading and the size of the fragment (derived from the signal obtained after probing the filter with the mtDNA probe) for the 1.6 (diamonds) and 1.0 kb (circles) ApaI fragments from cell line P1 cyb (filled symbols) and from cell line P2 cyb (open symbols). Each time point represents three loadings from a single experiment. (E) Comparison between percentage A3243G mutant in newly synthesised mtDNA and steady-state mtDNA. Results are shown for P1 cyb (diamonds) and P2 cyb (circles). If the proportion of A3243G mtDNA mutants in newly synthesised mtDNA reflected the proportion in steady-state mtDNA, then all points should lie along the line I = S. The deviation from expected behaviour is significant for both cell types: the points for P1 cyb (diamonds) lie significantly above the line and the points for P2 cyb (circles) lie significantly below it (P = 0.036 and 0.006, respectively).
Figure 3C demonstrates that signal intensity was roughly proportional to fragment size up to 4–5 kb within a 30 min incubation. There was no consistent change in the percentage of mutant mtDNA when end-labelled probes were used (not shown). Thus the potential for regional variation in labelling of different regions of the mitochondrial probes does not appear to influence calculations of the percentage of mutant in steady-state mtDNA. Calculations of the ratio of percentage mutant in newly synthesised to steady-state mtDNA gave similar results whether or not both or only the larger mutant mtDNA bands were used. Taken together, these findings suggest that the relative signal intensities after a 30 min pulse can be used to estimate differential incorporation into mtDNA fragments <4 kb and, hence, potentially, the relative rate of mutant and wild-type mtDNA synthesis in the region where they can be distinguished using restriction fragment length polymorphisms.
Figure 3D demonstrates a regional difference in the rate of incorporation of [32P]dTTP into mtDNA. The rate of incorporation of dTTP into the 1.6 kb band was greater per unit length than into the 1.0 kb band in both cell lines P1 cyb and P2 cyb. The P1 cyb cell line incorporates [α-32P]dTTP ∼2-fold faster than the P2 cyb line. Furthermore, the gradient of the line is consistently greater for the 1.6 kb (D-loop-containing) ApaI fragment than the 1.0 kb fragment. As the 1.6 kb band maps to the large non-coding region of mtDNA and contains the origin of replication of the heavy strand (OH), this may represent multiple initiation events or rapid turnover of mtDNA near to the triple-stranded region known as the D-loop (20).
Reproducibility of comparisons between the rate of synthesis of wild-type and mutant mtDNA in heteroplasmic cell lines
The rates of synthesis of mutant and wild-type mtDNA were compared in heteroplasmic cybrid lines derived from patients with MELAS (P1 cyb and P2 cyb). The apparent percentage of mutant in newly labelled mtDNA (I) was calculated from the difference in incorporation between time points, correcting for minor differences in loading of each lane by using incorporation relative to the steady-state signal (I/S). In all cases the results were virtually identical when the absolute incorporation at each time point was used instead, because the signal at the 0 time point was negligible, again correcting for differences in loading. For example, in Figure 3B, run 1, the estimates for percentage mutant in newly synthesised mtDNA in the P1 cyb line were nearly identical, ranging from 40–43% (mean 42%). At every time point the steady-state proportion of mutant mtDNA was less than this, namely 35–38% (mean 37%). The percentage mutant in labelled P2 cyb mtDNA in run 1 was 61–67% (mean 64%), whereas steady-state percentage mutant calculated from mtDNA probe hybridisation was 77–81% (mean 79%). These differences in percentage mutant between newly synthesised and steady-state mtDNA are very reproducible on repeated experiments, including 1–26 duplicate runs (mean 10) on five cell lines carried out on between one and nine occasions. This is illustrated by Figure 3E: if the proportion of A3243G mtDNA mutants in newly synthesised mtDNA reflected the proportion in steady-state mtDNA, then all the points should lie along the line I = S. The deviation from expected behaviour is significant: the proportion of mutant mtDNA in the newly synthesised mtDNA was significantly greater than the steady-state level for line P1 cyb and significantly less than the steady-state for line P2 cyb (P = 0.036 and 0.006, respectively, Wilcoxon single rank test).
