

Abstract
The synthesis of the title compound, C12H15BrN2OS, involves the reaction of 4-bromobenzoyl chloride with potassium thiocyanate in dry acetone, followed by condensation of 4-bromobenzoyl isothiocyanate with diethylamine. The carbonyl and thiocarbonyl bond lengths indicate that these correspond to double bonds. The short C—N bond lengths reveal the effects of resonance in this part of the molecule. The conformation of the molecule with respect to the thiocarbonyl and carbonyl units is twisted, with torsion angles of −5.7 (3) and 87.2 (2)°. The N atom of the diethylamine group is sp 2-hybridized: the sum of the angles around the N atom is 359.97 (14)°. The two diethyl groups are twisted in + and − antiperiplanar conformations with angles of −179.89 and 179.92°. In the crystal structure, the molecules form infinite chains via an intermolecular N—H⋯O interaction.
Related literature
For the synthesis, see: Özer et al. (2009 ▶); Arslan, Flörke & Külcü (2003 ▶), and references therein. For general background, see: Koch (2001 ▶); El Aamrani et al. (1998 ▶, 1999 ▶); Arslan et al. (2006 ▶); Arslan, Flörke & Külcü (2007 ▶); Arslan, Flörke, Külcü & Binzet (2007 ▶); Yuan et al. (2001 ▶); Zhang et al. (2004 ▶); Weiqun et al. (2004 ▶). For related compounds, see: Arslan, Külcü & Flörke (2003 ▶); Arslan et al. (2004 ▶); Khawar Rauf et al. (2009a
▶,b
▶); Khawar Rauf, Bolte & Anwar (2009 ▶); Khawar Rauf, Bolte & Rauf (2009 ▶). For bond-length data, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C12H15BrN2OS
M r = 315.23
Monoclinic,
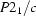
a = 6.9955 (9) Å
b = 18.680 (2) Å
c = 10.0816 (13) Å
β = 95.361 (3)°
V = 1311.7 (3) Å3
Z = 4
Mo Kα radiation
μ = 3.28 mm−1
T = 120 (2) K
0.38 × 0.37 × 0.11 mm
Data collection
Bruker SMART APEX diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2002 ▶) T min = 0.329, T max = 0.714
10831 measured reflections
3117 independent reflections
2730 reflections with I > 2σ(I)
R int = 0.027
Refinement
R[F 2 > 2σ(F 2)] = 0.025
wR(F 2) = 0.064
S = 1.06
3117 reflections
158 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.59 e Å−3
Δρmin = −0.26 e Å−3
Data collection: SMART (Bruker, 2002 ▶); cell refinement: SAINT (Bruker, 2002 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536809003183/at2713sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809003183/at2713Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O1i | 0.896 (5) | 2.016 (9) | 2.882 (2) | 162 (2) |
Symmetry code: (i) 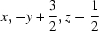 .
.
Acknowledgments
This work was supported by Mersin University Research Fund [project Nos. BAP-ECZ-F-TBB-(HA) 2004–3 and BAP-FEF-KB-(NK) 2006-3]. This study is part of the PhD thesis of GB.
supplementary crystallographic information
Comment
Transition metal complexes bearing thiourea ligand or its derivatives have been one of the highlights in coordination chemistry, which are used as reactant for extraction of some transition metal ions (Koch, 2001; El Aamrani et al., 1998, 1999). Moreover, the growing interest for thiourea derivative ligands and their metal complexes result from the important role they play in biological systems (Yuan et al., 2001; Zhang et al., 2004; Weiqun et al., 2004).
Recently, our research has focussed on the chemical and physical properties of thiourea derivatives and their metal complexes (Arslan et al., 2006; Arslan, Flörke & Külcü, 2007; Arslan, Flörke, Külcü & Binzet, 2007). In the present work, we report the crystal structure of 4-bromo-N-(diethylcarbamothioyl)benzamide, (I). The molecular structure of the title compound is depicted in Fig. 1.
The typical thiourea carbonyl [C6—O1 = 1.230 (2) Å] and thiocarbonyl (C1—S1 = 1.6638 (18) Å) double bonds as well as shortened C—N bond lengths (C1—N1 (1.435 (2) Å), C1—N2 (1.325 (2) Å) and C6—N1 (1.355 (2) Å)) are observed in the title compound. These bond lengths in the title compound are comparable to those of related structures (Khawar Rauf et al., 2009a,b; Khawar Rauf, Bolte & Anwar, 2009; Khawar Rauf, Bolte & Rauf, 2009; Arslan, Flörke & Külcü, 2003; Arslan et al., 2004). The other bond lengths in (I) show normal values (Allen et al., 1987).
