

Abstract
In the title compound, C9H9ClN2, a substituted imidazoline, the six- and five-membered rings are twisted from each other, making a dihedral angle of 17.07 (5)°. In the crystal structure, a short Cl⋯Cl [3.3540 (3) Å] interaction is observed. Neighbouring molecules are linked together by intermolecular N—H⋯N hydrogen bonds into a one-dimensional infinite chain along the [101] direction and short Cl⋯Cl contacts link the chains into a three-dimensional network. There is also a significant π-stacking interaction between the planar sections of the six- and five-membered rings.
Related literature
For bond-length data, see: Allen et al. (1987 ▶). For a related structure and the synthesis, see: Stibrany et al. (2004 ▶); Kia et al. (2008 ▶). For the biological and pharmacological activities of imidazoline derivatives, see, for example: Blancafort (1978 ▶); Chan (1993 ▶); Vizi (1986 ▶); Li et al. (1996 ▶); Ueno et al. (1995 ▶); Corey & Grogan (1999 ▶).
Experimental
Crystal data
C9H9ClN2
M r = 180.63
Orthorhombic,

a = 19.7329 (8) Å
b = 39.1479 (18) Å
c = 4.3493 (2) Å
V = 3359.8 (3) Å3
Z = 16
Mo Kα radiation
μ = 0.39 mm−1
T = 100.0 (1) K
0.51 × 0.50 × 0.09 mm
Data collection
Bruker APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2005 ▶) T min = 0.825, T max = 0.964
14166 measured reflections
3438 independent reflections
3224 reflections with I > 2σ(I)
R int = 0.025
Refinement
R[F 2 > 2σ(F 2)] = 0.027
wR(F 2) = 0.072
S = 1.10
3438 reflections
113 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.33 e Å−3
Δρmin = −0.15 e Å−3
Absolute structure: Flack (1983 ▶), 1429 Friedel pairs
Flack parameter: −0.05 (4)
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: SAINT (Bruker, 2005 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2003 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809001214/is2379sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809001214/is2379Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected interatomic distances (Å).
| Cl1⋯Cl1i | 3.3540 (3) |
| C1⋯C3ii | 3.3945 (12) |
| C1⋯C4ii | 3.3301 (15) |
| C4⋯C6iii | 3.3997 (15) |
| C5⋯C7iii | 3.3716 (12) |
Symmetry codes: (i) 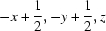 ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N1⋯N2iv | 0.896 (16) | 2.118 (16) | 3.0113 (11) | 174.5 (15) |
Symmetry code: (iv)  .
.
Acknowledgments
HKF and RK thank the Malaysian Government and Universiti Sains Malaysia for the Science Fund grant (No. 305/PFIZIK/613312). RK thanks Universiti Sains Malaysia for a post-doctoral research fellowship. HK thanks PNU for financial support. HKF also thanks Universiti Sains Malaysia for the Research University Golden Goose grant (No. 1001/PFIZIK/811012).
supplementary crystallographic information
Comment
Imidazoline derivatives are of great importance because they exhibit significant biological and pharmacological activities including antihypertensive (Blancafort, 1978), antihyperglycemic (Chan, 1993), antidepressive (Vizi, 1986), antihypercholesterolemic (Li et al., 1996) and antiinflammatory (Ueno et al., 1995) properties. These compounds are also used as catalysts and synthetic intermediates in some organic reactions (Corey & Grogan, 1999). Due to these important applications of imidazolines, here we report the crystal structure of the title compound, (I).
In the title compound (Fig. 1), bond lengths (Allen et al., 1987) and angles are within the normal ranges and are comparable with the related structures (Stibrany et al., 2004; Kia et al., 2008). The six- and five-membered rings are not coplanar and are twisted from each other by a dihedral angle of 18.07 (5)°. The interesting feature of the crystal structure is the short Cl···Cl [3.3540 (3) Å] (Table 1) which is shorter than the sum of the van der Waals radius of this atom. In the crystal structure (Fig. 2), neighbouring molecules are linked together by intermolecular N—H···N hydrogen bonds (Table 2) into 1-D infinite chains along the [1 0 1] direction and short Cl···Cl contacts link these chains into a 3-D network. There is also a significant π-stacking interaction between the planar sections associated with C1–C3–C4–C5–C6 and C7 of the six- and five-membered rings respectively (Table 1).