As the method is designed to compare the accumulation of newly synthesised relative to steady-state mtDNA in different restriction fragments of the same cell culture, all the quantitative results were based on comparisons within rather than between cell lines. Generally, cell line P2 cyb labelled 2- to 4-fold faster than line P1 cyb. We have not yet attempted to standardise between runs.
DISCUSSION
We have demonstrated incorporation of 32P-labelled nucleotides into mtDNA following permeabilisation of human cells with saponin and cold shock. The labelling of specific mtDNA restriction fragments (Fig. 2B) and failure to detect incorporation in ρ0 cell lines (Fig. 2C) demonstrates that this represents synthesis of mtDNA rather than nuclear pseudogenes.
The time course experiments (Fig. 3A and B) demonstrate that very little incorporation occurs in cells exposed to radiolabel and immediately placed on ice. Similarly, the predicted lag phase for shorter fragments was barely detectable over the range of times used, suggesting that incorporation is roughly linear and substrates for mtDNA synthesis are not limiting for at least 60 min. Figure 3A shows that when mtDNA was linearised there was a suggestion of a lag phase lasting 1–2 h, consistent with previous estimates of the time taken to replicate a single mtDNA molecule (21). The kinetics of incorporation manifested a plateau phase at later time points, after 5 h in Figure 3A. As the ratio of A3243G mutant mtDNA in newly synthesised relative to steady-state mtDNA did not change with time, any systematic error arising from using a small number of time points in the first hour is likely to be small.
The concentrations of saponin used in this study (30–40 µg/ml) were much lower than those in previous studies of nuclear replication (130 µg/ml) in which cell and organellar membranes were disrupted, but S phase-dependent, aphidicolin-sensitive DNA polymerase activity remained largely intact. They are also lower than those used for studies of skeletal muscle fibres and cardiac myocytes (50–100 µg/ml) in which mitochondrial function is preserved (11). Trypan blue exclusion studies on adherent monolayer cultures of HeLa cells (M.Hollinshead and D.Vaux, unpublished results) have shown that treatment with 10 µg/ml saponin permeabilises a significant proportion of, and with 25 µg/ml essentially all, cells. The use of 30 µg/ml saponin during this study would therefore be expected to permeabilise effectively all the cells in the culture. Mitochondria are still competent for transcription following treatment with saponin at a concentration of 40 µg/ml (M.Hollinshead and D.Vaux, unpublished results), which is slightly higher than the 30 µg/ml used in this study.
The results were reproducible for comparison of newly synthesised and steady-state mtDNA within a cell line (Fig. 3E). If the proportion of A3243G mutant in newly synthesised mtDNA reflected the proportion in steady-state mtDNA, then all the points in Figure 3E should lie along the line I = S. The figure shows that all the points for line P1 cyb lie above that line and nearly all the points for P2 cyb lie below it. This demonstrates that significantly more mutant mtDNA than expected is synthesised in line P1 cyb and signficantly less in line P2 cyb. The variation in percentage mutant in steady-state mtDNA in these experiments may reflect stochastic fluctuations or segregation in culture. In addition, there may be incomplete restriction enzyme digestion. As both of these factors operate on the percentage mutant in both steady-state and newly synthesised mtDNA, they cannot account for the significant differences from equality that we observed. Consistent with these data, when segregation occurs in cybrid lines derived from the ρ0 line 206, such as P1 cyb, the proportion of A3243G mutant mtDNA increases (17,22); in cybrid lines derived from the ρ0 line B2, such as P2 cyb, it decreases (17). Differences between cell lines were generally similar in different runs. Methods to enable comparisons between runs will be developed.
