

Abstract
In the molecule of the title compound, C11H14N2O4, a bifurcated intra/intermolecular N—H⋯(O,O) hydrogen bond occurs.The intramolecular component results in a non-planar six-membered ring with a flattened-boat conformation. In the crystal structure, the intermolecular interaction links the molecules into chains parallel to the b axis.
Related literature
For a related structure, see: Ates-Alagoz et al. (2001 ▶). For bond-length data, see: Allen et al. (1987 ▶). For ring-puckering parameters, see: Cremer & Pople (1975 ▶).
Experimental
Crystal data
C11H14N2O4
M r = 238.24
Monoclinic,
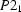
a = 4.2360 (8) Å
b = 16.180 (3) Å
c = 8.4890 (17) Å
β = 95.80 (3)°
V = 578.8 (2) Å3
Z = 2
Mo Kα radiation
μ = 0.11 mm−1
T = 294 (2) K
0.30 × 0.20 × 0.10 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Absorption correction: ψ scan (North et al., 1968 ▶) T min = 0.969, T max = 0.990
1213 measured reflections
1066 independent reflections
841 reflections with I > 2σ(I)
R int = 0.018
3 standard reflections frequency: 120 min intensity decay: none
Refinement
R[F 2 > 2σ(F 2)] = 0.077
wR(F 2) = 0.173
S = 1.01
1066 reflections
154 parameters
4 restraints
H-atom parameters constrained
Δρmax = 0.24 e Å−3
Δρmin = −0.30 e Å−3
Data collection: CAD-4 Software (Enraf–Nonius, 1989 ▶); cell refinement: CAD-4 Software; data reduction: XCAD4 (Harms & Wocadlo, 1995 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808043602/hk2598sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808043602/hk2598Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯O2 | 0.86 | 2.00 | 2.645 (10) | 131 |
| N1—H1A⋯O3i | 0.86 | 2.45 | 3.053 (10) | 128 |
Symmetry code: (i) 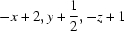 .
.
Acknowledgments
The authors thank Dr Shan Liu, Nanjing University of Technology, for useful discussions and the Center of Testing and Analysis, Nanjing University, for support.
supplementary crystallographic information
Comment
Some derivatives of benzoic acid are important chemical materials. We report herein the crystal structure of the title compound.
In the molecule of the title compound (Fig 1), the bond lengths (Allen et al., 1987) and angles are within normal ranges. Ring A (C3-C8) is, of course, planar. The intramolecular N-H···O hydrogen bond (Table 1) results in a nonplanar six-membered ring B (O2/N1/N2/C3/C4/H1A), having total puckering amplitude, QT, of 0.163 (2) Å, flattened-boat conformation [φ = 52.00 (3)° and θ = 19.29 (4)°] (Cremer & Pople, 1975).
In the crystal structure, intermolecular N-H···O hydrogen bonds (Table 1) link the molecules into chains parallel to the b axis (Fig. 2), in which they may be effective in the stabilization of the structure.
Experimental
For the preparation of the title compound, ethyl 4-chloro-3-nitrobenzoate (5.3 g, 0.023 mol) was refluxed in ethyl amine (20 ml) and tetrahydrofuran (50 ml) for 2 h. Then, solvents were evaporated and water was added to give yellow precipate. It was collected by filtration and washed with cold ethanol (2 X 15 ml) to afford the yellow solid (yield; 4.4 g, 80%) (Ates-Alagoz et al., 2001). Crystals suitable for X-ray analysis were obtained by slow evaporation of an ethanol solution.
Refinement
H atoms were positioned geometrically, with N-H = 0.86 Å (for NH) and C-H = 0.93, 0.97 and 0.96 Å for aromatic, methylene and methyl H, respectively, and constrained to ride on their parent atoms, with Uiso(H) = xUeq(C,N), where x = 1.5 for methyl H and x = 1.2 for all other H atoms.
Figures
Fig. 1.

The molecular structure of the title molecule, with the atom-numbering scheme. Hydrogen bond is shown as dashed line.
Fig. 2.

A partial packing diagram of the title compound. Hydrogen bonds are shown as dashed lines.
