

Abstract
The title Schiff base compound, C16H14ClN, adopts E configurations with respect to both the C=C and C=N bonds. The dihedral angle between the two aromatic rings is 53.27 (4)°, while the plane through the C=C—C=N system is inclined at 9.06 (8)° to the benzene ring and 44.92 (5)° to the chlorobenzene ring. In the crystal structure, weak C—H⋯Cl and C—H⋯N hydrogen bonds stack the molecules down the a axis.
Related literature
For background to the use of Schiff bases as ligands see: Khalaji et al. (2008a
▶,b
▶); and for their bio-activity, see: Karthikeyan et al. (2006 ▶); Xiong et al. (2008 ▶); Sriram et al. (2006 ▶). For related structures, see: Khalaji et al. (2007 ▶); Khalaji & Harrison (2008 ▶); Khalaji et al. (2008c
▶). For reference structural data, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C16H14ClN
M r = 255.73
Orthorhombic,
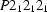
a = 7.2486 (10) Å
b = 11.6637 (17) Å
c = 15.598 (2) Å
V = 1318.7 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.27 mm−1
T = 89 (2) K
0.36 × 0.24 × 0.03 mm
Data collection
Bruker APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2006 ▶) T min = 0.841, T max = 0.992
21077 measured reflections
4077 independent reflections
3517 reflections with I > 2σ(I)
R int = 0.058
Refinement
R[F 2 > 2σ(F 2)] = 0.039
wR(F 2) = 0.109
S = 1.06
4077 reflections
164 parameters
H-atom parameters constrained
Δρmax = 0.30 e Å−3
Δρmin = −0.36 e Å−3
Absolute structure: Flack (1983 ▶), 1742 Friedel pairs
Flack parameter: 0.01 (6)
Data collection: APEX2 (Bruker, 2006 ▶); cell refinement: APEX2 and SAINT (Bruker, 2006 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶) and TITAN (Hunter & Simpson, 1999 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶) and Mercury (Macrae et al., 2006 ▶); software used to prepare material for publication: SHELXL97, enCIFer (Allen et al., 2004 ▶), PLATON (Spek, 2003 ▶) and publCIF (Westrip, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809001871/pk2147sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809001871/pk2147Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C7—H7⋯N1i | 0.95 | 2.67 | 3.524 (2) | 150 |
| C13—H13⋯Cl1ii | 0.95 | 2.92 | 3.7311 (17) | 144 |
Symmetry codes: (i) 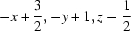 ; (ii)
; (ii) 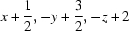 .
.
Acknowledgments
We thank the University of Otago for purchase of the diffractometer.
supplementary crystallographic information
Comment
Schiff-bases are well known chelating ligands in coordination chemistry (Khalaji et al., 2008a,b), and exhibit a wide range of biological activities (Karthikeyan et al., 2006) including anti-HIV activity (Xiong et al., 2008; Sriram et al., 2006). As a continuation of our work on the synthesis and structural characterization of Schiff-base compounds (Khalaji et al., 2007; Khalaji & Harrison, 2008; Khalaji et al., 2008c), we report here the structure of the title compound, C16H14NCl, (I), Fig 1.
The title Schiff-base compound, C16H14NCl, adopts E configurations with respect to both the C2=C4 and C1=N1 bonds. Bond lengths in the molecule are normal (Allen, et al., 1987) and similar to those found in related compounds (Khalaji et al., 2007; Khalaji & Harrison, 2008; Khalaji et al., 2008c). The dihedral angle between the two aromatic rings is 53.27 (4)° while the plane through the C2=C4–C1=N1 system is inclined at 9.06 (8)° to the C5···C10 ring and 44.92 (5)° to the C11···C16 ring.
In the crystal structure, weak C13—H13···Cl1 and C7—H7···N1 hydrogen bonds stack the molecules down the a axis.
Experimental
The title compound was prepared in 76% yield from 4-chloroaniline and α-methylcinnamaldehyde as reported elsewhere (Khalaji et al. 2007) and recrystallized from methanol.
Refinement
The H atom bound to N1 was located in a difference electron density map and refined freely with an isotropic displacement parameter. All other H-atoms were refined using a riding model with d(C—H) = 0.95 Å, Uiso= 1.2Ueq (C) for aromatic and 0.98 Å, Uiso = 1.5Ueq (C) for CH3 H atoms.
Figures
Fig. 1.

The structure of (I) with displacement ellipsoids for the non-hydrogen atoms drawn at the 50% probability level.
Fig. 2.