If incorporation of 32P-labelled nucleotide into mtDNA reflects de novo mtDNA synthesis, then this technique will be useful for studies of mtDNA replication. However, it is not clear whether the specific incorporation represents DNA replication, repair or both. As few DNA repair activities comparable to those identified for nuclear DNA have been identified in mitochondria, it is likely that mtDNA replication is the major DNA polymerase activity being measured with this technique, although this cannot be proved formally. Two aspects of our data support this. First, the pattern of incorporation over 60 min is more suggestive of mtDNA replication than repair (23), as there were no peaks at early time points (Fig. 3B). Secondly, Figure 3D demonstrates regional differences in the rate of labelling of mtDNA, which we attribute to rapid turnover of mtDNA in the region around the D-loop (20,24). While current data regarding mtDNA replication predict this, there is no a priori reason to suppose that there would be regional differences of such magnitude in patch repair. There are, however, a number of factors that might result in increased DNA repair under the conditions used. First, there might be a substantial increase in the rate of DNA damage because of the quantity of radioactivity added. However, when this technique was used to study incorporation of 32P-labelled nucleotides into nuclear DNA using comparable levels of radioactivity, 85% of the DNA polymerase acitivity was S phase-dependent and strongly inhibited by aphidicolin and, hence, likely to represent DNA polymerase α activity (9,25). While mtDNA may be more vulnerable to radiochemical damage because of the lack of histones, this would have to be much greater than occurs with nuclear DNA for replication to be masked completely by the DNA polymerase activity of mtDNA repair.
This method quantitates incorporation of radiolabel into mtDNA restriction fragments of wild-type and mutant mtDNA rather than into completed mtDNA molecules (7). The specific incorporation of [α-32P]dTTP into mtDNA that we have documented may represent only DNA polymerisation of replication complexes already assembled at the beginning of the experiment or may involve re-initiation. It is not clear whether initiation or elongation is rate limiting for mtDNA replication. Hence, we cannot be certain that our measurements represent differences in the rates of replication of wild-type and mutant mtDNA.
We have demonstrated a method that can be used to quantitate cytoplasmic DNA synthesis rates in cultured cells. While this may be applicable to studies of viral replication, it is most relevant to studies of mtDNA synthesis, in both normal cell lines and in those derived from patients with mitochondrial disease. First, the classical description of mammalian mtDNA replication involves unidirectional DNA synthesis from two distinct strand-specific origins. A round of replication begins at the heavy strand origin (the D-loop) where transcripts from an upstream promoter serve as the primers for DNA synthesis (20,21,26). There is now evidence for a second mode of replication (27) of mammalian mtDNA and the current method will be applied to dissecting the quantitative importance of these two modes in cell lines.
Secondly, it is most readily applicable to quantitating the relative rates of incorporation of 32P-labelled dTTP into wild-type and mutant mtDNA in cell lines containing heteroplasmic mutants that alter a restriction fragment, such as the A3243G mutation or mtDNA deletions. Variation in the proportion of mutant mtDNA between tissues is common in patients with heteroplasmic mtDNA disease and preferential accumulation of mutant mtDNA in affected tissues may account for the progressive and tissue-specific nature of these disorders. However, the mechanisms underlying the progressive changes are poorly understood. Sequential muscle biopsies on a patient with a proximal myopathy revealed that mutant mtDNA molecules accumulated in preference to wild-type mtDNA until they became the predominant mtDNA (28). In many cases the levels of mutant mtDNA vary in different tissues, being highest in highly oxidative, post-mitotic tissues such as brain, heart and skeletal muscle. These tissues are often the most severely affected by mitochondrial disease, suggesting that there are both stochastic (29) and tissue-specific factors which are important in determining the extent of mutant mtDNA accumulation (30). Experimental evidence for changes in distribution of mutant mtDNA comes from studies of cybrid cell lines in which mtDNA derived from patients can be studied against a ‘standardised’ nuclear background derived from a mtDNA-free (ρ0) cell line. The proportion of mutant mtDNA in such cybrid lines may change substantially with time (17,22). This may be influenced by factors such as: (i) the replication rate; (ii) an effect on cell growth of mutant compared with wild-type mtDNA; (iii) the distribution of mutant and wild-type mtDNA within and between cells (4). We are studying the molecular mechanisms which underlie these changes in tissue culture, as they may be important for the variable tissue distribution of mutants observed in patients. We have demonstrated that the proportion of A3243G mutant mtDNA was reproducibly higher in newly synthesised than steady-state mtDNA in the P1 cyb line; the converse is true for the P2 cyb line. These differences could reflect the relative replication rates of wild-type and mutant mtDNA and are consistent with studies of mtDNA segregation in such lines. They may be attributable to: (i) the nuclear background of the parent ρ0 line; (ii) differences in the mitochondrial haplotypes of the patients from whom they were derived; (iii) differences in the proportion of mutant mtDNA in the two lines.