Crystal data
| C11H14N2O4 | F(000) = 252 |
| Mr = 238.24 | Dx = 1.367 Mg m−3 |
| Monoclinic, P21 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2yb | Cell parameters from 25 reflections |
| a = 4.2360 (8) Å | θ = 10–12° |
| b = 16.180 (3) Å | µ = 0.11 mm−1 |
| c = 8.4890 (17) Å | T = 294 K |
| β = 95.80 (3)° | Block, colorless |
| V = 578.8 (2) Å3 | 0.30 × 0.20 × 0.10 mm |
| Z = 2 |
Data collection
| Enraf–Nonius CAD-4 diffractometer | 841 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.018 |
| graphite | θmax = 25.2°, θmin = 2.4° |
| ω/2θ scans | h = −5→5 |
| Absorption correction: ψ scan (North et al., 1968) | k = 0→19 |
| Tmin = 0.969, Tmax = 0.990 | l = 0→10 |
| 1213 measured reflections | 3 standard reflections every 120 min |
| 1066 independent reflections | intensity decay: none |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.077 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.173 | H-atom parameters constrained |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.05P)2 + 1.25P] where P = (Fo2 + 2Fc2)/3 |
| 1066 reflections | (Δ/σ)max < 0.001 |
| 154 parameters | Δρmax = 0.24 e Å−3 |
| 4 restraints | Δρmin = −0.30 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.7012 (15) | 1.1050 (4) | 0.7940 (7) | 0.0772 (18) | |
| O2 | 0.9134 (16) | 1.1622 (4) | 0.5988 (8) | 0.083 (2) | |
| O3 | 0.9764 (13) | 0.7330 (4) | 0.7615 (6) | 0.0589 (15) | |
| O4 | 0.7965 (13) | 0.8254 (3) | 0.9244 (6) | 0.0564 (14) | |
| N1 | 1.2210 (17) | 1.0712 (5) | 0.4054 (8) | 0.065 (2) | |
| H1A | 1.1591 | 1.1204 | 0.4256 | 0.078* | |
| N2 | 0.8667 (17) | 1.0957 (5) | 0.6810 (9) | 0.0659 (19) | |
| C1 | 1.212 (2) | 1.0329 (6) | 0.1174 (10) | 0.071 (2) | |
| H1B | 1.3443 | 1.0259 | 0.0328 | 0.106* | |
| H1C | 1.0571 | 1.0750 | 0.0891 | 0.106* | |
| H1D | 1.1069 | 0.9818 | 0.1355 | 0.106* | |
| C2 | 1.414 (2) | 1.0581 (6) | 0.2654 (10) | 0.071 (3) | |
| H2A | 1.5246 | 1.1089 | 0.2453 | 0.085* | |
| H2B | 1.5719 | 1.0157 | 0.2925 | 0.085* | |
| C3 | 1.1440 (15) | 1.0085 (4) | 0.4988 (7) | 0.0383 (15) | |
| C4 | 0.9754 (15) | 1.0194 (5) | 0.6315 (8) | 0.0416 (16) | |
| C5 | 0.9015 (15) | 0.9543 (4) | 0.7232 (8) | 0.0401 (17) | |
| H5A | 0.7858 | 0.9646 | 0.8086 | 0.048* | |
| C6 | 0.9927 (16) | 0.8715 (4) | 0.6946 (8) | 0.0404 (16) | |
| C7 | 1.1639 (14) | 0.8632 (4) | 0.5582 (7) | 0.0393 (16) | |
| H7A | 1.2333 | 0.8105 | 0.5348 | 0.047* | |
| C8 | 1.2314 (16) | 0.9232 (4) | 0.4633 (8) | 0.0377 (16) | |
| H8A | 1.3342 | 0.9118 | 0.3740 | 0.045* | |
| C9 | 0.9235 (16) | 0.8047 (4) | 0.7942 (8) | 0.0376 (15) | |
| C10 | 0.7087 (18) | 0.7563 (5) | 1.0314 (8) | 0.0483 (19) | |