Crystal packing of (I) viewed down the a axis with hydrogen bonds drawn as dashed lines. H atoms not involved in hydrogen bonding have been omitted.
Crystal data
| C16H14ClN | F(000) = 536 |
| Mr = 255.73 | Dx = 1.288 Mg m−3 |
| Orthorhombic, P212121 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2ac 2ab | Cell parameters from 5396 reflections |
| a = 7.2486 (10) Å | θ = 2.6–28.8° |
| b = 11.6637 (17) Å | µ = 0.27 mm−1 |
| c = 15.598 (2) Å | T = 89 K |
| V = 1318.7 (3) Å3 | Rectangular plate, pale yellow |
| Z = 4 | 0.36 × 0.24 × 0.03 mm |
Data collection
| Bruker APEXII CCD area-detector diffractometer | 4077 independent reflections |
| Radiation source: fine-focus sealed tube | 3517 reflections with I > 2σ(I) |
| graphite | Rint = 0.058 |
| ω scans | θmax = 30.7°, θmin = 3.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2006) | h = −10→10 |
| Tmin = 0.841, Tmax = 0.992 | k = −16→16 |
| 21077 measured reflections | l = −21→22 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.039 | H-atom parameters constrained |
| wR(F2) = 0.109 | w = 1/[σ2(Fo2) + (0.0649P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.06 | (Δ/σ)max = 0.001 |
| 4077 reflections | Δρmax = 0.30 e Å−3 |
| 164 parameters | Δρmin = −0.36 e Å−3 |
| 0 restraints | Absolute structure: Flack (1983), 1742 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Flack parameter: 0.01 (6) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.5152 (2) | 0.52967 (12) | 0.68870 (9) | 0.0202 (3) | |
| C1 | 0.4972 (2) | 0.61272 (13) | 0.63497 (9) | 0.0183 (3) | |
| H1 | 0.4550 | 0.6848 | 0.6553 | 0.022* | |
| C2 | 0.5394 (2) | 0.60026 (13) | 0.54384 (9) | 0.0172 (3) | |
| C3 | 0.6040 (3) | 0.48397 (13) | 0.51348 (10) | 0.0224 (3) | |
| H3A | 0.5303 | 0.4601 | 0.4639 | 0.034* | |
| H3B | 0.5892 | 0.4280 | 0.5598 | 0.034* | |
| H3C | 0.7343 | 0.4884 | 0.4970 | 0.034* | |
| C4 | 0.5161 (2) | 0.69557 (13) | 0.49535 (10) | 0.0178 (3) | |
| H4 | 0.4725 | 0.7602 | 0.5264 | 0.021* | |
| C5 | 0.5458 (2) | 0.71712 (13) | 0.40389 (10) | 0.0166 (3) | |
| C6 | 0.6332 (3) | 0.64156 (14) | 0.34605 (10) | 0.0227 (3) | |
| H6 | 0.6771 | 0.5695 | 0.3659 | 0.027* | |
| C7 | 0.6560 (2) | 0.67119 (14) | 0.26031 (10) | 0.0228 (3) | |
| H7 | 0.7151 | 0.6192 | 0.2223 | 0.027* | |
| C8 | 0.5931 (2) | 0.77606 (14) | 0.22974 (10) | 0.0228 (3) | |
| H8 | 0.6072 | 0.7953 | 0.1709 | 0.027* | |
| C9 | 0.5089 (3) | 0.85283 (15) | 0.28615 (11) | 0.0243 (4) | |
| H9 | 0.4672 | 0.9252 | 0.2660 | 0.029* | |
| C10 | 0.4861 (2) | 0.82324 (14) | 0.37165 (10) | 0.0205 (3) | |
| H10 | 0.4286 | 0.8762 | 0.4094 | 0.025* | |
| C11 | 0.4819 (2) | 0.55276 (13) | 0.77639 (10) | 0.0182 (3) | |
| C12 | 0.5526 (2) | 0.64996 (13) | 0.81773 (10) | 0.0203 (3) | |
| H12 | 0.6233 | 0.7040 | 0.7862 | 0.024* | |
| C13 | 0.5200 (2) | 0.66792 (13) | 0.90454 (10) | 0.0207 (3) | |