Finally, in mtDNA depletion qualitatively normal mtDNA is quantitatively reduced in affected tissues (31), presumably as a result of defects in nuclear genes essential for mtDNA replication, repair or maintenance (2). We are currently adapting the method to compare the rates of mtDNA synthesis in cell lines derived from patients with depletion and controls, with a view to carrying out a complementation analysis.
In conclusion, this method will be useful in investigating the pathogenesis of mtDNA diseases in cell lines derived from patients with a range of disorders.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the patients and their families for cooperation, Drs D. J. Vaux, I. J. Holt, M. Hollinshead, K. J. Morten, D. Marchington, D. Jackson and Professor P. R. Cook for helpful comments and Professor E. R. Moxon for his support. Financial support was from The Wellcome Trust, the Royal Society and the MRC. J.P. is a Royal Society University Research Fellow. C.F. held a Wellcome Prize Studentship.
References
- 1.Poulton J. (1996) New genetics of mitochondrial DNA diseases. Br. J. Hosp. Med., 55, 712–716. [PubMed] [Google Scholar]
- 2.Nishino I., Spinazzola,A. and Hirano,M. (1999) Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science, 283, 689–692. [DOI] [PubMed] [Google Scholar]
- 3.Kaukonen J., Juselius,J.K., Tiranti,V., Kyttala,A., Zeviani,M., Comi,G.P., Keranen,S., Peltonen,L. and Suomalainen,A. (2000) Role of Adenine Nucleotide Translocator 1 in mtDNA maintenance. Science, 289, 782–785. [DOI] [PubMed] [Google Scholar]
- 4.Lightowlers R., Chinnery,P., Turnbull,D. and Howell,N. (1997) Mammalian mitochondrial genetics: hereditary, heteroplasmy and disease. Trends Genet., 13, 450–455. [DOI] [PubMed] [Google Scholar]
- 5.Pica-Mattoccia L. and Attardi,G. (1972) Expression of the mitochondrial genome in HeLa cells: IX. Replication of mitochondrial DNA in relationship to the cell cycle in HeLa cells. J. Mol. Biol., 64, 465–484. [DOI] [PubMed] [Google Scholar]
- 6.Robberson D.L. and Clayton,D.A. (1973) Pulse-labeled components in the replication of mitochondrial deoxyribonucleic acid. J. Biol. Chem., 248, 4512–4514. [PubMed] [Google Scholar]
- 7.Moraes C.T. and Schon,E.A. (1995) Replication of a heteroplasmic population of normal and partially-deleted human mitochondrial genomes. Prog. Cell Res., 5, 209–215. [Google Scholar]
- 8.Enriquez J.A., Ramos,J., Perez-Martos,A., Lopez-Perez,M.J. and Montoya,J. (1994) Highly efficient DNA synthesis in isolated mitochondria from rat liver. Nucleic Acids Res., 22, 1861–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson D. and Cook,P. (1986) A cell-cycle-dependent DNA polymerase activity that replicates intact DNA in chromatin. J. Mol. Biol., 192, 65–76. [DOI] [PubMed] [Google Scholar]
- 10.Wansink D., Schul,W., van der Kraan,I., van Steensel,B., van Dreil,R. and de Jong,L. (1993) Fluorescent labelling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J. Cell Biol., 122, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saks V.A., Veksler,V.I., Kuznetsov,A.V., Kay,L., Sikk,P., Tiivel,T., Tranqui,L., Olivares,J., Winkler,K., Wiedemann,F. and Kunz,W.S. (1998) Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol. Cell. Biochem., 184, 81–100. [PubMed] [Google Scholar]
- 12.Schulz I. (1990) Permeabilizing cells: some methods and applications for the study of intracellular processes. Methods Enzymol., 192, 280–300. [DOI] [PubMed] [Google Scholar]
- 13.Berger N. and Johnson,E. (1976) DNA synthesis in permeabilized mouse L cells. Biochim. Biophys. Acta, 18, 1–17 [DOI] [PubMed] [Google Scholar]
- 14.Kornberg A. and Baker,T.A. (1992) DNA Replication, 2nd Edn. Freeman, New York, NY.