The conformation of the title molecule with respect to the thiocarbonyl and carbonyl moieties is twisted, as reflected by the C1—N1—C6—O1, C6—N1—C1—N2, and C6—N1—C1—S1 torsion angles of -5.7 (3) °, 87.2 (2) ° and -94.57 (18) °, respectively. The dihedral angle between the 4-bromophenyl ring and the plane O1/N1/C7/C6 is 10.10 (3) °, and the dihedral angle between the 4-bromophenyl ring and the plane S1/C1/N1/N2 is 86.98 (3) °. The atom N2 is sp2-hybridized, because of the sum of the angles around atom N2 is 359.97 (14) °. The two diethyl groups are twisted in a + and - antiperiplanar conformation with -179.89 ° and 179.92 °.
Iintermolecular N—H···O (x, -y+1.5, z-0.5) hydrogen bonds (Table 1) link the molecules into endless chains, as shown in Fig. 2.
Experimental
The title compound was prepared with a procedure similar to that reported in the literature (Arslan, Külcü & Flörke, 2003; Özer et al., 2009). A solution of 4-bromobenzoyl chloride (0.01 mol) in acetone (50 cm3) was added dropwise to a suspension of potassium thiocyanate (0.01 mol) in acetone (30 cm3). The reaction mixture was heated under reflux for 30 min, and then cooled to room temperature. A solution of diethylamine (0.01 mol) in acetone (10 cm3) was added and the resulting mixture was stirred for 2 h. Hydrochloric acid (0.1 N, 300 cm3) was added to the solution, which was then filtered. The solid product was washed with water and purifed by recrystalization from an ethanol:dichloromethane mixture (1:2). Analysis calculated for C12H15N2OSBr: C 45.7, H 4.8, N 8.9%. Found: C 45.7, H 4.9, N 8.7%.
Refinement
H atoms bound to C atoms were placed geometrically and allowed to ride on their parent atoms, with C—H = 0.95-0.99 Å and Uiso(H) = 1.2 or 1.5 Ueq(C). The nitrogen-bound H atom was located in a difference Fourier map and refined freely.
Figures
Fig. 1.

The molecular structure of (I). Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

Part of the structure showing the formation of endless chains involving N—H···O hydrogen bonds.
Crystal data
| C12H15BrN2OS | F(000) = 640 |
| Mr = 315.23 | Dx = 1.596 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 891 reflections |
| a = 6.9955 (9) Å | θ = 2.3–28.2° |
| b = 18.680 (2) Å | µ = 3.28 mm−1 |
| c = 10.0816 (13) Å | T = 120 K |
| β = 95.361 (3)° | Prism, colourless |
| V = 1311.7 (3) Å3 | 0.38 × 0.37 × 0.11 mm |
| Z = 4 |
Data collection
| Bruker SMART APEX diffractometer | 3117 independent reflections |
| Radiation source: sealed tube | 2730 reflections with I > 2σ(I) |
| graphite | Rint = 0.027 |
| φ and ω scans | θmax = 27.9°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2002) | h = −9→7 |
| Tmin = 0.329, Tmax = 0.714 | k = −24→24 |
| 10831 measured reflections | l = −13→13 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.025 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.064 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0328P)2 + 0.2678P] where P = (Fo2 + 2Fc2)/3 |