| H13 | 0.5671 | 0.7342 | 0.9323 | 0.025* | |
| C14 | 0.4183 (2) | 0.58807 (13) | 0.94995 (10) | 0.0193 (3) | |
| Cl1 | 0.38085 (6) | 0.60910 (4) | 1.05938 (2) | 0.02595 (11) | |
| C15 | 0.3494 (2) | 0.48964 (13) | 0.91081 (11) | 0.0206 (3) | |
| H15 | 0.2800 | 0.4354 | 0.9428 | 0.025* | |
| C16 | 0.3841 (2) | 0.47222 (13) | 0.82400 (10) | 0.0199 (3) | |
| H16 | 0.3404 | 0.4045 | 0.7969 | 0.024* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0204 (7) | 0.0212 (6) | 0.0190 (6) | 0.0000 (5) | 0.0007 (5) | 0.0002 (5) |
| C1 | 0.0174 (7) | 0.0186 (6) | 0.0189 (7) | 0.0011 (6) | 0.0001 (6) | −0.0018 (6) |
| C2 | 0.0162 (7) | 0.0171 (6) | 0.0182 (7) | 0.0014 (6) | −0.0009 (5) | −0.0021 (6) |
| C3 | 0.0278 (9) | 0.0173 (6) | 0.0222 (8) | 0.0029 (7) | 0.0032 (7) | −0.0004 (6) |
| C4 | 0.0186 (8) | 0.0163 (7) | 0.0185 (7) | 0.0000 (6) | 0.0011 (6) | −0.0038 (5) |
| C5 | 0.0145 (7) | 0.0161 (6) | 0.0191 (7) | −0.0025 (6) | 0.0003 (6) | −0.0018 (5) |
| C6 | 0.0291 (9) | 0.0164 (6) | 0.0225 (8) | 0.0000 (7) | 0.0035 (7) | −0.0014 (5) |
| C7 | 0.0280 (9) | 0.0206 (7) | 0.0199 (7) | −0.0031 (7) | 0.0053 (6) | −0.0036 (6) |
| C8 | 0.0244 (9) | 0.0264 (7) | 0.0175 (7) | −0.0045 (7) | −0.0010 (6) | 0.0008 (6) |
| C9 | 0.0257 (9) | 0.0238 (7) | 0.0233 (8) | 0.0035 (7) | 0.0006 (7) | 0.0054 (6) |
| C10 | 0.0204 (8) | 0.0198 (7) | 0.0213 (7) | 0.0028 (6) | 0.0019 (6) | 0.0004 (6) |
| C11 | 0.0174 (7) | 0.0190 (7) | 0.0183 (7) | 0.0034 (6) | −0.0007 (6) | 0.0014 (6) |
| C12 | 0.0209 (8) | 0.0183 (6) | 0.0218 (7) | −0.0003 (6) | −0.0028 (6) | 0.0042 (6) |
| C13 | 0.0233 (8) | 0.0174 (7) | 0.0214 (7) | −0.0008 (6) | −0.0039 (6) | −0.0012 (6) |
| C14 | 0.0181 (7) | 0.0217 (7) | 0.0180 (7) | 0.0051 (6) | −0.0004 (6) | 0.0021 (6) |
| Cl1 | 0.0302 (2) | 0.02978 (19) | 0.01791 (17) | 0.00604 (18) | 0.00123 (15) | −0.00069 (15) |
| C15 | 0.0197 (8) | 0.0196 (7) | 0.0225 (7) | 0.0009 (6) | 0.0016 (6) | 0.0030 (6) |
| C16 | 0.0199 (8) | 0.0172 (6) | 0.0228 (7) | 0.0000 (6) | −0.0005 (6) | −0.0006 (6) |
Geometric parameters (Å, °)
| N1—C1 | 1.287 (2) | C8—C9 | 1.396 (2) |
| N1—C11 | 1.415 (2) | C8—H8 | 0.9500 |
| C1—C2 | 1.4612 (19) | C9—C10 | 1.387 (2) |
| C1—H1 | 0.9500 | C9—H9 | 0.9500 |
| C2—C4 | 1.355 (2) | C10—H10 | 0.9500 |
| C2—C3 | 1.511 (2) | C11—C16 | 1.392 (2) |
| C3—H3A | 0.9800 | C11—C12 | 1.401 (2) |
| C3—H3B | 0.9800 | C12—C13 | 1.390 (2) |
| C3—H3C | 0.9800 | C12—H12 | 0.9500 |
| C4—C5 | 1.464 (2) | C13—C14 | 1.383 (2) |
| C4—H4 | 0.9500 | C13—H13 | 0.9500 |
| C5—C10 | 1.404 (2) | C14—C15 | 1.393 (2) |
| C5—C6 | 1.412 (2) | C14—Cl1 | 1.7456 (16) |
| C6—C7 | 1.391 (2) | C15—C16 | 1.392 (2) |
| C6—H6 | 0.9500 | C15—H15 | 0.9500 |
| C7—C8 | 1.390 (2) | C16—H16 | 0.9500 |
| C7—H7 | 0.9500 | ||
| C1—N1—C11 | 117.96 (14) | C7—C8—H8 | 120.3 |
| N1—C1—C2 | 122.50 (14) | C9—C8—H8 | 120.3 |
| N1—C1—H1 | 118.8 | C10—C9—C8 | 119.89 (15) |