| H10A | 0.8916 | 0.7222 | 1.0650 | 0.058* | |
| H10B | 0.5432 | 0.7217 | 0.9787 | 0.058* | |
| C11 | 0.590 (2) | 0.8020 (6) | 1.1726 (9) | 0.060 (2) | |
| H11A | 0.5211 | 0.7625 | 1.2463 | 0.090* | |
| H11B | 0.4157 | 0.8372 | 1.1357 | 0.090* | |
| H11C | 0.7590 | 0.8348 | 1.2240 | 0.090* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.095 (4) | 0.068 (4) | 0.073 (4) | 0.010 (4) | 0.028 (4) | 0.005 (4) |
| O2 | 0.094 (5) | 0.079 (5) | 0.078 (5) | −0.004 (4) | 0.019 (4) | 0.003 (4) |
| O3 | 0.068 (3) | 0.052 (4) | 0.059 (3) | 0.004 (3) | 0.015 (3) | 0.002 (3) |
| O4 | 0.070 (3) | 0.049 (3) | 0.051 (3) | 0.005 (3) | 0.009 (3) | −0.002 (3) |
| N1 | 0.072 (4) | 0.063 (5) | 0.059 (4) | −0.011 (4) | 0.005 (4) | −0.008 (4) |
| N2 | 0.068 (4) | 0.068 (5) | 0.061 (4) | 0.001 (4) | 0.002 (4) | −0.003 (4) |
| C1 | 0.083 (6) | 0.070 (6) | 0.061 (5) | 0.008 (5) | 0.012 (4) | 0.004 (5) |
| C2 | 0.071 (5) | 0.075 (7) | 0.067 (6) | 0.008 (5) | 0.012 (4) | −0.005 (5) |
| C3 | 0.043 (3) | 0.037 (4) | 0.033 (3) | −0.006 (3) | −0.003 (3) | 0.003 (3) |
| C4 | 0.040 (3) | 0.045 (4) | 0.039 (4) | −0.002 (3) | 0.003 (3) | 0.004 (3) |
| C5 | 0.039 (3) | 0.043 (4) | 0.038 (4) | 0.001 (3) | 0.002 (3) | −0.001 (3) |
| C6 | 0.044 (4) | 0.034 (4) | 0.043 (4) | 0.002 (3) | 0.006 (3) | −0.002 (3) |
| C7 | 0.044 (4) | 0.037 (4) | 0.037 (4) | 0.004 (3) | 0.005 (3) | −0.002 (3) |
| C8 | 0.050 (4) | 0.029 (4) | 0.035 (4) | −0.005 (3) | 0.009 (3) | 0.005 (3) |
| C9 | 0.047 (4) | 0.029 (4) | 0.037 (4) | 0.002 (3) | 0.004 (3) | −0.001 (3) |
| C10 | 0.055 (4) | 0.052 (5) | 0.038 (4) | 0.002 (4) | 0.005 (3) | 0.006 (4) |
| C11 | 0.062 (5) | 0.064 (5) | 0.053 (5) | 0.003 (4) | 0.007 (4) | −0.002 (4) |
Geometric parameters (Å, °)
| O1—N2 | 1.253 (9) | C3—C8 | 1.467 (9) |
| O2—N2 | 1.308 (10) | C4—C5 | 1.365 (10) |
| O3—C9 | 1.219 (9) | C5—C6 | 1.422 (9) |
| O4—C9 | 1.320 (8) | C5—H5A | 0.9300 |
| O4—C10 | 1.510 (8) | C6—C9 | 1.420 (9) |
| N1—C2 | 1.523 (11) | C6—C7 | 1.434 (9) |
| N1—C3 | 1.349 (10) | C7—C8 | 1.311 (9) |
| N1—H1A | 0.8600 | C7—H7A | 0.9300 |
| N2—C4 | 1.397 (10) | C8—H8A | 0.9300 |
| C1—C2 | 1.502 (12) | C10—C11 | 1.535 (10) |
| C1—H1B | 0.9600 | C10—H10A | 0.9700 |
| C1—H1C | 0.9600 | C10—H10B | 0.9700 |
| C1—H1D | 0.9600 | C11—H11A | 0.9600 |
| C2—H2A | 0.9700 | C11—H11B | 0.9600 |
| C2—H2B | 0.9700 | C11—H11C | 0.9600 |
| C3—C4 | 1.405 (9) | ||
| C2—N1—H1A | 118.9 | C4—C5—H5A | 118.4 |
| C3—N1—C2 | 122.3 (8) | C6—C5—H5A | 118.4 |
| C3—N1—H1A | 118.9 | C9—C6—C5 | 122.6 (6) |
| O1—N2—O2 | 115.9 (8) | C9—C6—C7 | 124.2 (6) |
| O1—N2—C4 | 124.3 (8) | C5—C6—C7 | 113.2 (6) |