| 3117 reflections | (Δ/σ)max = 0.001 |
| 158 parameters | Δρmax = 0.59 e Å−3 |
| 1 restraint | Δρmin = −0.26 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.18884 (3) | 0.416437 (9) | 0.341437 (19) | 0.02229 (7) | |
| S1 | 0.41112 (7) | 0.90370 (2) | 0.43285 (5) | 0.02424 (11) | |
| O1 | 0.4748 (2) | 0.73637 (7) | 0.62427 (13) | 0.0265 (3) | |
| N1 | 0.5090 (2) | 0.76602 (7) | 0.41132 (14) | 0.0176 (3) | |
| H1 | 0.478 (3) | 0.7585 (12) | 0.3242 (7) | 0.035 (6)* | |
| N2 | 0.7563 (2) | 0.84313 (7) | 0.48486 (15) | 0.0184 (3) | |
| C1 | 0.5705 (3) | 0.83737 (9) | 0.44671 (17) | 0.0180 (4) | |
| C2 | 0.8900 (3) | 0.78163 (9) | 0.49166 (19) | 0.0230 (4) | |
| H2A | 0.8245 | 0.7391 | 0.5248 | 0.028* | |
| H2B | 1.0018 | 0.7926 | 0.5561 | 0.028* | |
| C3 | 0.9600 (3) | 0.76427 (10) | 0.3581 (2) | 0.0290 (4) | |
| H3A | 1.0476 | 0.7233 | 0.3674 | 0.043* | |
| H3B | 1.0277 | 0.8058 | 0.3258 | 0.043* | |
| H3C | 0.8501 | 0.7525 | 0.2943 | 0.043* | |
| C4 | 0.8433 (3) | 0.91336 (9) | 0.51931 (19) | 0.0219 (4) | |
| H4A | 0.7698 | 0.9512 | 0.4681 | 0.026* | |
| H4B | 0.9764 | 0.9142 | 0.4935 | 0.026* | |
| C5 | 0.8459 (3) | 0.92934 (10) | 0.66664 (19) | 0.0265 (4) | |
| H5A | 0.9048 | 0.9763 | 0.6855 | 0.040* | |
| H5B | 0.9204 | 0.8925 | 0.7176 | 0.040* | |
| H5C | 0.7141 | 0.9296 | 0.6922 | 0.040* | |
| C6 | 0.4567 (3) | 0.72010 (9) | 0.50554 (17) | 0.0172 (3) | |
| C7 | 0.3850 (2) | 0.64814 (9) | 0.45935 (17) | 0.0166 (3) | |
| C8 | 0.3120 (3) | 0.60335 (10) | 0.55234 (18) | 0.0197 (4) | |
| H8A | 0.3030 | 0.6200 | 0.6406 | 0.024* | |
| C9 | 0.2519 (3) | 0.53439 (9) | 0.51763 (18) | 0.0203 (4) | |
| H9A | 0.2013 | 0.5039 | 0.5812 | 0.024* | |
| C10 | 0.2670 (2) | 0.51082 (9) | 0.38901 (18) | 0.0180 (3) | |
| C11 | 0.3388 (3) | 0.55432 (9) | 0.29424 (18) | 0.0204 (4) | |
| H11A | 0.3481 | 0.5372 | 0.2063 | 0.025* | |
| C12 | 0.3970 (3) | 0.62334 (9) | 0.32955 (18) | 0.0190 (4) | |
| H12A | 0.4453 | 0.6539 | 0.2651 | 0.023* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.02598 (11) | 0.01446 (10) | 0.02654 (11) | −0.00248 (6) | 0.00300 (7) | −0.00079 (6) |
| S1 | 0.0274 (3) | 0.0178 (2) | 0.0274 (3) | 0.00464 (17) | 0.00185 (19) | −0.00387 (17) |
| O1 | 0.0459 (9) | 0.0198 (6) | 0.0141 (6) | −0.0039 (6) | 0.0046 (6) | −0.0019 (5) |
| N1 | 0.0241 (8) | 0.0153 (7) | 0.0131 (7) | −0.0018 (6) | 0.0007 (6) | −0.0016 (5) |
| N2 | 0.0240 (8) | 0.0130 (7) | 0.0184 (8) | −0.0006 (6) | 0.0028 (6) | −0.0016 (5) |
| C1 | 0.0277 (10) | 0.0139 (8) | 0.0127 (8) | −0.0025 (7) | 0.0040 (7) | −0.0005 (6) |
| C2 | 0.0242 (10) | 0.0171 (8) | 0.0270 (10) | 0.0023 (7) | −0.0025 (8) | 0.0019 (7) |
| C3 | 0.0293 (11) | 0.0264 (10) | 0.0313 (11) | 0.0090 (8) | 0.0034 (8) | −0.0025 (8) |