Experimental
The synthetic method was based on the previous work (Stibrany et al., 2004), except that 10 mmol of 3-chloro-2-cyanobenzene and 40 mmol of ethylenediamine were used. Single crystals suitable for X-ray diffraction were obtained by evaporation of an acetonitrile solution at room temperature.
Refinement
The H atom bound to N1 was located in a difference Fourier map and refined freely. Other H atoms were positioned geometrically and refined in a riding model approximation, with C—H = 0.93–0.97 Å, and with Uiso(H) = 1.2Ueq(C).
Figures
Fig. 1.

The molecular structure of (I) with atom labels and 50% probability ellipsoids for non-H atoms.
Fig. 2.

The crystal packing of (I), viewed down the c-axis showing linking of molecules through intermolecular N—H···N hydrogen bonds and short Cl···Cl interactions. The intermolecular interactions are shown as dashed lines.
Crystal data
| C9H9ClN2 | F(000) = 1504 |
| Mr = 180.63 | Dx = 1.428 Mg m−3 |
| Orthorhombic, Fdd2 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: F 2 -2d | Cell parameters from 8962 reflections |
| a = 19.7329 (8) Å | θ = 2.9–36.7° |
| b = 39.1479 (18) Å | µ = 0.39 mm−1 |
| c = 4.3493 (2) Å | T = 100 K |
| V = 3359.8 (3) Å3 | Block, colourless |
| Z = 16 | 0.51 × 0.50 × 0.09 mm |
Data collection
| Bruker APEXII CCD area-detector diffractometer | 3438 independent reflections |
| Radiation source: fine-focus sealed tube | 3224 reflections with I > 2σ(I) |
| graphite | Rint = 0.025 |
| φ and ω scans | θmax = 35.0°, θmin = 2.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2005) | h = −31→25 |
| Tmin = 0.825, Tmax = 0.964 | k = −60→60 |
| 14166 measured reflections | l = −6→6 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.027 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.072 | w = 1/[σ2(Fo2) + (0.037P)2 + 1.1492P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.10 | (Δ/σ)max = 0.001 |
| 3438 reflections | Δρmax = 0.33 e Å−3 |
| 113 parameters | Δρmin = −0.15 e Å−3 |
| 1 restraint | Absolute structure: Flack (1983), 1429 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Flack parameter: −0.05 (4) |
Special details
| Experimental. The low-temperature data was collected with the Oxford Cyrosystem Cobra low-temperature attachment. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.165233 (12) | 0.246951 (6) | 0.02330 (7) | 0.02846 (7) | |
| N1 | 0.00209 (4) | 0.10678 (2) | 0.1715 (2) | 0.01782 (15) | |
| N2 | 0.11053 (4) | 0.11676 (2) | 0.3205 (2) | 0.01792 (15) | |
| C1 | 0.10570 (4) | 0.18532 (2) | 0.0803 (2) | 0.01732 (16) | |
| H1A | 0.1405 | 0.1803 | 0.2172 | 0.021* | |
| C2 | 0.10314 (4) | 0.21685 (2) | −0.0634 (2) | 0.01837 (17) | |