| O2—N2—C4 | 119.6 (7) | C8—C7—C6 | 126.0 (7) |
| C9—O4—C10 | 117.5 (6) | C8—C7—H7A | 117.0 |
| C2—C1—H1B | 109.5 | C6—C7—H7A | 117.0 |
| C2—C1—H1C | 109.5 | C7—C8—C3 | 119.6 (6) |
| H1B—C1—H1C | 109.5 | C7—C8—H8A | 120.2 |
| C2—C1—H1D | 109.5 | C3—C8—H8A | 120.2 |
| H1B—C1—H1D | 109.5 | O3—C9—O4 | 122.1 (7) |
| H1C—C1—H1D | 109.5 | O3—C9—C6 | 122.3 (6) |
| C1—C2—N1 | 112.7 (7) | O4—C9—C6 | 115.6 (6) |
| C1—C2—H2A | 109.0 | O4—C10—C11 | 103.4 (6) |
| N1—C2—H2A | 109.0 | O4—C10—H10A | 111.1 |
| C1—C2—H2B | 109.0 | C11—C10—H10A | 111.1 |
| N1—C2—H2B | 109.0 | O4—C10—H10B | 111.1 |
| H2A—C2—H2B | 107.8 | C11—C10—H10B | 111.1 |
| N1—C3—C4 | 123.3 (7) | H10A—C10—H10B | 109.0 |
| N1—C3—C8 | 120.4 (6) | C10—C11—H11A | 109.5 |
| C4—C3—C8 | 116.3 (6) | C10—C11—H11B | 109.5 |
| C5—C4—N2 | 114.2 (6) | H11A—C11—H11B | 109.5 |
| C5—C4—C3 | 121.6 (7) | C10—C11—H11C | 109.5 |
| N2—C4—C3 | 124.2 (7) | H11A—C11—H11C | 109.5 |
| C4—C5—C6 | 123.2 (6) | H11B—C11—H11C | 109.5 |
| C3—N1—C2—C1 | 84.9 (10) | C4—C5—C6—C7 | −1.2 (9) |
| C2—N1—C3—C4 | 177.9 (6) | C9—C6—C7—C8 | 179.8 (7) |
| C2—N1—C3—C8 | −3.1 (11) | C5—C6—C7—C8 | −1.2 (9) |
| O1—N2—C4—C5 | −3.6 (10) | C6—C7—C8—C3 | 3.4 (10) |
| O2—N2—C4—C5 | −177.5 (7) | N1—C3—C8—C7 | 177.8 (7) |
| O1—N2—C4—C3 | 175.8 (7) | C4—C3—C8—C7 | −3.1 (9) |
| O2—N2—C4—C3 | 1.9 (10) | C10—O4—C9—O3 | −1.7 (10) |
| N1—C3—C4—C5 | 179.9 (7) | C10—O4—C9—C6 | 178.0 (6) |
| C8—C3—C4—C5 | 0.9 (8) | C5—C6—C9—O3 | 173.2 (7) |
| N1—C3—C4—N2 | 0.6 (10) | C7—C6—C9—O3 | −7.9 (11) |
| C8—C3—C4—N2 | −178.5 (6) | C5—C6—C9—O4 | −6.5 (10) |
| N2—C4—C5—C6 | −179.3 (6) | C7—C6—C9—O4 | 172.4 (6) |
| C3—C4—C5—C6 | 1.3 (9) | C9—O4—C10—C11 | 176.7 (6) |
| C4—C5—C6—C9 | 177.8 (6) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O2 | 0.86 | 2.00 | 2.645 (10) | 131 |
| N1—H1A···O3i | 0.86 | 2.45 | 3.053 (10) | 128 |
Symmetry codes: (i) −x+2, y+1/2, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HK2598).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Ates-Alagoz, Z. & Buyukbingol, E. (2001). Heterocycl. Commun.7, 455-460.
- Cremer, D. & Pople, J. A. (1975). J. Am. Chem. Soc 97, 1354–1358.
- Enraf–Nonius (1989). CAD-4 Software Enraf–Nonius, Delft, The Netherlands.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Harms, K. & Wocadlo, S. (1995). XCAD4 University of Marburg, Germany.
- North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968). Acta Cryst. A24, 351–359.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808043602/hk2598sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808043602/hk2598Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report