| C4 | 0.0259 (10) | 0.0172 (8) | 0.0230 (10) | −0.0052 (7) | 0.0040 (7) | −0.0023 (7) |
| C5 | 0.0317 (11) | 0.0246 (9) | 0.0228 (10) | −0.0065 (8) | 0.0009 (8) | −0.0063 (7) |
| C6 | 0.0206 (9) | 0.0157 (8) | 0.0155 (9) | 0.0023 (6) | 0.0021 (7) | −0.0007 (6) |
| C7 | 0.0174 (8) | 0.0150 (7) | 0.0172 (9) | 0.0017 (6) | 0.0007 (7) | −0.0006 (6) |
| C8 | 0.0244 (9) | 0.0205 (8) | 0.0146 (9) | 0.0001 (7) | 0.0035 (7) | −0.0009 (7) |
| C9 | 0.0211 (9) | 0.0197 (8) | 0.0207 (9) | −0.0022 (7) | 0.0042 (7) | 0.0041 (7) |
| C10 | 0.0165 (9) | 0.0138 (7) | 0.0233 (9) | 0.0007 (6) | 0.0002 (7) | −0.0008 (6) |
| C11 | 0.0275 (10) | 0.0178 (8) | 0.0164 (9) | −0.0017 (7) | 0.0038 (7) | −0.0023 (7) |
| C12 | 0.0243 (9) | 0.0164 (8) | 0.0168 (9) | −0.0011 (7) | 0.0051 (7) | 0.0016 (6) |
Geometric parameters (Å, °)
| Br1—C10 | 1.8944 (17) | C4—H4A | 0.9900 |
| S1—C1 | 1.6638 (18) | C4—H4B | 0.9900 |
| O1—C6 | 1.230 (2) | C5—H5A | 0.9800 |
| N1—C6 | 1.355 (2) | C5—H5B | 0.9800 |
| N1—C1 | 1.435 (2) | C5—H5C | 0.9800 |
| N1—H1 | 0.896 (5) | C6—C7 | 1.494 (2) |
| N2—C1 | 1.325 (2) | C7—C8 | 1.389 (2) |
| N2—C4 | 1.474 (2) | C7—C12 | 1.398 (2) |
| N2—C2 | 1.479 (2) | C8—C9 | 1.390 (2) |
| C2—C3 | 1.511 (3) | C8—H8A | 0.9500 |
| C2—H2A | 0.9900 | C9—C10 | 1.383 (3) |
| C2—H2B | 0.9900 | C9—H9A | 0.9500 |
| C3—H3A | 0.9800 | C10—C11 | 1.384 (2) |
| C3—H3B | 0.9800 | C11—C12 | 1.389 (2) |
| C3—H3C | 0.9800 | C11—H11A | 0.9500 |
| C4—C5 | 1.513 (3) | C12—H12A | 0.9500 |
| C6—N1—C1 | 120.52 (14) | C4—C5—H5A | 109.5 |
| C6—N1—H1 | 122.0 (15) | C4—C5—H5B | 109.5 |
| C1—N1—H1 | 115.4 (15) | H5A—C5—H5B | 109.5 |
| C1—N2—C4 | 120.85 (14) | C4—C5—H5C | 109.5 |
| C1—N2—C2 | 123.33 (14) | H5A—C5—H5C | 109.5 |
| C4—N2—C2 | 115.79 (15) | H5B—C5—H5C | 109.5 |
| N2—C1—N1 | 114.24 (15) | O1—C6—N1 | 121.04 (16) |
| N2—C1—S1 | 126.53 (13) | O1—C6—C7 | 121.79 (16) |
| N1—C1—S1 | 119.20 (13) | N1—C6—C7 | 117.11 (15) |
| N2—C2—C3 | 112.43 (15) | C8—C7—C12 | 119.34 (16) |
| N2—C2—H2A | 109.1 | C8—C7—C6 | 117.74 (15) |
| C3—C2—H2A | 109.1 | C12—C7—C6 | 122.84 (16) |
| N2—C2—H2B | 109.1 | C7—C8—C9 | 120.67 (16) |
| C3—C2—H2B | 109.1 | C7—C8—H8A | 119.7 |
| H2A—C2—H2B | 107.8 | C9—C8—H8A | 119.7 |
| C2—C3—H3A | 109.5 | C10—C9—C8 | 118.91 (16) |
| C2—C3—H3B | 109.5 | C10—C9—H9A | 120.5 |
| H3A—C3—H3B | 109.5 | C8—C9—H9A | 120.5 |
| C2—C3—H3C | 109.5 | C9—C10—C11 | 121.67 (16) |
| H3A—C3—H3C | 109.5 | C9—C10—Br1 | 119.22 (13) |
| H3B—C3—H3C | 109.5 | C11—C10—Br1 | 119.11 (13) |
| N2—C4—C5 | 111.95 (15) | C10—C11—C12 | 119.01 (16) |
| N2—C4—H4A | 109.2 | C10—C11—H11A | 120.5 |
| C5—C4—H4A | 109.2 | C12—C11—H11A | 120.5 |
| N2—C4—H4B | 109.2 | C11—C12—C7 | 120.39 (16) |
| C5—C4—H4B | 109.2 | C11—C12—H12A | 119.8 |
| H4A—C4—H4B | 107.9 | C7—C12—H12A | 119.8 |
| C4—N2—C1—N1 | 177.55 (14) | N1—C6—C7—C8 | −173.35 (16) |
| C2—N2—C1—N1 | −0.4 (2) | O1—C6—C7—C12 | −167.57 (18) |