| C3 | 0.05234 (5) | 0.22529 (2) | −0.2708 (2) | 0.01901 (17) | |
| H3A | 0.0517 | 0.2465 | −0.3667 | 0.023* | |
| C4 | 0.00232 (5) | 0.20104 (3) | −0.3313 (3) | 0.01959 (17) | |
| H4A | −0.0323 | 0.2062 | −0.4686 | 0.024* | |
| C5 | 0.00358 (5) | 0.16926 (2) | −0.1890 (2) | 0.01739 (16) | |
| H5A | −0.0301 | 0.1533 | −0.2313 | 0.021* | |
| C6 | 0.05544 (4) | 0.16118 (2) | 0.0174 (2) | 0.01498 (14) | |
| C7 | 0.05755 (4) | 0.12791 (2) | 0.1766 (2) | 0.01505 (15) | |
| C8 | 0.01553 (5) | 0.07848 (3) | 0.3841 (3) | 0.02052 (18) | |
| H8A | 0.0053 | 0.0566 | 0.2907 | 0.025* | |
| H8B | −0.0102 | 0.0809 | 0.5728 | 0.025* | |
| C9 | 0.09225 (5) | 0.08268 (3) | 0.4421 (3) | 0.02013 (18) | |
| H9A | 0.1021 | 0.0813 | 0.6602 | 0.024* | |
| H9B | 0.1175 | 0.0650 | 0.3364 | 0.024* | |
| H1N1 | −0.0392 (8) | 0.1159 (4) | 0.148 (4) | 0.036 (4)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.01974 (10) | 0.01899 (11) | 0.04665 (16) | −0.00554 (8) | −0.00483 (11) | 0.00392 (11) |
| N1 | 0.0124 (3) | 0.0167 (4) | 0.0244 (4) | −0.0016 (3) | −0.0022 (3) | 0.0014 (3) |
| N2 | 0.0132 (3) | 0.0161 (3) | 0.0245 (4) | 0.0007 (3) | −0.0013 (3) | 0.0019 (3) |
| C1 | 0.0122 (3) | 0.0165 (4) | 0.0232 (4) | 0.0008 (3) | −0.0002 (3) | −0.0003 (3) |
| C2 | 0.0139 (3) | 0.0166 (4) | 0.0246 (4) | −0.0010 (3) | 0.0019 (3) | −0.0004 (3) |
| C3 | 0.0189 (4) | 0.0171 (4) | 0.0210 (4) | 0.0013 (3) | 0.0025 (3) | 0.0013 (3) |
| C4 | 0.0180 (4) | 0.0211 (4) | 0.0196 (4) | 0.0016 (3) | −0.0018 (3) | 0.0000 (3) |
| C5 | 0.0148 (3) | 0.0192 (4) | 0.0181 (4) | 0.0000 (3) | −0.0006 (3) | −0.0007 (3) |
| C6 | 0.0120 (3) | 0.0149 (4) | 0.0180 (4) | 0.0012 (3) | 0.0018 (3) | −0.0014 (3) |
| C7 | 0.0117 (3) | 0.0158 (4) | 0.0177 (4) | −0.0003 (3) | 0.0011 (3) | −0.0016 (3) |
| C8 | 0.0164 (4) | 0.0188 (4) | 0.0264 (4) | −0.0021 (3) | −0.0011 (3) | 0.0042 (3) |
| C9 | 0.0159 (4) | 0.0184 (4) | 0.0260 (5) | 0.0002 (3) | −0.0008 (3) | 0.0039 (3) |
Geometric parameters (Å, °)
| Cl1—C2 | 1.7413 (10) | C3—H3A | 0.9300 |
| N1—C7 | 1.3719 (12) | C4—C5 | 1.3897 (14) |
| N1—C8 | 1.4675 (13) | C4—H4A | 0.9300 |
| N1—H1N1 | 0.896 (16) | C5—C6 | 1.3976 (13) |
| N2—C7 | 1.2942 (12) | C5—H5A | 0.9300 |
| N2—C9 | 1.4799 (13) | C6—C7 | 1.4759 (13) |
| C1—C2 | 1.3844 (14) | C8—C9 | 1.5436 (13) |
| C1—C6 | 1.3969 (12) | C8—H8A | 0.9700 |
| C1—H1A | 0.9300 | C8—H8B | 0.9700 |
| C2—C3 | 1.3884 (14) | C9—H9A | 0.9700 |
| C3—C4 | 1.3944 (14) | C9—H9B | 0.9700 |
| Cl1···Cl1i | 3.3540 (3) | C4···C6iii | 3.3997 (15) |
| C1···C3ii | 3.3945 (12) | C5···C7iii | 3.3716 (12) |
| C1···C4ii | 3.3301 (15) | ||
| C7—N1—C8 | 107.50 (7) | C1—C6—C5 | 119.51 (8) |
| C7—N1—H1N1 | 119.1 (10) | C1—C6—C7 | 119.03 (8) |