- 15.Goto Y.-I., Nonaka,I. and Horai,S. (1990) A mutation in the tRNA leu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature, 348, 651–653. [DOI] [PubMed] [Google Scholar]
- 16.Matthews P., Brown,R., Morten,K., Marchington,D., Poulton,J. and Brown,G. (1995) Intracellular heteroplasmy for disease-associated point mutations in mtDNA: implications for disease expression and evidence for mitotic segregation of heteroplasmic units of mtDNA. Hum. Genet., 96, 261. [DOI] [PubMed] [Google Scholar]
- 17.Dunbar D., Moonie,P., Jacobs,H. and Holt,I. (1995) Different cellular backgrounds confer a marked advantage to either mutant or wild-type mitochondrial genomes. Proc. Natl Acad. Sci. USA, 92, 6562–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King M.P. and Attardi G. (1988) Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell, 52, 811–819. [DOI] [PubMed] [Google Scholar]
- 19.Cheng S., Higuchi,R. and Stoneking,M. (1994) Complete mitochondrial genome amplification [letter]. Nature Genet., 7, 350–351. [DOI] [PubMed] [Google Scholar]
- 20.Clayton D.A. (1991) Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell. Biol., 7, 453–478. [DOI] [PubMed] [Google Scholar]
- 21.Clayton,D.A. (1982) Replication of animal mitochondrial DNA. Cell, 28, 693–705. [DOI] [PubMed] [Google Scholar]
- 22.Yoneda M., Chomyn,A., Martinuzzi,A., Hurko,O. and Attardi,G. (1992) Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc. Natl Acad. Sci. USA, 89, 11164–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson D., Hassan,A., Errington,R. and Cook,P. (1994) Sites in human nuclei where damage induced by ultravioloet light is repaired. J. Cell Sci., 107, 1753–1760. [DOI] [PubMed] [Google Scholar]
- 24.Madsen C.S., Ghivizzani,S.C. and Hauswirth W.W. (1993) Protein binding to a single termination-associated sequence in the mitochondrial DNA D-loop region. Mol. Cell. Biol., 13, 2162–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson D. and Cook,P. (1986) Different populations of DNA polymerase alpha in HeLa cells. J. Mol. Biol., 192, 77–86. [DOI] [PubMed] [Google Scholar]
- 26.Wong T.W. and Clayton,D.A. (1985) In vitro replication of human mitochondrial DNA: accurate initiation at the origin of light-strand synthesis. Cell, 42, 951–958. [DOI] [PubMed] [Google Scholar]
- 27.Holt I., Lorimer,H. and Jacobs,H. (2000) Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell, 100, 515–524. [DOI] [PubMed] [Google Scholar]
- 28.Weber K., Wilson,J., Taylor,L., Brierley,E., Johnson,M. and Turnbull,D. (1997) A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am. J. Hum. Genet., 60, 373–380. [PMC free article] [PubMed] [Google Scholar]
- 29.Shoubridge E.A. (1995) Segregation of mitochondrial DNAs carrying a pathogenic point mutation (tRNA(leu3243)) in cybrid cells. Biochem. Biophys. Res. Commun., 213, 189–195. [DOI] [PubMed] [Google Scholar]
- 30.Jenuth J., Peterson,A. and Shoubridge,E. (1997) Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nature Genet., 16, 93–95. [DOI] [PubMed] [Google Scholar]
- 31.Moraes C.T., Shanske,S., Tritschler,H.J., Aprille,J.R., Andreetta,F., Bonilla,E., Schon,E.A. and DiMauro,S. (1991) mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am. J. Hum. Genet., 48, 492–501. [PMC free article] [PubMed] [Google Scholar]