| C4—N2—C1—S1 | −0.6 (3) | N1—C6—C7—C12 | 9.7 (2) |
| C2—N2—C1—S1 | −178.51 (14) | C12—C7—C8—C9 | 0.3 (3) |
| C6—N1—C1—N2 | 87.2 (2) | C6—C7—C8—C9 | −176.75 (16) |
| C6—N1—C1—S1 | −94.57 (18) | C7—C8—C9—C10 | 0.4 (3) |
| C1—N2—C2—C3 | 82.9 (2) | C8—C9—C10—C11 | −0.5 (3) |
| C4—N2—C2—C3 | −95.12 (19) | C8—C9—C10—Br1 | 179.13 (13) |
| C1—N2—C4—C5 | 92.2 (2) | C9—C10—C11—C12 | 0.0 (3) |
| C2—N2—C4—C5 | −89.7 (2) | Br1—C10—C11—C12 | −179.64 (13) |
| C1—N1—C6—O1 | −5.7 (3) | C10—C11—C12—C7 | 0.7 (3) |
| C1—N1—C6—C7 | 176.95 (15) | C8—C7—C12—C11 | −0.8 (3) |
| O1—C6—C7—C8 | 9.3 (3) | C6—C7—C12—C11 | 176.06 (17) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O1i | 0.90 (1) | 2.02 (1) | 2.882 (2) | 162 (2) |
Symmetry codes: (i) x, −y+3/2, z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: AT2713).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Arslan, H., Flörke, U. & Külcü, N. (2003). Acta Cryst. E59, o641–o642.
- Arslan, H., Flörke, U. & Külcü, N. (2004). Turk. J. Chem.28, 673–678.
- Arslan, H., Flörke, U. & Külcü, N. (2007). Spectrochim. Acta A, 67, 936–943. [DOI] [PubMed]
- Arslan, H., Flörke, U., Külcü, N. & Binzet, G. (2007). Spectrochim. Acta A, 68, 1347–1355. [DOI] [PubMed]
- Arslan, H., Külcü, N. & Flörke, U. (2003). Transition Met. Chem.28, 816–819.
- Arslan, H., Külcü, N. & Flörke, U. (2006). Spectrochim. Acta A, 64, 1065–1071. [DOI] [PubMed]
- Bruker (2002). SMART, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- El Aamrani, F. Z., Kumar, A., Beyer, L., Cortina, J. L. & Sastre, A. M. (1998). Solvent Extr. Ion Exch.16, 1389–1406.
- El Aamrani, F. Z., Kumar, A., Cortina, J. L. & Sastre, A. M. (1999). Anal. Chim. Acta, 382, 205–231.
- Khawar Rauf, M., Bolte, M. & Anwar, S. (2009). Acta Cryst. E65, o249. [DOI] [PMC free article] [PubMed]
- Khawar Rauf, M., Bolte, M. & Badshah, A. (2009a). Acta Cryst. E65, o143. [DOI] [PMC free article] [PubMed]
- Khawar Rauf, M., Bolte, M. & Badshah, A. (2009b). Acta Cryst. E65, o240. [DOI] [PMC free article] [PubMed]
- Khawar Rauf, M., Bolte, M. & Rauf, A. (2009). Acta Cryst. E65, o234. [DOI] [PMC free article] [PubMed]
- Koch, K. R. (2001). Coord. Chem. Rev.216, 473-488.
- Özer, C. K., Arslan, H., VanDerveer, D. & Binzet, G. (2009). J. Coord. Chem.62, 266–276.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Weiqun, Z., Baolong, L., Liming, Z., Jiangang, D., Yong, Z., Lude, L. & Xujie, Y. (2004). J. Mol. Struct.690, 145–150.
- Yuan, Y. F., Wang, J. T., Gimeno, M. C., Laguna, A. & Jones, P. G. (2001). Inorg. Chim. Acta, 324, 309–317.
- Zhang, Y. M., Wei, T. B., Xian, L. & Gao, L. M. (2004). Phosphorus Sulfur Silicon Relat. Elem 179, 2007–2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536809003183/at2713sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809003183/at2713Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report