| C8—N1—H1N1 | 122.5 (11) | C5—C6—C7 | 121.45 (8) |
| C7—N2—C9 | 106.25 (7) | N2—C7—N1 | 116.68 (8) |
| C2—C1—C6 | 119.26 (9) | N2—C7—C6 | 123.17 (8) |
| C2—C1—H1A | 120.4 | N1—C7—C6 | 120.13 (8) |
| C6—C1—H1A | 120.4 | N1—C8—C9 | 101.53 (7) |
| C1—C2—C3 | 122.13 (9) | N1—C8—H8A | 111.5 |
| C1—C2—Cl1 | 118.68 (7) | C9—C8—H8A | 111.5 |
| C3—C2—Cl1 | 119.19 (8) | N1—C8—H8B | 111.5 |
| C2—C3—C4 | 118.15 (9) | C9—C8—H8B | 111.5 |
| C2—C3—H3A | 120.9 | H8A—C8—H8B | 109.3 |
| C4—C3—H3A | 120.9 | N2—C9—C8 | 106.06 (8) |
| C5—C4—C3 | 120.84 (9) | N2—C9—H9A | 110.5 |
| C5—C4—H4A | 119.6 | C8—C9—H9A | 110.5 |
| C3—C4—H4A | 119.6 | N2—C9—H9B | 110.5 |
| C4—C5—C6 | 120.12 (8) | C8—C9—H9B | 110.5 |
| C4—C5—H5A | 119.9 | H9A—C9—H9B | 108.7 |
| C6—C5—H5A | 119.9 | ||
| C6—C1—C2—C3 | −0.48 (14) | C9—N2—C7—C6 | −179.11 (8) |
| C6—C1—C2—Cl1 | 178.76 (7) | C8—N1—C7—N2 | 9.55 (12) |
| C1—C2—C3—C4 | 0.66 (14) | C8—N1—C7—C6 | −171.73 (8) |
| Cl1—C2—C3—C4 | −178.58 (8) | C1—C6—C7—N2 | −15.38 (13) |
| C2—C3—C4—C5 | −0.37 (15) | C5—C6—C7—N2 | 166.02 (9) |
| C3—C4—C5—C6 | −0.09 (15) | C1—C6—C7—N1 | 165.98 (9) |
| C2—C1—C6—C5 | 0.00 (13) | C5—C6—C7—N1 | −12.62 (13) |
| C2—C1—C6—C7 | −178.62 (8) | C7—N1—C8—C9 | −13.33 (10) |
| C4—C5—C6—C1 | 0.28 (14) | C7—N2—C9—C8 | −8.35 (11) |
| C4—C5—C6—C7 | 178.87 (9) | N1—C8—C9—N2 | 13.08 (10) |
| C9—N2—C7—N1 | −0.43 (11) |
Symmetry codes: (i) −x+1/2, −y+1/2, z; (ii) x, y, z+1; (iii) x, y, z−1.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N1···N2iv | 0.896 (16) | 2.118 (16) | 3.0113 (11) | 174.5 (15) |
Symmetry codes: (iv) x−1/4, −y+1/4, z−1/4.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IS2379).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Blancafort, P. (1978). Drugs Future, 3, 592–592.
- Bruker (2005). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Chan, S. (1993). Clin. Sci.85, 671–677. [DOI] [PubMed]
- Corey, E. J. & Grogan, M. J. (1999). Org. Lett.1, 157–160. [DOI] [PubMed]
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Kia, R., Fun, H.-K. & Kargar, H. (2008). Acta Cryst. E64, o2406. [DOI] [PMC free article] [PubMed]
- Li, H. Y., Drummond, S., De Lucca, I. & Boswell, G. A. (1996). Tetrahedron, 52, 11153–11162.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Stibrany, R. T., Schugar, H. J. & Potenza, J. A. (2004). Acta Cryst. E60, o527–o529.
- Ueno, M., Imaizumi, K., Sugita, T., Takata, I. & Takeshita, M. (1995). Int. J. Immunopharmacol.17, 597–603. [DOI] [PubMed]
- Vizi, E. S. (1986). Med. Res. Rev.6, 431–449. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809001214/is2379sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809001214/is2379Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report