| C2—C1—H1 | 118.8 | C10—C9—H9 | 120.1 |
| C4—C2—C1 | 115.80 (14) | C8—C9—H9 | 120.1 |
| C4—C2—C3 | 126.88 (13) | C9—C10—C5 | 121.79 (15) |
| C1—C2—C3 | 117.32 (13) | C9—C10—H10 | 119.1 |
| C2—C3—H3A | 109.5 | C5—C10—H10 | 119.1 |
| C2—C3—H3B | 109.5 | C16—C11—C12 | 119.14 (15) |
| H3A—C3—H3B | 109.5 | C16—C11—N1 | 118.32 (14) |
| C2—C3—H3C | 109.5 | C12—C11—N1 | 122.44 (15) |
| H3A—C3—H3C | 109.5 | C13—C12—C11 | 120.52 (15) |
| H3B—C3—H3C | 109.5 | C13—C12—H12 | 119.7 |
| C2—C4—C5 | 131.77 (14) | C11—C12—H12 | 119.7 |
| C2—C4—H4 | 114.1 | C14—C13—C12 | 119.20 (15) |
| C5—C4—H4 | 114.1 | C14—C13—H13 | 120.4 |
| C10—C5—C6 | 117.38 (14) | C12—C13—H13 | 120.4 |
| C10—C5—C4 | 117.05 (14) | C13—C14—C15 | 121.45 (15) |
| C6—C5—C4 | 125.55 (14) | C13—C14—Cl1 | 119.28 (12) |
| C7—C6—C5 | 120.84 (15) | C15—C14—Cl1 | 119.25 (12) |
| C7—C6—H6 | 119.6 | C16—C15—C14 | 118.82 (15) |
| C5—C6—H6 | 119.6 | C16—C15—H15 | 120.6 |
| C8—C7—C6 | 120.63 (15) | C14—C15—H15 | 120.6 |
| C8—C7—H7 | 119.7 | C11—C16—C15 | 120.83 (14) |
| C6—C7—H7 | 119.7 | C11—C16—H16 | 119.6 |
| C7—C8—C9 | 119.46 (15) | C15—C16—H16 | 119.6 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C7—H7···N1i | 0.95 | 2.67 | 3.524 (2) | 150 |
| C13—H13···Cl1ii | 0.95 | 2.92 | 3.7311 (17) | 144 |
Symmetry codes: (i) −x+3/2, −y+1, z−1/2; (ii) x+1/2, −y+3/2, −z+2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: PK2147).
References
- Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004). J. Appl. Cryst.37, 335–338.
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bruker (2006). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Hunter, K. A. & Simpson, J. (1999). TITAN2000 University of Otago, New Zealand.
- Karthikeyan, M. S., Prasad, D. J., Poojary, B., Bhat, K. S., Holla, B. S. & Kumari, N. S. (2006). Bioorg. Med. Chem.14, 7482–7489. [DOI] [PubMed]
- Khalaji, A. D., Amirnasr, M. & Harrison, W. T. A. (2008a). Anal. Sci.24, x89–x90.
- Khalaji, A. D. & Harrison, W. T. A. (2008). Anal. Sci.24, x3–x4.
- Khalaji, A. D., Slawin, A. M. Z. & Woollins, J. D. (2007). Acta Cryst. E63, o4257.
- Khalaji, A. D., Welter, R., Amirnasr, M. & Barry, A. H. (2008b). Anal. Sci.24, x137–x138.
- Khalaji, A. D., Welter, R., Amirnasr, M. & Barry, A. H. (2008c). Anal. Sci.24, x139–x140.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst.39, 453–457.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
- Sriram, D., Yogeeswari, P., Sirisha, N. & Saraswat, V. (2006). Bioorg. Med. Chem. Lett.16, 2127–2129. [DOI] [PubMed]
- Westrip, S. P. (2009). publCIF In preparation.
- Xiong, Y.-Z., Chen, F.-E., Balzarini, J., Clercq, E. D. & Pannecouque, C. (2008). Eur. J. Med. Chem.43, 1230–1236. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809001871/pk2147sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809001871/pk2147Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report